Modular synthesis of poly(perylene bisimides) using click chemistry: a comparative study†
Andreas S.
Lang
and
Mukundan
Thelakkat
*
Applied Functional Polymers, Department of Macromolecular Chemistry I, University of Bayreuth, Universitätsstr. 30, 95444, Bayreuth, Germany. E-mail: mukundan.thelakkat@uni-bayreuth.de; Tel: +49 921 553108
First published on 13th July 2011
Abstract
A systematic study of the influence of the chemical substitution pattern of side chain polymers carrying perylene bisimide (PBI) pendants is presented. To achieve a high comparability, the modular approach of copper-catalyzed azide–alkyne cycloaddition (CuAAC, “click” chemistry) was chosen to attach six differently substituted PBI azide moieties to a poly(propargyloxystyrene) backbone to obtain poly(perylene bisimides), PPBIs. The N, N′ substituted PPBIs differ structurally in two ways. On the one hand, the N-swallow-tail substituent was varied between hydrophilic oligoethyleneglycol and hydrophobic alkyl groups. On the other hand, the length of the N′-alkyl spacer, connecting PBI moiety and polymer backbone, was varied from (CH2)6 to (CH2)8 and to (CH2)11. The polymer analogous reactions between PBI-azides and poly(propargyloxystyrene) were monitored by 1H NMR and were found to be nearly quantitative in all cases. The resulting PPBIs exhibit Mn around 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and narrow PDIs of 1.1. Structure–property relationships of all polymers were elucidated by studying their thermal behaviour using DSC and their structural properties with XRD measurements.
000 g mol−1 and narrow PDIs of 1.1. Structure–property relationships of all polymers were elucidated by studying their thermal behaviour using DSC and their structural properties with XRD measurements.
Introduction
Recent synthetic developments in polymer science such as the combination of controlled radical polymerization (CRP) and copper-catalyzed azide–alkyne cycloaddition (CuAAC, “click” chemistry)1 have given scientists an elegant tool to create polymers in a diversity never known before. Generally, in this concept, an alkyne or azide functionalized monomer is polymerized using CRP followed by a subsequent polymer analogous copper-catalyzed “click” reaction with a respective azide or alkyne derivative. This allows the synthesis of manifold complex polymers out of functional “building bricks” and a single easy-to-obtain scaffold polymer. By this modular approach, the influence of the substitution pattern in a series of “click” polymers can be studied, without causing any differences in polymer length and molecular weight distribution. Thus, structure–property relationships can be determined more reliably. Narrow molecular weight distributions for such scaffold polymers can be accomplished by choosing one of the three CRP methods, atom transfer radical polymerization (ATRP),2,3 reversible addition–fragmentation chain transfer polymerization (RAFT)4,5 or nitroxide mediated radical polymerization (NMRP).6–8 For example, Haddleton et al. used these principles to build up libraries of glycopolymers by the combination of “click” chemistry and ATRP.3 Zhang et al. determined the influence of different spacer lengths of azobenzene-containing side-chain liquid crystalline polymers which were synthesized by “click” chemistry and free radical polymerization.9 In a previous work, we comparatively studied poly(perylene bisimides) (PPBI) prepared from direct NMRP of perylene bisimide acrylates and those synthesized by “click” chemistry between an azide functionalized PBI and poly(propargyl acrylate).6PBI derivatives represent an important class of light absorbing n-type semiconductor materials,10,11 exhibiting a relatively high electron affinity among large-band gap materials.12 Thus, PBIs are promising materials for the application in organic electronic devices.13–15 Polymers with PBI as pendant groups feature additional advantages of good film forming properties.16 Such polymers were already incorporated as one block in semiconductor donor–acceptor block copolymers for photovoltaic application.13,14,17–21 Nevertheless, the polymerization of PBI monomers by direct NMRP is not ideal and is limited to certain substituted monomers because this polymerization works only in very concentrated solution.6 The choice of chemical substitution of the pendant PBIs is thereby limited by the solubility of the monomers and the corresponding polymers under polymerization conditions. Also, transfer reactions were observed in the NMRP of PBI acrylates which lead to broad molecular weight distribution in the homopolymerization and to the formation of homopolymer in the synthesis of block copolymers. These problems can be overcome by a combination of “click” chemistry and NMRP in which case differently substituted PBIs can be attached to a scaffold polymer very easily.
Here, we report the synthesis of six differently substituted PBI polymers, utilizing “click” chemistry as synthetic route. Two types of pendant PBIs were attached; those carrying oligoethylene glycol (OEG) and alkyl swallow-tail substituents. Each type of PPBI was synthesized with three different alkyl spacers of (CH2)6, (CH2)8 and (CH2)11. We studied the polymers regarding thermal, structural and optical properties. Thus, the polymers carrying hydrophilic OEG swallow-tails were systematically compared with polymers with PBIs bearing alkyl swallow-tails.
Experimental section
General
The free nitroxide N-tert-butyl-α-isopropyl-α-phenylnitroxide 3 was synthesized according to published procedures.22N-(1-Heptyloctyl)-perylene-3,4,9,10-tetracaboxylic acid bisimide 10 and N-(1,3-bis(2-(2-(2-methoxyl)ethoxyl)ethoxyl)propyl) perylene-3,4,9,10-tetracaboxylic acid bisimide 11 were prepared according to a published procedure.23 Propargyl bromide (80% in toluene), 4-acetoxy styrene (96%), anisole (99%), 1,6-dibromohexane (97%), 1,8-dibromooctane (97%), dimethylformamide (99.8%), silver nitrate (≥99.0%) and triphenylphosphine (≥95.0) were purchased from Sigma-Aldrich. Tetrabutylammonium fluoride (>98%), 1,8-diazabicyclo[5.4.0]undec-7-ene (≥99.0%), trimethylsilyl chloride (98%) and PMDETA (≥98%) were purchased from Fluka. CuBr (98%) was bought from Acros. All reagents were used without further purification unless otherwise noted. 1H NMR (300 MHz) spectra were recorded on a Brucker AC 300 spectrometer in CDCl3 and calibrated to CHCl3 signal (7.26 ppm for 1H). UV-vis spectra of solutions in CHCl3 with a concentration of 10−5 mol·L−1 were recorded on a Hitachi 3000 spectrophotometer and photoluminescence spectra were acquired on a Shimadzu RF 5301 PC spectrofluorophotometer upon excitation at 490 nm. SEC measurements were carried out in THF with 0.25% tetrabutylammonium bromide with two Varian MIXED-C columns (300 × 7.5 mm) at room temperature and at a flow rate of 0.5 mL min−1 using UV (Waters model 486) with 254 nm detector wavelength and refractive index (Waters model 410) detectors. Polystyrene in combination with o-DCB as an internal standard was used for calibration. Differential scanning calorimetry experiments were conducted at heating rates of 40 K·min−1 under N2 atmosphere with a Perkin Elmer Diamond DSC, calibrated with indium. The endothermic maximum was taken as Tm. Thermogravimetry measurements were conducted on a Mettler Toledo TGA/SDTA 851e under N2 atmosphere at a heating rate of 10 K min−1. Temperature of decomposition (Tonset) was calculated from the onset of the respective curve. X-Ray diffraction experiments for PPBI 2, PPBI 5, PPBI 6 and PBI-Sty were performed on a Huber GuinierDiffractometer 6000 equipped with a Huber quartz monochromater 611 with Cu-Kα1: 1.54051 Å. The samples PPBI 1, PPBI 3 and PPBI 4 were measured at the beamline ID2 at the European Synchrotron Radiation Facility in Grenoble. The energy of the photons was 12.540 keV (λ ≈ 0.1 nm).Synthesis
:ethyl acetate 50:1 as eluent, to give the two isomers of the product as colourless crystals and colourless oil which weighed 4.20 g (66.2%).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH).
CH), 1.79–0.92 (br s, 3H, CH, CH2). Mn = 7400 g mol−1; PDI = 1.11. IR (ATR): ν (cm−1) 3286 (CC–H), 2919, 2120 (CC), 1607, 1507.
:THF from 1:0 to 0:1 as eluent. The fractions containing pure 7 were evaporated and the pure product was obtained as a slightly yellowish liquid and weighed 11.0 g (87.7%). 1H NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.27 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.51–1.35 (4H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:ethyl acetate from 1:0 to 5:1. The fractions containing pure 8 were evaporated and the product was obtained as a slightly yellowish liquid and weighed 3.7 g (72.5%). 1H NMR (300 MHz, CHCl3): δ (ppm) 1H NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.25 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.51–1.24 (8H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:ethyl acetate from 1:0 to 8:1. The pure product weighed 1.4 g (12.7%). NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.25 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.48–1.21 (12H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:acetone of 95:5. The fractions containing pure PBI-N31 were evaporated and gave the product as a red solid weighing 2.00 g (82.6%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.72–8.51 (m, 8H, ArH), 5.26–5.13 (m, 1H, N–CH), 4.21 (t, 2H, J = 7.6, N–CH2), 3.30 (t, 2H, J = 6.9, CH2–N3), 2.35–2.18 (m, 2H, 2αCHH), 1.97–1.73 (m, 4H, 2αCHH, N–CH2CH2), 1.72–1.60 (m, 2H, CH2CH2–N3), 1.54–1.18 (m, 24H, 12CH2), 0.84 (t, 6H, J = 6.8, 2CH3). IR (ATR): ν (cm−1) 2923, 2855, 2092 (–N3), 1964, 1650.
:H2O of 3:1 and filtered. The pure product was obtained as a red powder and weighed 1.20 g (96.0%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.76–8.34 (m, 8H, ArH), 5.27–5.08 (m, 1H, N–CH), 4.17 (t, 2H, J = 7.5 Hz, N–CH2), 3.25 (t, 2H, J = 6.9 Hz, CH2–N3), 2.33–2.15 (m, 2H, 2αCHH), 1.96–1.80 (m, 2H, 2αCHH), 1.80–1.67 (m, 1H, CH2CH2–N3), 1.50–1.07 (m, 28H, 14CH2), 0.82 (t, 6H, J = 6.6 Hz, CH3). IR (ATR): ν (cm−1) 2924, 2857, 2092 (–N3), 1960, 1652.
:CHCl3 of 1:1 as eluent over a short silica column. The product was obtained as a red powder and weighed 457 mg (62.0%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.65–8.50 (8H, m, ArH), 5.75–5.65 (1H, m, N–CH), 4.24–4.14 (4H, m, 2αCHH, N–CH2), 4.00–3.90 (2H, m, 2αCHH), 3.75–3.50 (12H, m, 2OCH2CH2OCH2), 3.44–3.38 (4H, m, 2CH2OCH3), 3.32–3.24 (8H, m, CH2–N3, 2OCH3) 1.86–1.72 (2H, m, CH2CH2–N3), 1.70–1.42 (10H, m, 5CH2). IR (ATR): ν (cm−1) 2923, 2845, 2095 (–N3), 1964, 1650, 1594.
:CHCl3 1:1 as eluent. The product was obtained as a red powder and weighed 161 mg (36.5%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.66–8.44 (m, 8H, ArH), 5.76–5.64 (m, 1H, N–CH), 4.25–4.14 (m, 4H, 2αCHH, N–CH2), 4.03–3.90 (m, 2H, 2αCHH), 3.78–3.52 (m, 12H, 2OCH2CH2OCH2), 3.46–3.38 (m, 4H, 2CH2OCH3), 3.24 (m, 8H, 2OCH3, CH2–N3), 2.10–1.18 (m, 18H, 9CH2). IR (ATR): ν (cm−1) 2921, 2858, 2093 (–N3), 1963, 1650, 1593.
CH).
CH), 1.79–0.92 (br s, 3H, CH, CH2). Mn = 7400 g mol−1; PDI = 1.11. IR (ATR): ν (cm−1) 3286 (CC–H), 2919, 2120 (CC), 1607, 1507.
:THF from 1:0 to 0:1 as eluent. The fractions containing pure 7 were evaporated and the pure product was obtained as a slightly yellowish liquid and weighed 11.0 g (87.7%). 1H NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.27 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.51–1.35 (4H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:ethyl acetate from 1:0 to 5:1. The fractions containing pure 8 were evaporated and the product was obtained as a slightly yellowish liquid and weighed 3.7 g (72.5%). 1H NMR (300 MHz, CHCl3): δ (ppm) 1H NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.25 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.51–1.24 (8H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:ethyl acetate from 1:0 to 8:1. The pure product weighed 1.4 g (12.7%). NMR (300 MHz, CHCl3): δ (ppm) 3.40 (2H, t, J = 6.8 Hz), 3.25 (2H, t, J = 6.5 Hz), 1.92–1.80 (2H, m), 1.66–1.55 (2H, m), 1.48–1.21 (12H, m). IR (ATR): ν (cm−1) 2092 (–N3).
:acetone of 95:5. The fractions containing pure PBI-N31 were evaporated and gave the product as a red solid weighing 2.00 g (82.6%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.72–8.51 (m, 8H, ArH), 5.26–5.13 (m, 1H, N–CH), 4.21 (t, 2H, J = 7.6, N–CH2), 3.30 (t, 2H, J = 6.9, CH2–N3), 2.35–2.18 (m, 2H, 2αCHH), 1.97–1.73 (m, 4H, 2αCHH, N–CH2CH2), 1.72–1.60 (m, 2H, CH2CH2–N3), 1.54–1.18 (m, 24H, 12CH2), 0.84 (t, 6H, J = 6.8, 2CH3). IR (ATR): ν (cm−1) 2923, 2855, 2092 (–N3), 1964, 1650.
:H2O of 3:1 and filtered. The pure product was obtained as a red powder and weighed 1.20 g (96.0%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.76–8.34 (m, 8H, ArH), 5.27–5.08 (m, 1H, N–CH), 4.17 (t, 2H, J = 7.5 Hz, N–CH2), 3.25 (t, 2H, J = 6.9 Hz, CH2–N3), 2.33–2.15 (m, 2H, 2αCHH), 1.96–1.80 (m, 2H, 2αCHH), 1.80–1.67 (m, 1H, CH2CH2–N3), 1.50–1.07 (m, 28H, 14CH2), 0.82 (t, 6H, J = 6.6 Hz, CH3). IR (ATR): ν (cm−1) 2924, 2857, 2092 (–N3), 1960, 1652.
:CHCl3 of 1:1 as eluent over a short silica column. The product was obtained as a red powder and weighed 457 mg (62.0%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.65–8.50 (8H, m, ArH), 5.75–5.65 (1H, m, N–CH), 4.24–4.14 (4H, m, 2αCHH, N–CH2), 4.00–3.90 (2H, m, 2αCHH), 3.75–3.50 (12H, m, 2OCH2CH2OCH2), 3.44–3.38 (4H, m, 2CH2OCH3), 3.32–3.24 (8H, m, CH2–N3, 2OCH3) 1.86–1.72 (2H, m, CH2CH2–N3), 1.70–1.42 (10H, m, 5CH2). IR (ATR): ν (cm−1) 2923, 2845, 2095 (–N3), 1964, 1650, 1594.
:CHCl3 1:1 as eluent. The product was obtained as a red powder and weighed 161 mg (36.5%). 1H NMR (300 MHz, CHCl3): δ (ppm) 8.66–8.44 (m, 8H, ArH), 5.76–5.64 (m, 1H, N–CH), 4.25–4.14 (m, 4H, 2αCHH, N–CH2), 4.03–3.90 (m, 2H, 2αCHH), 3.78–3.52 (m, 12H, 2OCH2CH2OCH2), 3.46–3.38 (m, 4H, 2CH2OCH3), 3.24 (m, 8H, 2OCH3, CH2–N3), 2.10–1.18 (m, 18H, 9CH2). IR (ATR): ν (cm−1) 2921, 2858, 2093 (–N3), 1963, 1650, 1593.
Stock solution of CuBr/PMDETA in anisole
CuBr (50 mg, 0.35 mmol) was added to 5 mL anisole in a Schlenk tube. The solvent was degassed by purging with nitrogen for 20 minutes. Subsequently, degassed (by purging with nitrogen for 20 minutes) PMDETA (60 mg, 0.35 mmol) was added under inert gas atmosphere and the mixture was stirred for an hour. The solution was stored at RT and was taken out by syringe through a septum, when needed.General procedure for “click”-reaction to PPBI 1–6
Poly(propargyloxystyrene) (30 mg, 0.19 mmol) was dissolved in 20 mL of anisole together with 1.2 equivalents of the respective perylene azide. After degassing by purging with nitrogen for 10 minutes, 3 drops of stock solution of PMDETA/CuBr in anisole were added by syringe through a septum. The reaction mixture was stirred overnight, filtered over silica and precipitated in MeOH. To remove the excess of perylene azide, the red polymers were extracted in a Soxhlet apparatus with methyl ethyl ketone or acetone. Special remarks for the single compounds and data can be found in the following entries.700 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 182 °C, Tm = 298 °C.
200 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 166 °C, Tm = 216 °C.
300 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 153 °C.
600 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 142 °C.
700 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 123 °C.
600 g mol−1; PDI = 1.09; Tonset = 330 °C, Tg = 120 °C.
Results and discussion
The chemical structures and the synthetic route for the “clicked” polymers PPBI 1–6 are shown in Scheme 1. Poly(propargyloxystyrene) 5 was chosen as an alkyne carrying scaffold polymer because it is easy to synthesize and provides polymers with narrow PDIs. The polymerization of the protected monomer 2 by nitroxide mediated radical polymerization (NMRP) and the accessibility of poly(propargyloxystyrene) for “click” reactions was demonstrated by Voit et al. previously.8,27,28 The alkyne group of the monomer has to be protected with a trimethylsilyl group because otherwise crosslinking would occur during the polymerization. We conducted the polymerization of 2 in o-dichlorobenzene at 125 °C in the presence of 0.1 equivalents of free nitroxide 3 to shift the NMRP equilibrium more to the dormant species and thereby to control the polymerization in a better way. The protected polymer 4 could be obtained with Mn,SEC = 9300 g mol−1 and a PDI of 1.11 corresponding to 44 repeating units. After deprotection of 4 with tetrabutylammonium fluoride, the expected scaffold alkyne polymer 5 with Mn,SEC = 7400 g mol−1 and a PDI of 1.11 was obtained.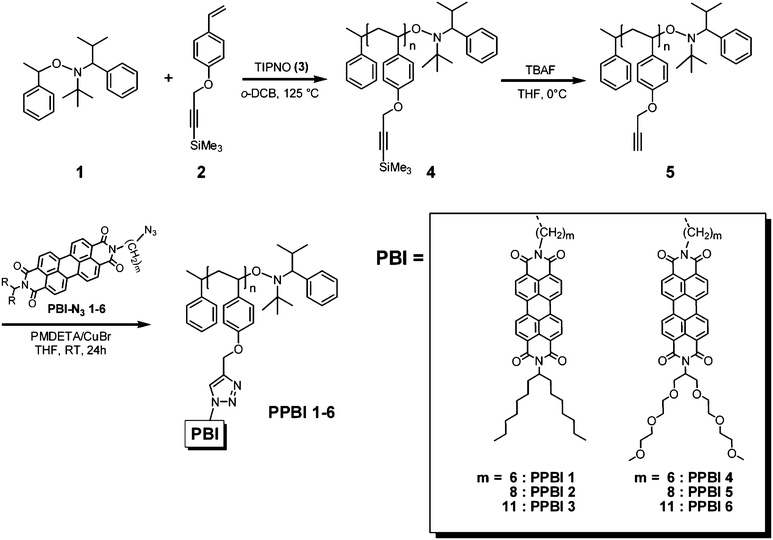 | ||
| Scheme 1 Synthetic scheme for alkyne functionalized scaffold polymer 5 and “clicked” poly(perylene bisimides), PPBIs 1–6. | ||
The “click” reaction of the azides PBI-N31–6 (1.2 molar eq.) with the alkyne carrying polymer 5 was conducted in anisole at RT. A stock solution of PMDETA/CuBr in anisole was prepared and used as a ligand/catalyst system. The approach of using a stock solution, which can be stored under inert gas atmosphere for long time, is more exact and convenient than adding ligand and copper species separately. Anisole is preferable as solvent, because it dissolves both PBI-azides and PPBIs, in contrast to THF, which is only a moderate solvent for PPBIs. After the “click”-reaction, the polymers were cleaned by removing the excess of PBI azides by Soxhlet extraction with methyl ethyl ketone or acetone (see Experimental part).
All PPBIs exhibited monomodal distribution and very similar PDIs (1.09) as the scaffold polymer 5. Fig. 1 shows exemplarily the size exclusion chromatography (SEC) traces of PPBI 1 (hydrophobic) and PPBI 4 (hydrophilic) in comparison to protected polymer 4 and deprotected scaffold polymer 5. The detailed SEC data of the polymers are presented in Table 1. Slightly smaller molecular weights were obtained for polymers with longer spacers than for those with shorter ones. We ascribe this to a difference in solution behaviour of the PPBIs in THF (SEC solvent), which is only a moderate solvent for PPBIs as aforementioned. Since the protected polymer 4 resembles the PS-standard used for SEC calibration, we could calculate the number of repeating units in 4 and 5 as 44. Thus, theoretical molecular weights of all PPBIs for 100% conversion in “click” reaction were calculated from this value (Table 1). It is evident that the molecular weights of PPBIs, determined by SEC are overestimated compared to the theoretical molecular weights. This is well known in main chain and side chain rigid rod polymers.29
 | ||
| Fig. 1 Normalized UV-signal of SEC traces (detection at 254 nm) of protected polymer 4 (black line), deprotected polymer 5 (red line) and exemplarily one hydrophobic PPBI 1 (green line) and one hydrophilic PPBI 4 (blue line) polymer (both with (CH2)6 spacer). A clear increase in molecular weight for PPBI 1 and PPBI 4 after the “click” reaction and monomodal narrow distributions of molecular weight can be observed. The upper curves are vertically displaced for clarity. | ||
| Polymer | M n/g mol−1 (SEC)a | M w/Mn (SEC)a | M n/g mol−1 (theor.)b | T onset/°Cc | T g/°Cd | T m/°Cd | ΔH/J g−1d | Spacer | Notes |
|---|---|---|---|---|---|---|---|---|---|
| a Measured with size exclusion chromatography in tetrahydrofuran and calibrated to polystyrene standards. b Theoretical molecular weights calculated from 44 repeating units of 5 assuming 100% conversion. c Measured by thermogravimetric analysis under N2 atmosphere. d Measured by differential scanning calorimetry under N2 atmosphere. | |||||||||
| 4 | 9300 | 1.11 | n.o. | Poly(TMS-propargyloxystyrene) | |||||
| 5 | 7400 | 1.11 | 386 | 59 | n.o. | Poly(propargyloxystyrene) | |||
| PPBI 1 | 59700 |
1.09 | 39200 |
330 | 182 | 298 | 1.8 | (CH2)6 | Hydrophobic PPBI |
| PPBI 2 | 59200 |
1.09 | 40500 |
330 | 166 | 216 | 0.4 | (CH2)8 | Hydrophobic PPBI |
| PPBI 3 | 58300 |
1.09 | 42300 |
330 | 153 | n.o. | (CH2)11 | Hydrophobic PPBI | |
| PPBI 4 | 59600 |
1.09 | 42200 |
330 | 142 | n.o. | (CH2)6 | Hydrophilic PPBI | |
| PPBI 5 | 58700 |
1.09 | 43500 |
330 | 123 | n.o. | (CH2)8 | Hydrophilic PPBI | |
| PPBI 6 | 55600 |
1.09 | 45300 |
330 | 120 | n.o. | (CH2)11 | Hydrophilic PPBI |
All polymers were analyzed by FTIR spectroscopy. The alkyne CC vibration at 2120 cm−1 and the CC–H vibration at 3286 cm−1 present in polymer 5 cannot be observed in the PPBIs 1–6 anymore. Also the strong azide vibration which is present in all PBI azides at around 2090 cm−1 cannot be observed in any of the PPBIs 1–6, proving that no unreacted azide is left in the polymer after purification.
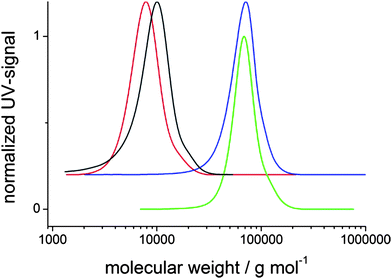
The polymers were also analyzed by 1H NMR to estimate the conversion in “click” reaction. Fig. 2 shows the 1H NMR spectrum of alkyne polymer 5 and exemplarily the spectra of hydrophobic PPBI 1 and of hydrophilic PPBI 4. After the “click” reaction, the signals of the prominent alkyne proton at 2.5 ppm, as well as that of the OCH2 group at 4.6 ppm of polymer 5 disappear. In the “clicked” polymers, the OCH2 signals are shifted to 5.1 ppm. The signal of the newly formed triazole proton, usually occurring around 7.6 ppm, cannot be seen in the spectra because it is superimposed by the broad signals of the aromatic protons of perylene, but can be determined by integration between 8.4 and 7.4 ppm. The CH2N3 protons of the PBI azides at 3.3 ppm cannot be observed any more in the PPBIs, proving the absence of remaining PBI-N3 in the polymer. The corresponding protons in the “clicked” polymers are now located adjacent to the newly built triazole unit and occur at 4.0 ppm.
 | ||
| Fig. 2 1H NMR spectra (in CDCl3) of alkyne polymer 5 and exemplarily of a PPBI with hydrophobic alkyl swallow-tail (PPBI 1) and one with hydrophilic OEG swallow-tail (PPBI 4). Signals from residual alkyne groups cannot be observed in the 1H NMR spectrum of the PPBIs. | ||
The fact that (a) no acetylene protons are present and (b) all OCH2(CC) protons are converted to OCH2(triazole) indicates a quantitative conversion within the margins of the experimental error in 1H NMR. The optical properties of the PPBIs were investigated by UV/vis and photoluminescence measurements (ESI, Fig. S3–S6†). All polymers show similar absorption spectra in CHCl3 solution (c = 10−5 mol·L−1) with the characteristic absorption maxima of PBI at 527 nm, 491 nm and 464 nm. These peaks correspond to the finger-print vibration transitions (0–0, 0–1, 0–2) for the S0–S1 electronic transition in PBI. Both hydrophobic and hydrophilic polymers show a featureless red emission which is typical for strongly π–π-stacked PBIs. A very weak peak at 534 nm can be attributed to a very small amount of non-aggregated chromophores in the polymer. While hydrophilic and hydrophobic polymers behave quite similar in their absorption, the fluorescence measurements reveal a red shift of the fluorescence peak at 611 nm in hydrophobic polymers to 624 nm in hydrophilic polymers. This could be attributed to a smaller delocalization length in the excited state for the hydrophobic polymers.30
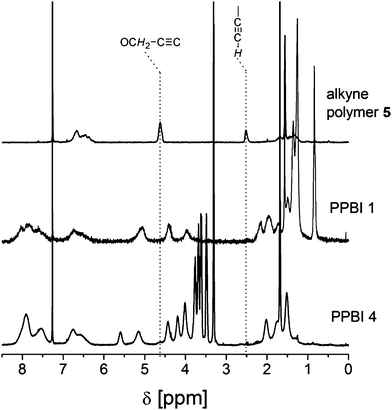
To determine the influence of the different substituents on the thermal and structural properties, the phase transitions of polymers PPBI 1–6 were investigated by DSC measurements and the structural features using X-ray diffraction experiments. The DSC thermograms and the X-ray diffractograms in SAXS regime of the hydrophobic polymers PPBI 1–3 and hydrophilic polymers PPBI 4–6 are shown in Fig. 3 and 5 respectively.
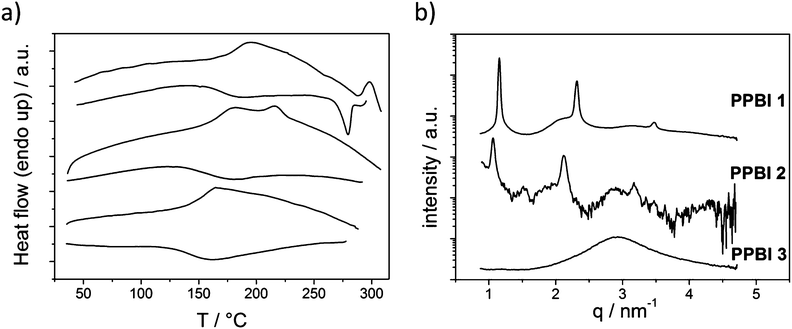 | ||
| Fig. 3 (a) DSC traces (upper: second heating and lower: cooling cycle) of hydrophobic polymers PPBI 1, 2 and 3 (from top to bottom) recorded at 40 K min.−1 (b) XRD pattern of PPBI 1, 2 and 3 at RT for the SAXS regime. | ||
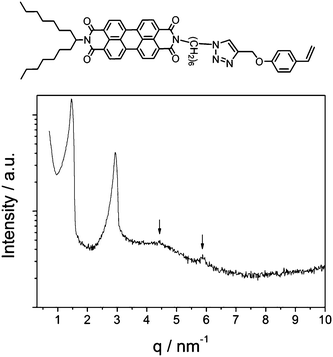 | ||
| Fig. 4 Chemical structure and XRD pattern of model monomer PBI-Sty 6. | ||
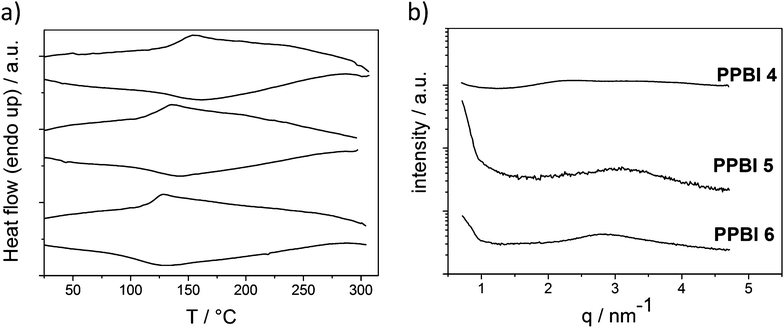 | ||
| Fig. 5 (a) DSC traces (upper: second heating cycle and lower: cooling cycle) of hydrophilic PPBI 4, 5 and 6 (from top to bottom) recorded at 40 K min.−1 (b) XRD pattern of PPBI 4, 5 and 6 at RT. | ||
First, the results of the hydrophobic polymers PPBI 1–3 with alkyl swallow-tail and (CH2)6, (CH2)8 and (CH2)11 spacers, respectively, are discussed below.
PPBI 1 with (CH2)6 spacer exhibits a Tg at 182 °C and an endothermic transition at 298 °C (ΔH = 1.8 J g−1). As observed in temperature-dependent XRD measurements (Fig. S12†), the transition at 298 °C is a melting point. The XRD diffraction at RT shows a liquid crystalline lamellar ordering with reflections at 1.15 nm−1, 2.31 nm−1 and 3.47 nm−1 which is stable between RT and 298 °C. This corresponds to a lamellar distance of 5.46 nm. Since the length of the fully stretched PBI pendant groups is estimated to be 4.28 nm, we suggest a SmC bilayer structure for PPBI 1 where the PBI moieties are tilted. The tilt angle depends on the degree of interdigitation/mixing up of the swallow tail substituents (see ESI, Fig. S14†). The broad halos below the assigned reflections are attributed to amorphous parts of the polymer.
PPBI 2 with (CH2)8 spacer shows a Tg at 166 °C and an endothermic transitions at 216 °C. As observed in temperature dependent XRD measurements, the polymer melts into isotropic state at 216 °C (Fig. S13†). XRD diffraction at RT shows a lamellar ordering with reflections at 1.06 nm−1, 2.12 nm−1 and 3.18 nm−1 which is stable between RT and 216 °C. This corresponds to a lamellar distance of 5.93 nm. The PBI pendants are estimated to have a length of 4.51 nm, assuming fully stretched conformation. Therefore we also suggest a SmC bilayer structure as in PPBI 1. The broad halos below the assigned reflections are attributed to amorphous parts of the polymer. Contrary to PPBI 1 and PPBI 2, PPBI 3 with (CH2)11 spacer shows only a Tg at 153 °C and no melting peak in DSC. Also, XRD measurements show no reflections in the low q region and thereby confirm an amorphous state at RT.
A comparison of the three polymers PPBI 1–3 shows a clear dependence of the melting points on the spacer length of the PBI moiety, with which it is attached at the backbone. In the case of polymer PPBI 3 with (CH2)11 spacer the flexibility and softness is so large that it results in a completely amorphous behaviour. Among the polymers PPBI 1 and PPBI 2, an increase of the spacer length shifts the melting point to lower temperatures. The Tg of the three polymers also decreases with increasing length of the alkyl spacer. This effect is well known in side chain liquid crystalline polymers and is attributed to an increased plasticization of the backbone by longer alkyl spacers.31 Comparison of the XRD data of polymers PPBI 1 and PPBI 2 shows an increase of the lamellar distance (from 5.46 to 5.93 nm) which fits very well to the increase of the spacer length by two methylene units. For PPBI 1, birefringence could be observed in polarized light microscopy (Fig. 6). The observed textures were very small and could not be unequivocally attributed to typical fan shaped or focal conic smectic textures. PPBI 2 did not show any birefringence or characteristic textures also after thermal annealing shortly under the Tc or on slow cooling at 1 K min−1. This can be explained by the high viscosity of the polymer in this state and taking into consideration the very low ΔH of 0.4 J g−1 for this polymer. As expected, the amorphous polymer PPBI 3 does not show any birefringence.
 | ||
| Fig. 6 Optical microscopic images of (a) PPBI 1 and (b) PBI-Sty 6 at RT under crossed polarizers exhibiting birefringence. (c) and (d) show optical microscopic images of PPBI 2 and PPBI 4 respectively with a λ/4 plate. No texture could be observed for PPBI 2 even at prolonged annealing or on slow cooling at 1 K min−1. | ||
To elucidate the origin of the LC behaviour of the polymers PPBI 1 and PPBI 2, a model compound (PBI-Sty 6) of PPBI 1 was synthesized by “click” reaction of propargyloxystyrene and PBI-N31 under the same conditions as for the synthesis of the PPBIs. The compound was studied in XRD at RT (Fig. 4). Interestingly, model compound PBI-Sty 6 showed also lamellar behaviour, indicated by reflections at q values of 1.46 nm−1, 2.93 nm−1, 4.46 nm−1 and 5.88 nm−1 which are in a ratio of 1:2:3:4. This corresponds to a lamellar distance of 4.29 nm, which correlates very well with the estimated length of the fully stretched model monomer PBI-Sty 6 (4.28 nm). While the polymers PPBI 1 and PPBI 2 showed SmC behaviour, model monomer PBI-Sty 6 clearly exhibits a SmA phase. Low molecular, symmetrically swallow-tail-substituted PBIs often exhibit columnar hexagonal ordering due to their space-filling nature of substituents.23 As the PBI PBI-Sty 6 and the PBIs in the polymers are unsymmetrically substituted with a linear spacer and one swallow-tail-substituent, smectic behaviour is not unexpected for these molecules. For PBI-Sty 6, birefringence could be observed in polarized light microscopy (Fig. 6). The observed textures could best be described as focal conic.
In contrast to the hydrophobic substituted PPBIs, the hydrophilic polymers PPBI 4–6 show no melting peaks in DSC. Also, XRD measurements at RT show no sharp reflections at small q values but only a broad halo, confirming the amorphous nature of these polymers. PPBI 4 exhibits a Tg at 142 °C, PPBI 5 a Tg at 123 °C and PPBI 6 a Tg at 120 °C. There was no birefringence or texture observed in polarized light microscopy for the amorphous polymers PPBI 4–6, as expected. It seems that the flexible OEG swallow-tail hinders the formation of ordered liquid crystalline or crystalline structures. The tendency of OEG swallow-tail substituted PBIs to form lower molecular order compared to their alkyl swallow-tail substituted analogues can be attributed to the higher flexibility and space-filling properties of OEG and is known for low molecular PBIs.23 The Tg of the three polymers decreases with increasing length of the alkyl spacer. As in the hydrophobic substituted PPBIs, this effect is attributed to an increased plasticization of the backbone by longer alkyl spacers.31
The Tgs observed in the side chain polymers (between 120 °C and 180 °C) are comparable to those observed in poly perylene acrylates with (CH2)6 spacers or polymers from PBI-N3 clicked to an alkyne bearing polyacrylate.6 The presence of a glassy state with LC order at RT, especially for polymer PPBI 1 and PPBI 2 offers possibilities of achieving order in thin films by tempering methods.
Conclusions
We showed that by the modular “click” chemistry approach in combination with nitroxide mediated radical polymerization, a variation in the substitution of side chain polymers is easily possible. “Click” chemistry has been used to synthesize three hydrophilic and three hydrophobic PPBIs with OEG- and alkyl swallow-tails, respectively. In each series, the length of the spacer between the nitrogen of the triazole unit and the nitrogen of the PBI was varied from (CH2)6 to (CH2)8 and to (CH2)11. Longer spacers shift the Tg to lower temperatures due to the higher plasticization of the backbone. The structural analysis revealed that the shorter spacers with six and eight methyl units (PPBI 1 and 2) with alkyl swallow-tail substituents lead to liquid crystalline behaviour. Based on the first XRD results, a lamellar SmC structure is suggested. When the alkyl spacer is too long, as in PPBI 3, no melting transition could be observed and the polymer becomes amorphous in nature. The introduction of OEG swallow-tails at the PPBI leads to amorphous polymers for any spacer length (PPBI 4–6). Especially the presence of a glassy state with LC order at RT allows tailoring the required film properties in these polymers. Polar groups in perylene bisimide containing polymers are suitable for the phase separation in block copolymers due to an additional polar–apolar driving force and thereby an increased χ parameter. The applicability of these polymers as acceptor materials in suitable devices is under study.Acknowledgements
Financial support from German Research Council DFG (Projects GRK 1640 and SPP 1355) are kindly acknowledgedNotes and references
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS.
- D. Quémener, M. L. Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155–173 CrossRef.
- X. Zhang, X. Lian, L. Liu, J. Zhang and H. Zhao, Macromolecules, 2008, 41, 7863–7869 CrossRef CAS.
- A. S. Lang, A. Neubig, M. Sommer and M. Thelakkat, Macromolecules, 2010, 43, 7001–7010 CrossRef CAS.
- M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS.
- S. Fleischmann, H. Komber and B. Voit, Macromolecules, 2008, 41, 5255–5264 CrossRef CAS.
- Z. Li, Y. Zhang, L. Zhu, T. Shen and H. Zhang, Polym. Chem., 2010, 1, 1501–1511 RSC.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- H. Langhals, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS.
- G. Horowitz, F. Kouki, P. Spearman, D. Fichou, C. Nogues, X. Pan and F. Garnier, Adv. Mater., 1996, 8, 242–245 CrossRef CAS.
- S. M. Lindner, S. Hüttner, A. Chiche, M. Thelakkat and G. Krausch, Angew. Chem., Int. Ed., 2006, 45, 3364–3368 CrossRef CAS.
- M. Sommer, S. M. Lindner and M. Thelakkat, Adv. Funct. Mater., 2007, 17, 1493–1500 CrossRef CAS.
- G. D. Sharma, P. Balraju, J. A. Mikroyannidis and M. M. Stylianakis, Sol. Energy Mater. Sol. Cells, 2009, 93, 2025–2028 CrossRef CAS.
- S. Hüttner, M. Sommer and M. Thelakkat, Appl. Phys. Lett., 2008, 92, 093302 CrossRef.
- S. M. Lindner and M. Thelakkat, Macromolecules, 2004, 37, 8832–8835 CrossRef CAS.
- Q. Zhang, A. Cirpan, T. P. Russell and T. Emrick, Macromolecules, 2009, 42, 1079–1082 CrossRef CAS.
- S. Rajaram, P. B. Armstrong, B. J. Kim and J. M. J. Fréchet, Chem. Mater., 2009, 21, 1775–1777 CrossRef CAS.
- Y. Tao, B. McCulloch, S. Kim and R. A. Segalman, Soft Matter, 2009, 5, 4219–4230 RSC.
- M. Sommer, A. S. Lang and M. Thelakkat, Angew. Chem., Int. Ed., 2008, 47, 7901–7904 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- A. Wicklein, A. Lang, M. Muth and M. Thelakkat, J. Am. Chem. Soc., 2009, 131, 14442–14453 CrossRef CAS.
- E. Kaul, V. Senkovskyy, R. Tkachov, V. Bocharova, H. Komber, M. Stamm and A. Kiriy, Macromolecules, 2009, 43, 77–81 CrossRef.
- B. Jagadish, R. Sankaranarayanan, L. Xu, R. Richards, J. Vagner, V. J. Hruby, R. J. Gillies and E. A. Mash, Bioorg. Med. Chem. Lett., 2007, 17, 3310–3313 CrossRef CAS.
- E. J. O'Neil, K. M. DiVittorio and B. D. Smith, Org. Lett., 2006, 9, 199–202 CrossRef.
- S. Fleischmann, H. Komber, D. Appelhans and B. I. Voit, Macromol. Chem. Phys., 2007, 208, 1050–1060 CrossRef CAS.
- S. Fleischmann, A. Kiriy, V. Bocharova, C. Tock, H. Komber and B. Voit, Macromol. Rapid Commun., 2009, 30, 1457–1462 CrossRef CAS.
- J. Liu, R. S. Loewe and R. D. McCullough, Macromolecules, 1999, 32, 5777–5785 CrossRef CAS.
- J. J. Han, A. D. Shaller, W. Wang and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 6974–6982 CrossRef CAS.
- A. A. Craig and C. T. Imrie, Macromolecules, 1999, 32, 6215–6220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra, UV/vis, additional XRD data and photoluminescence spectra. See DOI: 10.1039/c1py00191d |
| This journal is © The Royal Society of Chemistry 2011 |