Phosphoric and phosphoramidic acids as bifunctional catalysts for the ring-opening polymerization of ε-caprolactone: a combined experimental and theoretical study†
Damien
Delcroix
ab,
Aline
Couffin
ab,
Nicolas
Susperregui
cd,
Christophe
Navarro
e,
Laurent
Maron
*cd,
Blanca
Martin-Vaca
*ab and
Didier
Bourissou
*ab
aUniversity of Toulouse, UPS, LHFA, 118 route de Narbonne, F-31062, Toulouse, France. E-mail: dbouriss@chimie.ups-tlse.fr; bmv@chimie.ups-tlse.fr; Fax: +33 561558204; Tel: +33 561557737
bCNRS, LHFA UMR 5069, F-31062, Toulouse, France
cUniversity of Toulouse, INSA, UPS, LPCNO, 135 avenue de Rangueil, F-31077, Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr; Fax: +33 561556065; Tel: +33 561559664
dCNRS, LPCNO UMR 5215, F-31077, Toulouse, France
eArkema, Lacq Research Center, PO Box 34, 64170, Lacq, France
First published on 29th July 2011
Abstract
The ring-opening polymerization of ε-caprolactone catalyzed by the phosphoric acid (PhO)2P(O)OH (PA) or the phosphoramidic acid (PhO)2P(O)NH(SO2CF3) (PAA) has been investigated experimentally and computationally. The two catalysts promote polymerization under mild conditions, with PAA being slightly more active than PA. The controlled character of the polymerization has been examined thoroughly, and the polymerization conditions were optimized so as to afford well-defined polycaprolactones of molar masses up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and narrow molar mass distributions. Density Functional Theory calculations support a bifunctional mechanism for these organo-catalyzed ROPs, with participation of both the acidic proton and basic P
000 g mol−1 and narrow molar mass distributions. Density Functional Theory calculations support a bifunctional mechanism for these organo-catalyzed ROPs, with participation of both the acidic proton and basic P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (/SO) moiety of the phosphoric and phosphoramidic acids.
O (/SO) moiety of the phosphoric and phosphoramidic acids.
Introduction
Spectacular progress has been achieved in the past decade in organo-catalyzed ring-opening polymerization (ROP), enabling the preparation of polyesters under mild, metal-free conditions.1 A variety of organo-catalytic systems have been demonstrated to promote efficiently the controlled ROP of lactones and dilactones. In contrast with metal alkoxides that invariably promote ROPvia a coordination–insertion mechanism,2 various pathways can be distinguished with organo-catalysts.Hedrick et al. and Waymouth et al. reported the ability of Lewis/Brønsted bases such as DMAP (4-dimethylamino-pyridine),3 NHC (N-heterocyclic carbenes),4 and DBU (1,8-diazabicyclo[5.4.0]-undec-7-ene)5 to catalyze the ROP of lactidevia either nucleophilic activation of the monomer or basic activation of the initiating/propagating agent. Later on, catalytic systems combining a tertiary amine with a hydrogen bond donor were described, the most representative being TBD (1,5,7-triazabicyclo[4.4.0]dec-5-ene)6 and the association of a tertiary amine with a thiourea.7,8 With these bifunctional systems, efficient and selective ROP can be achieved under mild conditions thanks to the concomitant basic activation of the initiating/propagating agent and electrophilic activation of the monomer.
Over the last few years, Brønsted acid-catalyzed ROP of lactones and dilactones has also attracted renewed interest.9 Following the pioneering work of Endo et al.10 and Jerome et al.11 on the controlled polymerization of δ-valerolactone and ε-caprolactone using HCl as a catalyst, sulfonic acids were shown to promote efficiently the ROP of lactide, ε-caprolactone and β-butyrolactone under mild conditions.12,13 Most remarkably, methane sulfonic acid (MSA) was found to compete in activity with trifluoromethane sulfonic acid (HOTf), despite a difference in acidity of more than 10 pKa units.12b Also noteworthy is the bifunctional mode of action substantiated computationally for the sulfonic acids, with participation of both the acidic proton and the basic SO moiety.14
With the aim of further exploring the potential of Brønsted acid catalysis in ROP, we recently became interested in phosphoric and phosphoramidic acids.15 The structure of these acids is easily and highly tunable, which could be advantageous to optimize activity and selectivity. In addition, the propensity of phosphoric acids to act as bifunctional catalystsvia both their acidic proton and basic PO moiety has been recently exploited synthetically16 and substantiated theoretically17 in various coupling reactions. Such a bifunctional behavior clearly draws some parallel with what we proposed recently for sulfonic acids in ROP. Phosphoric and phosphoramidic acids were thus considered as good candidates to study and develop further hydrogen-bonding catalyzed ROP.
In this study, both the phosphoric acid (PhO)2P(O)OH (PA) and phosphoramidic acid (PhO)2P(O)NH(SO2CF3) (PAA) (Fig. 1) have been evaluated as catalysts for the ROP of ε-caprolactone (ε-CL). The introduction of the NH(SO2CF3) group at phosphorus was anticipated to increase activity towards ROP, by analogy with that usually encountered in coupling reactions.18 In addition, PAA was considered as a hybrid system between the phosphoric acid PA and bistriflamide Tf2NH, which has been used recently to promote the ROP of δ-valero and ε-caprolactone.19 Here we report a comparative experimental study of PA and PAA as catalysts for the ROP of ε-caprolactone. The polymerization conditions have been optimized to achieve good control and obtain well-defined PCL of molar masses up to 15000 g mol−1. In addition, the mode of action of the two catalysts has been explored computationally. These results as a whole, parallel and complete the experimental study reported by Kakuchi et al.20 during the preparation of this manuscript.
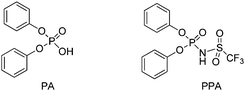 | ||
| Fig. 1 Structures of the phosphoric and phosphoramidic acids investigated as catalysts for the ROP of ε-caprolactone. | ||
Experimental
Materials
All reactions were performed under an inert atmosphere of argon, using standard Schlenk techniques. Solvents, toluene (>99.9%) and dichloromethane (>99.95%), were dried prior to use with a Braun solvent-purificator system. ε-Caprolactone (99.5%, Aldrich) was purified by distillation over CaH2 and stored under argon. The phosphoric acid (PhO)2P(O)OH (99%, Aldrich) was dried under vacuum in the presence of phosphorus pentoxide and stored in a glovebox. n-Pentanol (99%) was dried over sodium and distilled before use. The phosphoramidic acid (PhO)2P(O)NH(SO2CF3) was prepared following published methods.18c,21Characterization of the polymers
000000 g mol−1) and two STYRAGEL columns (HR1, 100–5000 g mol−1 and HR 4E, 50–100000 g mol−1) were used. Calibrations were performed using polystyrene standards (400–100000 g mol−1) and a correction factor of 0.56 was applied.22
:1:1 respectively. The mixed solution was hand-spotted on a stainless steel MALDI target and left to dry. The spectrum was recorded in the reflectron mode. Baseline corrections and data analyses were performed using MassLynx version 4.1 and Polymerix Software, Sierra Analytics, Version 2.0.0.
Methods
Computational details
Calculations were carried out with the Gaussian 0323 suite of programs at the DFT level of theory using the hybrid functional B3PW91.24Sulfur, nitrogen, carbon, oxygen and hydrogen atoms were described with a 6-31G(d,p) double-ζ basis set.25 Phosphorus and fluorine atoms were treated with a Stuttgart–Dresden pseudopotential in combination with its adapted basis set,26 augmented by a set of polarization function d.27 Geometry optimizations were carried out without any symmetry restrictions, and the nature of the extrema was verified with analytical frequency calculations. The intrinsic reaction coordinate was followed using the IRC technique for all located transition states. The reference energy has been set to zero for the most stable ternary adduct of reactants.Results and discussion
Experimental study
The activities of the phosphoric acid (PhO)2P(O)OH (PA) and phosphoramidic acid (PhO)2P(O)NH(SO2CF3) (PAA) towards the ROP of ε-CL were first evaluated under the following conditions (Scheme 1): 40 equivalents of monomer were reacted with 1 equivalent of n-pentanol (as initiator) in toluene solution ([ε-CL]0 = 0.9 mol L−1) at 30 °C in the presence of 1 equivalent of catalyst (Runs 3 and 7, Table 1). Monomer conversion >98% was achieved in 4 h with PA and 1.5 h with PAA. The replacement of the OH for a NHSO2CF3 group at phosphorus thus induces a noticeable increase in activity, in line with the higher acidity of PAA (pKa ≈ −3) over PA (pKa ≈ 2).18 Both acids appear significantly more active than HCl·OEt2 (for which 19 h were needed to achieve complete conversion of 40 equiv. in the same conditions), and PAA reproduces the best results obtained with sulfonic acids, namely HOTf and AMS (for which complete conversion of 40 equiv. of ε-CL was observed after 1.5 h).12b| Run | [M]/[I] | Timeb/h | M n c/g mol−1 | M n(th) d/g mol−1 | M w/Mnc |
|---|---|---|---|---|---|
| a Polymerizations of 0.9 mol L−1 solutions of ε-caprolactone carried out at 30 °C in toluene. b Polymerization time necessary to achieve monomer conversion >98% according to 1H NMR spectroscopy. c Obtained from size exclusion chromatography analysis in tetrahydrofuran using polystyrene standards and a correction factor of 0.56.22 d Calculated from the molar mass of ε-caprolactone (114 g mol−1) × the monomer/initiator ratio plus the molar mass of the initiator (88 g mol−1). | |||||
| 1 | 10 | 1 | 1190 | 1230 | 1.15 |
| 2 | 20 | 2 | 1990 | 2370 | 1.11 |
| 3 | 40 | 4 | 4320 | 4650 | 1.09 |
| 4 | 80 | 9 | 5860 | 9210 | 1.07 |
| 5 | 10 | 0.3 | 1200 | 1230 | 1.14 |
| 6 | 20 | 0.75 | 2230 | 2370 | 1.07 |
| 7 | 40 | 1.5 | 4600 | 4650 | 1.06 |
| 8 | 80 | 5.5 | 8000 | 9210 | 1.07 |
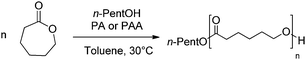 | ||
| Scheme 1 ROP of ε-CL initiated by n-PentOH and catalyzed by PA or PAA. | ||
According to SEC analysis, the number-average molar mass Mn of the obtained PCL (4320–4600 g mol−1) fits nicely with the targeted one (4648 g mol−1) and the molar mass distribution is narrow (Mw/Mn = 1.06–1.09). Moreover, 1H NMR analyses (Fig. 2a for PAA, Fig. S1a† for PA) show end-group fidelity with quantitative incorporation of the protic initiator in the polymer chains. Calibration of the terminal CH3 of the pentanoate moiety at 3H (h) leads to an integration of the polymer signals in perfect agreement with the initial [monomer]/[initiator] ratio (DPNMR = 40), and the signal corresponding to the –CH2OH chain-end integrates for 2H (a), indicating that all the polymer chains have been actually initiated by n-pentanol. Consistently, MALDI-TOF MS analyses (Fig. 2b for PAA, Fig. S1b† for PA) depict in both cases a very major population of PCL with molecular formula of molar mass M = n × 114(Mε-CL) + 88(MPentOH) + 23(MNa+) g mol−1, corresponding to linear polymer chains initiated by n-pentanol. A small amount of low molar mass PCL featuring carboxylic acid chain ends is also identified in the spectra.
![(a) 1H NMR spectrum (CDCl3, 300 MHz) and (b) MALDI-TOF MS (region m/z 1000 to 6000 g mol−1) of a PCL ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 30 °C).](/image/article/2011/PY/c1py00210d/c1py00210d-f2.gif) | ||
| Fig. 2 (a) 1H NMR spectrum (CDCl3, 300 MHz) and (b) MALDI-TOF MS (region m/z 1000 to 6000 g mol−1) of a PCL ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 30 °C). | ||
The course of the polymerization was monitored by SEC and NMR. For the two catalysts, a linear relationship was found between the molar mass of the PCL and the monomer conversion (Fig. 3a for the polymerisation of 40 equiv of ε-CL with PAA, Fig. S2a† with PA), indicating that all the polymer chains grow at the same relative rate. This is consistent with the narrow molar mass distributions of the obtained PCL and suggests controlled character for the polymerization. The plot of ln([ε-CL]0/[ε-CL]) versus time shows an upward curvature (Fig. 3b for PAA, Fig. S2b† for PA). Such a deviation from the expected first-order dependence on monomer concentration has already been noticed by Kubisa and Basko13 for the polymerization of ε-CL with MSA as catalyst. When the basicity of the cyclic ester of the monomer is higher than that of the acyclic ester within the polymer,28 the ratio between the activated monomer concentration and the monomer concentration increases as the monomer is consumed, resulting in an apparent speed up of the polymerization.13
![(a) Plot of Mn(SEC) (estimated by SEC applying a correction factor of 0.56) vs.ε-CL conversion ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 30 °C). (b) Semi-logarithmic kinetic plot ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 25 °C).](/image/article/2011/PY/c1py00210d/c1py00210d-f3.gif) | ||
| Fig. 3 (a) Plot of Mn(SEC) (estimated by SEC applying a correction factor of 0.56) vs.ε-CL conversion ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 30 °C). (b) Semi-logarithmic kinetic plot ([ε-CL]0/[n-PentOH]0/[PAA] = 40/1/1, toluene, 25 °C). | ||
In order to evaluate and compare further the behavior of PA and PAA, the initial monomer to initiator ratio was varied from 10 to 80 while keeping a catalyst to initiator ratio of 1 (Table 1, Fig. 4). PCL of increasing Mn(SEC) (up to 8000 g mol−1) and narrow distributions (<1.15) were obtained. For M/I ratios from 10 to 40, the Mn(SEC) values increase linearly and match those targeted for the two acids. However, some deviation was observed for M/I = 80. The Mn(SEC) value obtained with PA (5860 g mol−1) is substantially lower than that expected (Mn(th) = 9210 g mol−1), indicating the occurrence of undesirable side-reactions. Kakuchi made a similar observation in his recent report,20 and PCL of relatively high molar mass and narrow distribution could only be obtained by quenching the polymerization at 60–80% monomer conversion. With PAA, the 80 equivalents of ε-CL are polymerized within 5.5 h (compared to 9 h with PA) and the Mn(SEC) value of the resulting PCL (8000 g mol−1) approaches that targeted, suggesting a better control of the polymerization.
![Plot of Mn(SEC)vs.ε-CL to initiator ratio (□ for PA and ◆ for PAA, toluene, 30 °C, [catalyst]/[initiator] = 1, [ε-CL]0 = 0.9 mol L−1). Broken line shows Mn(th) values calculated from the molar mass of ε-CL (114 g mol−1) × [ε-CL]0/[n-PentOH] plus the molar mass of n-PentOH (88 g mol−1).](/image/article/2011/PY/c1py00210d/c1py00210d-f4.gif) | ||
| Fig. 4 Plot of Mn(SEC)vs.ε-CL to initiator ratio (□ for PA and ◆ for PAA, toluene, 30 °C, [catalyst]/[initiator] = 1, [ε-CL]0 = 0.9 mol L−1). Broken line shows Mn(th) values calculated from the molar mass of ε-CL (114 g mol−1) × [ε-CL]0/[n-PentOH] plus the molar mass of n-PentOH (88 g mol−1). | ||
For both PA and PAA, the MALDI-TOF MS spectrum of the PCL prepared with an initial M/I ratio of 80 depicts, in addition to the expected major population for n-PentO–(ε-CL)n–OH chains, a minor population corresponding to polymer chains featuring carboxylic acid instead of pentyl ester end-groups (Fig. S4†). We recently made similar observations in the acid-catalyzed ROP of trimethylene carbonate, and by analogy, we propose here also some competition between activated-monomer and active-chain-end pathways. Note that the undesirable population is more abundant with PA than with PAA, in line with the lower Mn value obtained with the phosphoric over the phosphoramidic acid.21,29
From these results, it is clear that PAA slightly surpasses PA in terms of both activity and polymerization control. With the aim of improving further the polymerization control for monomer to initiator ratios ≥80, we then evaluated the influence of the polymerization conditions using PAA as catalyst and Run 8 as reference (Table 2). Increasing the catalyst to initiator ratio from 1 to 3 (Run 9) did not affect significantly the characteristics of the obtained PCL (identical Mn and Mw/Mn values), but markedly reduced the reaction time (from 5.5 h to only 1.5 h). This increase in polymerization rate parallels that observed with methanesulfonic acid MSA, but contrasts with the deceleration observed with HOTf.12b A clear improvement was achieved by increasing the initial monomer concentration from 0.9 to 2.7 mol L−1 (Run 10). The Mn(SEC) value increases substantially and now deviates from the targeted value by less than 4%. Working at high monomer concentration inherently results in an increase in viscosity of the reaction media. To circumvent this issue, the polymerization temperature was raised to 60 °C. For a catalyst to initiator ratio of 1 (Run 11), this results in slightly lower polymerization control, but when the PAA loading was increased (Run 12), the monomer was polymerized within less than 1 h and well-controlled and narrowly distributed PCL was obtained (Mn(SEC) = 9200 g mol−1 and Mw/Mn = 1.10). Increasing the monomer concentration, polymerization temperature and catalyst loading thus allows us to drive the polymerization to completion while keeping good polymerization control (Fig. 5). Under these optimized conditions, PCLs of higher molar mass, up to 15000 g mol−1, were prepared (Runs 13 and 14). Their molar masses Mn are close to those targeted and their distributions remain narrow (<1.22), indicating minimal extent of undesirable side-reactions. However there appears to be a limit to the molar mass that can be achieved. Under these conditions, increasing the monomer to initiator feed to 230 only led to a small increase of the molar mass (Mn(SEC) = 15700 g mol−1 and Mw/Mn = 1.13).
| Run | [M]/[I] | [Cat]/[I] | [M]0/mol L−1 | T/°C | Timeb/h | M n c/g mol−1 | M n(th) d/g mol−1 | M w/Mnc |
|---|---|---|---|---|---|---|---|---|
| a Reactions carried out in toluene. b Polymerization time necessary to achieve monomer conversion >98% according to 1H NMR spectroscopy. c Obtained from size exclusion chromatography analysis in tetrahydrofuran, using polystyrene standards and a correction factor of 0.56.22 d Calculated from the molar mass of ε-CL (114 g mol−1) × the monomer/initiator ratio plus the molar mass of the initiator (88 g mol−1). | ||||||||
| 8 | 80 | 1 | 0.9 | 30 | 5.5 | 8000 | 9210 | 1.07 |
| 9 | 80 | 3 | 0.9 | 30 | 1.5 | 8100 | 9210 | 1.05 |
| 10 | 80 | 1 | 2.7 | 30 | 5 | 8900 | 9210 | 1.08 |
| 11 | 80 | 1 | 2.7 | 60 | 1.75 | 7600 | 9210 | 1.19 |
| 12 | 80 | 3 | 2.7 | 60 | 0.75 | 9200 | 9210 | 1.10 |
| 13 | 120 | 3 | 2.7 | 60 | 1.75 | 11650 |
13770 |
1.15 |
| 14 | 160 | 3 | 2.7 | 60 | 2 | 15100 |
18330 |
1.22 |
![Plot of Mn (estimated by SEC and a correction factor of 0.56) vs.ε-CL to initiator ratio with PAA as catalyst (□ for [ε-CL]0 = 2.7 mol L−1, 60 °C, [catalyst]/[initiator] = 3 and ◆ for [ε-CL]0 = 0.9 mol L−1; 30 °C, [catalyst]/[initiator] = 1). Broken line shows Mn(th) values calculated from the molar mass of ε-CL (114 g mol−1) × [ε-CL]0/[n-PentOH] plus the molar mass of n-PentOH (88 g mol−1).](/image/article/2011/PY/c1py00210d/c1py00210d-f5.gif) | ||
| Fig. 5 Plot of Mn (estimated by SEC and a correction factor of 0.56) vs.ε-CL to initiator ratio with PAA as catalyst (□ for [ε-CL]0 = 2.7 mol L−1, 60 °C, [catalyst]/[initiator] = 3 and ◆ for [ε-CL]0 = 0.9 mol L−1; 30 °C, [catalyst]/[initiator] = 1). Broken line shows Mn(th) values calculated from the molar mass of ε-CL (114 g mol−1) × [ε-CL]0/[n-PentOH] plus the molar mass of n-PentOH (88 g mol−1). | ||
In parallel to these experimental studies, theoretical calculations have been performed to shed light into the mode of action of phosphoric acid PA and phosphoramidic acid PAA in the ROP of ε-CL.
Computational study
Over the last few years, computational studies have been increasingly used to gain insight into the mechanism of organo-catalyzed ROP.14,30–33 Accordingly, the propensity of TBD,30thiourea/sparteine derivatives,31 and even DMAP32 and sulfonic acids14 to act as bifunctional catalysts has been pointed out. Here, the precise role of PA and PAA has been theoretically investigated. Recently, bifunctional mechanisms involving both the acidic proton and the basic PO moiety of phosphoric acids have been substantiated computationally for various coupling reactions,17 but not for ring-opening polymerization, and to the best of our knowledge, the mode of action of hybrid catalysts such as PAA has not been investigated so far. A detailed DFT (density functional theory) study has been carried out on the model reaction of ring-opening of ε-CL with methanol (Scheme 2). Different modes of action have been considered for PA and PAA, but only the bifunctional pathways involving the acidic proton and the basic PO (or SO) moiety will be discussed here since they were found to be significantly more favorable, with transition states lying more than 10 kcal mol−1 lower in energy than the alternative routes.21
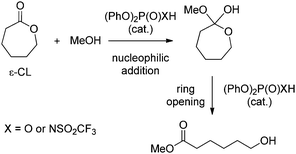 | ||
| Scheme 2 Model reaction used to study the role of phosphoric and phosphoramidic acid catalysts in the ROP of ε-caprolactone. | ||
O moiety of the acid (O2⋯H distance of 1.52 Å vs. 1.02 Å for the O1⋯H bond in methanol). Overall, the phosphoric acid acts as a proton shuttle via an eight-membered ring structure, similar to that encountered with sulfonic acids.14 It is worth noting that TS11 is lower in energy by at least 14 kcal mol−1 than the transition states located for alternative pathways.21 This underlines the importance of the cooperative activation of the ε-CL and initiating/propagating alcohol (methanol here). The formation of the tetrahedral intermediate is predicted to be endothermic by 9.6 kcal mol−1 and to require an activation barrier of 17.8 kcal mol−1.
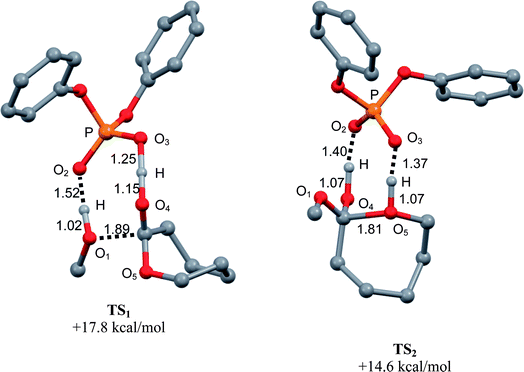 | ||
| Fig. 6 Optimized geometries of the transition states associated with the nucleophilic addition (TS11) and ring-opening (TS22) steps upon bifunctional activation by the phosphoric acid PA. Selected bond distances are given in Å, Gibbs energies relative to the most stable ternary adduct of reactants. | ||
The ring-opening then proceeds via transition state TS22 (Fig. 6, right). The cleavage of the endocyclic C–O bond is concomitant with proton transfer from the acid to the endocyclic oxygen (O5⋯H distance of 1.07 Å vs. 1.37 Å for the O3⋯H distance). In addition, the basic PO group is engaged in a weak hydrogen bond with the hydroxyl group (O2⋯H distance of 1.40 Å vs. 1.07 Å in the hydroxyl group), leading again to an eight-membered ring structure. The ring-opening viaTS22 requires a low activation barrier (5 kcal mol−1), and the bifunctional route is here also substantially more favorable than the alternative pathways (that involve transition states at least 11 kcal mol−1 higher in energy).21
O or the SO moiety have been considered. As for PA, the nucleophilic addition was found to proceed via concomitant activation of the ε-CL and methanol (eight-membered ring transition states TS33 and TS′33, Fig. 7, left). The proton transfer from the catalyst to the CO group of ε-CL is noticeably more advanced in TS33 and TS′33 (N⋯H distances of 1.45–1.63 Å vs. O4⋯H distance of 1.05–1.07 Å) than in TS11, in line with the slightly higher acidity of phosphoramidic acid PAA over the phosphoric acid PA.18 In the mean time, the hydrogen of methanol forms a weak hydrogen bond with the PO moiety in TS33 (O2⋯H distance of 1.60 Å vs. 1.00 Å for the O1⋯H bond in methanol) and with the SO moiety in TS′33 (O2⋯H distance of 1.53 Å vs. 1.05 Å for the O1⋯H bond in methanol). Overall, PAA behaves as a proton shuttle, with slightly higher acid activation of ε-CL and weaker basic activation of methanol than those encountered with PA. The corresponding activation barriers (20.6 kcal mol−1 for TS33 and 24.6 kcal mol−1 for TS′33) are close to each other and again, alternative pathways require much higher energy (at least 39.8 kcal mol−1).
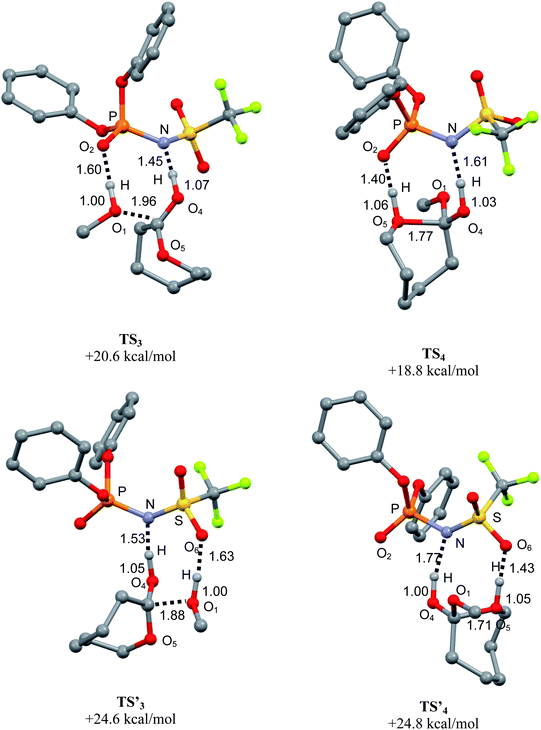 | ||
| Fig. 7 Optimized geometries of the transition states associated with the nucleophilic addition (TS′33) and ring-opening (TS′44) steps upon bifunctional activation by the phosphoramidic acid PAA (top: participation of the PO moiety, bottom: participation of the SO moiety). Selected bond distances are given in Å, Gibbs energies relative to the most stable ternary adduct of reactants. | ||
For the subsequent ring-opening step, two transition states TS44 and TS′44 involving either the PO or the SO moiety of PAA were located as well (Fig. 7, right). In both cases, the cleavage of the endocyclic C–O bond is advanced (distances of 1.71–1.77 Å) and the phosphoramidic acid acts as a proton shuttle via double hydrogen bonding. From an energetic point of view, the PAA adducts of the tetrahedral intermediate are located 15.4–20.4 kcal mol−1 above the reactants, and their ring-opening viaTS44 and TS′44 requires low activation barriers (3.4–4.4 kcal mol−1).
Despite the dissymmetry of the phosphoramidic acid catalyst PAA, the reaction profiles for the two bifunctional routes thus appear rather similar. For the nucleophilic addition as well as ring-opening, the participation of the PO was predicted to be slightly more favorable than that of the SO moiety, but both routes are likely to intervene. Given the precision of the method used and the approximations made, these computational studies cannot account for the slightly higher activity of PAA over PA, but they clearly emphasize the importance of cooperative activation, both systems typically behaving as proton shuttles. The ability of the trifluoromethanesulfonyl group of PAA to compete with the phosphoryl group for the basic activation of protic substrates is also pointed out.
Conclusions
The phosphoric acid PA and phosphoramidic acid PAA have been shown to be efficient catalysts for the ROP of ε-CL under mild conditions. The polymerization conditions have been optimized in order to gain access to well-defined PCL of molar mass up to 15000 g mol−1 and narrow distributions (<1.22). The mechanism of these Brønsted acid-catalyzed ROPs has been thoroughly investigated computationally. Accordingly, bifunctional pathways are significantly more favorable, with PA and PAA acting as proton shuttles in both the nucleophilic addition and ring-opening steps. These results expand the variety of Brønsted acids capable of promoting the ROP of ε-CL and further substantiate the role and interest of multiple hydrogen-bonding in ROP catalysis. Future work seeks to exploit the structural modularity of phosphorus-based acids to increase their catalytic activity.
Acknowledgements
We are grateful to the CNRS, the ANR (ANR-08-CP2D-01-BIOPOLYCAT) and the University Paul Sabatier (France) for financial support of this work. CINES and CALMIP are acknowledged for grant of computing time. L. M. thanks the Institut Universitaire de France.Notes and references
- (a) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS; (b) D. Bourissou, S. Moebs-Sanchez and B. Martin Vaca, C. R. Chim., 2007, 10, 775 CrossRef CAS; (c) A. P. Dove, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 357 Search PubMed; (d) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS.
- (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, in The Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, p. 255 Search PubMed; (d) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712 CrossRef CAS.
- (a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018 CrossRef CAS; (b) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, H. Li, E. C. Hagberg, R. M. Waymouth and J. L. Hedrick, in N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2006, p. 275 Search PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 Search PubMed.
- (a) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 Search PubMed; (b) A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749 Search PubMed.
- (a) A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798 Search PubMed; (b) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863 Search PubMed.
- For other hydrogen donor derivatives combined with a tertiary amine see: (a) S. Koeller, J. Kadota, A. Deffieux, F. Peruch, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, J. Am. Chem. Soc., 2009, 131, 15088 Search PubMed; (b) S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, Chem.–Eur. J., 2010, 16, 4196 Search PubMed; (c) A. Alba, A. Schopp, A. P. De Sousa Delgado, R. Cherif-Cheikh, B. Martin-Vaca and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 959 Search PubMed; (d) J. M. Becker, S. Tempelaar, M. J. Stanford, R. J. Pounder, J. A. Covington and A. P. Dove, Chem.–Eur. J., 2010, 16, 6099 Search PubMed.
- For early work in the absence of initiator, see: (a) H. R. Kricheldorf and R. Dunsing, Makromol. Chem., 1986, 187, 1611 Search PubMed; (b) H. R. Kricheldorf and I. Kreiser, Makromol. Chem., 1987, 188, 1861 Search PubMed.
- (a) Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316 Search PubMed; (b) F. Sanda, H. Sanada, Y. Shibasaki and T. Endo, Macromolecules, 2002, 33, 680 Search PubMed.
- X. Lou, C. Detrembleur and R. Jérôme, Macromolecules, 2002, 35, 1190 Search PubMed.
- (a) D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993 Search PubMed; (b) S. Gazeau-Bureau, D. Delcroix, B. Martin-Vaca, D. Bourissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782 Search PubMed; (c) D. Delcroix, B. Martin-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828 Search PubMed.
- (a) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7071 Search PubMed; (b) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3090 Search PubMed; (c) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7919 Search PubMed; (d) M. Basko and P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 265 Search PubMed.
- N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581 Search PubMed.
- D. Bourissou, B. Martin-Vaca, D. Delcroix and C. Navarro, Patent FR0958742, priority date: December, 8th 2009.
- (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 Search PubMed; (b) M. Terada, Chem. Commun., 2008, 4097 Search PubMed; (c) M. Terada, Synthesis, 2010, 1929 Search PubMed.
- (a) L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2008, 130, 8741 Search PubMed; (b) T. Marcelli, P. Hammar and F. Himo, Chem.–Eur. J., 2008, 14, 8562 Search PubMed; (c) L. Simón and J. M. Goodman, J. Am. Chem. Soc., 2009, 131, 4070 Search PubMed; (d) X. H. Chen, Q. Wei, S. W. Luo and L. Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819 Search PubMed; (e) N. Li, X. H. Chen, J. Song, S. W. Luo, W. Fan and L. Z. Gong, J. Am. Chem. Soc., 2009, 131, 15301 Search PubMed; (f) M. Yamanaka and T. Hirata, J. Org. Chem., 2009, 74, 3266 Search PubMed; (g) L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589 Search PubMed; (h) S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Chem.–Eur. J., 2010, 16, 3021 Search PubMed.
- (a) D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626 Search PubMed; (b) M. Rueping, W. Ieawsuwan, A. P. Antonchick and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2007, 46, 2097 Search PubMed; (c) M. Rueping, B. J. Nachtsheim, R. M. Koenigs and W. Ieawsuwan, Chem.–Eur. J., 2010, 16, 13116 Search PubMed.
- (a) R. Kakuchi, Y. Tsuji, K. Chiba, K. Fuchise, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7090 Search PubMed; (b) M. Oshimura, T. Tang and A. Takasu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1210 Search PubMed.
- The phosphoric acid (PhO)2P(O)OH has been evaluated in the ROP of ε-caprolactone and δ-valerolactone: K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999 Search PubMed.
- See ESI†.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E. 01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 Search PubMed; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244 Search PubMed.
- W. J. Hehre, R. Ditchfie and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 Search PubMed.
- A. Bergner, M. Dolg, W. Kuechle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431 Search PubMed.
- L. Maron and C. Teichteil, Chem. Phys., 1998, 237, 105 Search PubMed.
- Higher proton affinities have been proposed for esters in E conformation (as in medium-sized lactones) vs. Z conformation (as in the polymer chain), see: (a) K. B. Wiberg and R. F. Waldron, J. Am. Chem. Soc., 1991, 113, 7705 Search PubMed; (b) G. Bouchou, D. Drancourt and D. Leblanc, New J. Chem., 1995, 19, 1243 Search PubMed.
- Competitive initiation with adventitious water may also lead to PCL featuring carboxylic acid chain ends. This may also contribute to the deviation from the expected molar mass, but probably less than the AM/ACE competition. Indeed, no significant modification was observed upon increasing the monomer purity, and under similar conditions, the deviation of the molar mass was found to critically depend on the polymerization rate (PA versusPAA).
- (a) L. Simón and J. M. Goodman, J. Org. Chem., 2007, 72, 9656 Search PubMed; (b) A. Chuma, H. W. Horn, W. C. Swope, R. C. Pratt, L. Zhang, B. G. G. Lohmeijer, C. G. Wade, R. M. Waymouth, J. L. Hedrick and J. E. Rice, J. Am. Chem. Soc., 2008, 130, 6749 Search PubMed.
- R.-X. Zhu, R.-X. Wang, D.-J. Zhang and C.-B. Liu, Aust. J. Chem., 2009, 62, 157 Search PubMed.
- C. Bonduelle, B. Martín-Vaca, F. P. Cossío and D. Bourissou, Chem.–Eur. J., 2008, 14, 5304 Search PubMed.
- For a theoretical mechanistic study of the NHC-catalyzed ROP of lactide, see: C. Lai, H. M. Lee and C. Hu, Tetrahedron Lett., 2005, 46, 6265 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of PAA; 1H NMR and MALDI-TOF MS spectra of PCL; coordinates of all stationary point structures in xyz format. See DOI: 10.1039/c1py00210d |
| This journal is © The Royal Society of Chemistry 2011 |