Modular design for the controlled production of polymeric nanotubes from polymer/peptide conjugates†
Robert
Chapman
a,
Katrina A.
Jolliffe
*b and
Sébastien
Perrier
*a
aKey Centre for Polymers & Colloids, School of Chemistry, The University of Sydney, NSW 2006, Australia
bSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: kate.jolliffe@sydney.edu.au; sebastien.perrier@sydney.edu.au; Fax: +61 (2) 9351 3329; Tel: +61 (2) 9351 3366; Fax: +61 (2) 9351 3329; Tel: +61 (2) 9351 2297
First published on 27th May 2011
Abstract
We have established a new strategy to produce functional organic nanotubes of controlled structure from cyclic peptide/polymer conjugates. The structure guiding cyclic peptide motif was coupled to the polymeric chains viacopper-catalyzed azide–alkyne click reaction, to yield very well-defined conjugates. The resulting conjugates were then self-assembled into nanotubes in solution. We were able to control to some extent the length of the nanotubes, by modifying the length of the polymer chain as well as by mixing conjugates of different molecular weights together. In a similar fashion we were able to prepare functional tubes from a range of polymers including poly(butyl acrylate), poly(dimethyl amino ethyl acrylate), poly(acrylic acid), poly(styrene) and poly(hydroxyl ethyl acrylate), as well as tubes with mixed functionality, by self-assembling a mixture of conjugates of differing polymer functionality. This modular approach is a powerful technique to generate a large number of nanotubes of varying size and functionality, in a controlled and rapid process. We believe this new approach will permit the design of a wide range of functional organic nanotubes of controlled structure, in a simple and efficient process.
Introduction
Hollow nanotubular structures (nanotubes, NTs) are present in many natural and artificial systems. Biological nanotubes such as transmembrane channel proteins1,2 are among nature's most admirable devices, while mesoporous aluminosilicates3,4 and carbon nanotubes5,6 are examples of revolutionary synthetic nanotubular devices that have had a major impact on materials science.The range of potential applications of natural and synthetic (in)organic nanotubes has dramatically increased over the past few years, to include fields as diverse as microelectronics, sensors, catalysis, drug delivery, ion channels, separation technology and medicine.7 The geometry and large surface area of NTs make them an exciting prospect for applications in nanotechnology. However, to date their widespread use in practical applications has been hindered by the limited processes available for their controlled manufacture. Current approaches are unable to exhibit control over both the size of the tubular structures and the functionalization of the internal and external surfaces. The ability to isolate individual NTs rather than aggregates is also difficult to achieve with current methods. This poses problems for device fabrication, as the resulting NTs (e.g.carbon nanotubes) are not uniform in width or length, have limited solubility (and are therefore difficult to functionalize), cannot be made reproducibly and are expensive to prepare.8,9
Molecular self-assembly has emerged as the main bottom-up approach for the affordable production of bulk quantities of well-defined organic tubular nanostructures.7–12 The resulting organic structures can be used as produced, or employed as sacrificial templates for the production of inorganic nanotubes, via mineralisation or reduction of metallic salts. Building blocks include polymeric amphiphiles, carbohydrate-based molecular rings, DNA, proteins and peptides.
Recent progress in ligation techniques that permit efficient conjugation of synthetic polymers to peptides has increased the possibilities of engineering polymeric materials with complex nanostructure.13,14 In these constructs, the peptides provide the scaffold on which the polymeric materials are built. In particular, β-sheet forming peptides allow access to a range of novel nanostructures, including tapes, ribbons, fibrils, cylinders and tubes,14–19 that can be transferred to polymeric materials. The breadth of properties available in modern polymer science make these materials very versatile, opening up applications in biosensors, electronic devices and biomaterials.11 Among the various peptidic building blocks reported to date, cyclic peptides containing 6–12 amino acids of alternating D and L chirality have been shown to assemble into tubes through anti-parallel hydrogen bonding.20,21 The resulting NTs are attractive as they are easy to form, permit the introduction of functionality on their external surfaces, and have reproducible internal channel size. Rings with 8 amino acid residues tend to result in the strongest interactions, capable of forming tubes even when large substituents, including polymers22–27 and fullerenes,28 are attached to the periphery of the ring and lead to materials with exciting applications. For instance, Xu et al. have recently reported an elegant approach to the formation of subnanometre porous membranes containing high-density arrays of through channels, by growing cyclic peptide nanotubes directed in a structural framework set by the self-assembly of block copolymers.24
While there has been extensive research into the design of the cyclic peptide, problems including limited solubility, lack of control of NT length and aggregation in solution still remain. An effective approach to improve the solution behavior and applications of cyclic peptide nanotubes is their functionalization with polymeric chains. Recent work has demonstrated that polymeric chains can be grafted to a peptide NTs via a divergent synthesis strategy, in which a polymeric chain is grown from either a cyclic peptide unimer,25 or a preassembled peptide nanotube.26,27 Although the resulting materials have promising applications, this strategy makes it difficult to accurately characterize the polymer/peptide conjugate and resulting polymeric NTs. For instance, graft density, reaction yields, polymer functionality and molecular weight are difficult to assess. In addition, given the strong ability for cyclic peptides to assemble in most solvents, it is difficult to control the growth of the polymeric chains from a single cyclic peptide rather than from an aggregate, thus leading to heterogeneous rather than homogeneous polymerisation. As a result of these limitations, there is still only a limited understanding of the mechanism of NT assembly, and control over the process is quite poor. The alternative to this approach is a convergent synthesis strategy, in which the peptide and polymer are synthesized independently, and then coupled together in a highly efficient coupling reaction. This modular approach has the potential to offer a number of significant advantages: (1) the individual synthesis of the peptide and polymer allows for better characterization of the conjugate than would otherwise be possible, as each component can be fully characterized independently; (2) solvent selection for conjugation is typically more flexible than solvents for polymerization reactions; (3) graft densities during the conjugation reaction can be precisely calculated and controlled; (4) a convergent strategy enables the facile synthesis of a library of conjugates, as once the conditions of the conjugation reaction are established, a wide range of polymers can be coupled to the peptidevia combinatorial chemistry, without needing to tune the conditions for each reaction. Not only does this last feature allow for the engineering of NTs with diverse functionality, but it also permits an in-depth investigation into the effect that the type of polymer and its molecular weight have on the self-assembly of the conjugate, and the formation of the NTs. Although very promising, convergent synthesis of NTs has been scarcely employed, and its advantages in terms of modular design of NTs have been vastly under-exploited. Results to date have reported poor control over the conjugate structures, and the features of the modular approach have not been exploited to study NT formation.23,24,29 Here, we introduce a strategy based on convergent synthesis to make cyclic peptide/polymer conjugates via click chemistry, and obtain very well-defined conjugates. We study their self-assembly into nanotubes with a view to obtaining better control over the height and length of the tubes, and a better understanding of the self-assembly process by which they are formed. Finally, we illustrate the considerable potential of this modular approach by forming a range of shell-functionalized polymeric nanotubes.
Experimental
Materials
Fmoc-Lys-OH, Fmoc-Trp(Boc)-OH, Fmoc-D-Leu-OH, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium (HBTU) and 1-hydroxy-benzotriazole (HOBt) were obtained from Novabiochem and used as supplied. Butyl-trithiocarbonate propanoic acid (BTCPA) was obtained from Dulux and used as supplied, and azoisobutyronitrile (AIBN) was obtained from Aldrich and precipitated from methanol prior to use. Hünig's base (di(isopropyl)ethylamine, DIPEA), trifluoroacetic acid (TFA), sulfuryl chloride, and all solvents were ordered through Sigma-Aldrich and used as received. N- and tert-butyl acrylate (BA & t-BA) as well as dimethyl amino ethyl acrylate (DMAEA) were purchased with an MEHQ inhibitor present, which was removed by passing over a catechol trapping resin. Hydroxyethyl acrylate (HEA) was purified as described in the literature prior to use.30H2N-L-Trp(Boc)-D-Leu-L-Lys(N3)-D-Leu-L-Trp(Boc)-D-Leu-L-Lys(N3)-D-Leu-OH (2)
Under anhydrous conditions, 2-chlorotrityl chloride resin (1.0 g, 1.01 mmol g−1) was suspended in DCM (20 ml) in a fritted 100 ml round bottom flask and shaken for 30 min. After the DCM was filtered off, a solution of Fmoc-D-Leucine-OH (2.25 mmol, 0.80 g) and DIPEA (8.0 mmol, 1.03 g) in DCM (20 ml) was added to the resin, and the suspension was shaken for 2 h. The resin was then washed with 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 solution of DCM:MeOH:DIPEA (2 × 20 ml) to cap any unreacted sites, and then with DCM (3 × 20 ml), DMF (3 × 20 ml), and DCM (3 × 20 ml). The resin was dried under vacuum and used for further solid phase peptide synthesis. Loading was calculated to be ∼1.5 mmol g−1 unloaded resin by UV-Vis spectroscopy. To couple each amino acid, the Fmoc group was first removed with 20% piperidine in DMF (2 × 3 min), and the resin was washed with DMF (5 × 20 ml), DCM (3 × 20 ml) and DMF (2 × 20 ml). Fmoc-amino acid (1.2 equiv.) and HBTU (1.5 equiv.) were then dissolved in a solution of DIPEA (8 mmol, 1.03 g) in DMF (20 ml) and added to the reaction vessel. The mixture was agitated by rotation overnight and washed with DMF (5 × 20 ml) before deprotection and coupling of the next amino acid. After cleavage of the final Fmoc group, the resin was agitated with solution of hexafluoroisopropanol (HFIP):DCM (1:4, 3 × 15 ml, 3 × 10 min), and subsequently washed with DCM (3 × 20 ml). The combined washings were concentrated in vacuo to yield the product 2 (1.68 g, 82%) as a white solid.
:2:1 solution of DCM:MeOH:DIPEA (2 × 20 ml) to cap any unreacted sites, and then with DCM (3 × 20 ml), DMF (3 × 20 ml), and DCM (3 × 20 ml). The resin was dried under vacuum and used for further solid phase peptide synthesis. Loading was calculated to be ∼1.5 mmol g−1 unloaded resin by UV-Vis spectroscopy. To couple each amino acid, the Fmoc group was first removed with 20% piperidine in DMF (2 × 3 min), and the resin was washed with DMF (5 × 20 ml), DCM (3 × 20 ml) and DMF (2 × 20 ml). Fmoc-amino acid (1.2 equiv.) and HBTU (1.5 equiv.) were then dissolved in a solution of DIPEA (8 mmol, 1.03 g) in DMF (20 ml) and added to the reaction vessel. The mixture was agitated by rotation overnight and washed with DMF (5 × 20 ml) before deprotection and coupling of the next amino acid. After cleavage of the final Fmoc group, the resin was agitated with solution of hexafluoroisopropanol (HFIP):DCM (1:4, 3 × 15 ml, 3 × 10 min), and subsequently washed with DCM (3 × 20 ml). The combined washings were concentrated in vacuo to yield the product 2 (1.68 g, 82%) as a white solid.
cyclo[-L-Trp(Boc)-D-Leu-L-Lys(N3)-D-Leu-L-Trp(Boc)-D-Leu-L-Lys(N3)-D-Leu-] (3)
Linear peptide 2 (1.65 g, 1.24 mmol) was dissolved in DMF (1.0 l) under a N2 atmosphere, and cooled to 0 °C. HBTU (1.5 equiv.), HOBt (1.5 equiv.) and DIPEA (3 equiv.) were dissolved in DMF (20 ml) and added dropwise to the mixture. The mixture was stirred at room temperature and monitored by TLC. After 72 h, a further 1.5 equiv. of HBTU and HOBt, with DIPEA (3 equiv.), were added. After a total of 84 h, the solution was concentrated to near dryness and triturated from MeOH to yield a white precipitate. The precipitate was washed with MeOH (5×) to yield the Boc protected cyclic peptide (0.751 g, 46%). Boc groups were removed by dissolving the protected peptide in a cocktail of TFA/triisopropanol/thioanisole/water (85:5:5:5), and stirring for 2 h. Concentration of the mixture, followed by trituration with MeOH gave the desired product 3 in quantitative yields; m/z (APCI) = 1133.9 ([M + H]+, calc = 1133.7); 1H-NMR (300 MHz, d-TFA) δ ppm: 1.05–0.82 (m, 24H) 1.20–2.00 (m, 24H), 3.41 (t, J = 5.1 Hz, 4H), 4.85–4.78 (m, 4H), 4.91 (t, J = 6.56 Hz, 2H), 5.32 (t, J = 7.40 Hz, 2H), 7.53–7.37 (m, 4H), 7.65 (s, 2H), 7.76 (d, J = 7.22 Hz, 2H), 8.22 (d, J = 7.75 Hz, 2H).
General polymerisation procedure
The RAFT agent 4 was prepared according to the previous work by our group,29,31 and mixed with the deinhibited monomer, AIBN (0.1 mol:1 mol RAFT agent), and a solvent in the ratios given in Table 1 and degassed by cooling the flask to 0 °C and bubbling with N2 for 15 min. Polymerizations were conducted in an oil bath, under N2 at 1 atm. Samples of the crude were taken throughout the reaction for determination of conversion by 1H-NMR, and the remainder was purified by precipitation and subsequent washing from an ice cold mixture of 10% water in methanol. Butyl acrylate polymerizations were conducted at 70 °C with 40% (w/w) dioxane, hydroxyethyl acrylate (HEA) at 60 °C with 70% (w/w) tert-butanol, styrene in bulk at 65 °C, and dimethyl amino ethyl acrylate at 60 °C in 70% dioxane. tert-Butyl acrylate was polymerized at 80 °C in bulk, and then reacted overnight in 10% TFA/DCM at room temperature to cleave the tert-butyl group and generate poly(acrylic acid).
| # | Polymer | [M]:[RAFT] |
Conversion | 1H-NMR | SEC | ||
|---|---|---|---|---|---|---|---|
| DP | M n | M n b | PDI | ||||
| a p(t-BA) was hydrolysed to p(AA) before conjugation to the peptide. b SEC results uncorrected relative to PS standards. | |||||||
| 5 | p(BA)16 | 18:1 |
79% | 16 | 2320 | 2250 | 1.15 |
| 6 | p(BA)36 | 36:1 |
98% | 36 | 4900 | 4000 | 1.13 |
| 7 | p(BA)108 | 120:1 |
84% | 108 | 14150 |
11400 |
1.15 |
| 8 | p(HEA)15 | 20:1 |
73% | 15 | 2020 | 2350 | 1.16 |
| 9 | p(DMAEA)19 | 20:1 |
95% | 19 | 2990 | 3000 | 1.21 |
| 10 | p(t-BA)55a | 55:1 |
98% | 55 | 7330 | 7500 | 1.18 |
| 11 | p(S)20 | 20:1 |
98% | 20 | 2360 | 4350 | 1.12 |
General procedure for conjugation reactions
Cyclic peptide 3 (15 mg, 0.013 mmol) was suspended in TFE (2.0 g) by sonication for 10 min. To this was added a solution of the polymer (1.1 or 1.5 equiv.) and CuSO4 (4 equiv., 0.053 mmol, 13 mg) in DMF (2.0 g). Sodium ascorbate (0.165 mmol, 33 mg) was added, and the mixture was stirred in a microwave reactor at 100 °C for 15 min. The initial power input was set to 200 W and the vessel was cooled using N2 throughout the reaction to maximise the microwave power input, which averaged ∼50 W over the course of the reaction. In the case of pBA, pDMAEA, pAA and pS reactions, the product was purified by precipitation and subsequent washing in water, and collected by centrifugation (14500 rpm). For pHEA reactions the reaction mixture was purified by dialysis (2000 g mol−1membrane) in water for 5 days. Conjugates were purified either viacolumn chromatography on neutral alumina with TFA/DCM (1:9) leaving ∼1.5% copper (w/w, ICP), or by precipitation from an aqueous EDTA solution (0.065 M, pH 8.5), leaving 0% copper (w/w, ICP).
Merrifield scavenger resin
Merrifield resin (3.0 g, 1.1 mmol g−1) was dispersed in DMSO (50 ml) with NaN3 (1.07 g, 16.5 mmol) and stirred in an oil bath at 60 °C for 48 h. The functionalised resin was then filtered hot, and washed with hot DMSO (50 ml). The resin was redispersed in methanol (50 ml), heated at 60 °C for 30 min, filtered again and washed alternately with methanol (30 ml) and DCM (30 ml) five times. The product was dried in vacuo. ATR-IR: 2200 cm−1 (N3). Elemental analysis: 85.14% C, 6.80% N, 7.21% H (∼1.6 mmol N3 per g resin).Typical self-assembly procedure
The conjugate, for example pBA-conjugate, once purified was dissolved in TFA to ensure complete dissociation of any aggregates. The TFA was then removed and the conjugate was redissolved in DMF (1 mg ml−1). This mixture was then diluted slowly with THF (in the case of pBA and pS conjugates), methanol (in the case of pHEA conjugates) or water (in the case of pDMAEA and pAA conjugates), to a final ratio of 1:9.
Characterization
NMR experiments were performed on a 300 or 400 MHz Bruker NMR instrument, and mass spectrometry was performed using atmospheric pressure chemical ionization. Molecular weights and polydispersities of the polymers and conjugates were determined by 1H-NMR and size exclusion chromatography (SEC) with respect to polystyrene standards (1–40000 g mol−1). DMF + 0.1% LiBr was used as the SEC eluent, and analyses were run at 50 °C over PolarGel columns using a DRI detector. Water was used as a flowrate marker to correct retention times. Dynamic light scattering experiments to measure the effective hydrodynamic radius were performed with a 633 nm wavelength laser at 90° with a 400 μm spot size. Relative intensities were calculated using Brookhaven NNLS software. Samples were prepared in cylindrical quartz cells in a N2 flowbench, and solvents were filtered through a 20 nm PTFE filter prior to use to eliminate dust. Analysis was performed through a bath of filtered decalin to control the temperature at 25 °C. In mixed solvent systems a linear relationship was assumed for the calculation of viscosities and refractive indices. Transmission electron microscopy (TEM) images were taken on a JEOL JEM-1400 microscope at 120 kV. Sample grids were prepared by coating 400 mesh copper grids with a Pioloform film, and sputter coating with carbon (∼10 nm). Samples were drop cast, without staining, onto the grids from solution and dried in vacuo prior to imaging.
Results and discussion
Synthesis
A key requirement for our convergent synthesis is an efficient coupling reaction, which proceeds to high yields under mild conditions, without polluting the final product with excess catalysts or reactants.32 These prerequisites are remarkably matched by the copper catalyzed azide–alkyne cycloaddition (CuAAC), which is also orthogonal to amino acid functionalities and polymerization reactions.33 We incorporated the azide functionality onto the peptide backbone rather than into the polymer. Indeed, azide moieties are truly orthogonal to the reactions involved in peptide synthesis (as indeed to almost all biological reactions),34 but not orthogonal to all polymerization conditions. We have observed, for example, that azides may add onto the vinyl groups of monomers during RAFT polymerizations.35 Conveniently, the introduction of the azide functionality to the cyclic peptide is relatively straightforward, through substitution of the side-chain amine on an Fmoc-protected Lysine 1, which was achieved in good yields using Stick's diazo transfer reagent.36 Subsequent solid phase peptide synthesis (SPPS) by alternately coupling this modified amino acid (or Fmoc-L-Trp(Boc)-OH) with Fmoc-D-Leucine gave the octapeptide 2, which was cleaved from the resin and cyclized under similar conditions at high dilution to yield the cyclic peptide. This approach allows for the introduction of an azide group into the desired positions on the cyclic peptide with 100% efficiency (Scheme 1).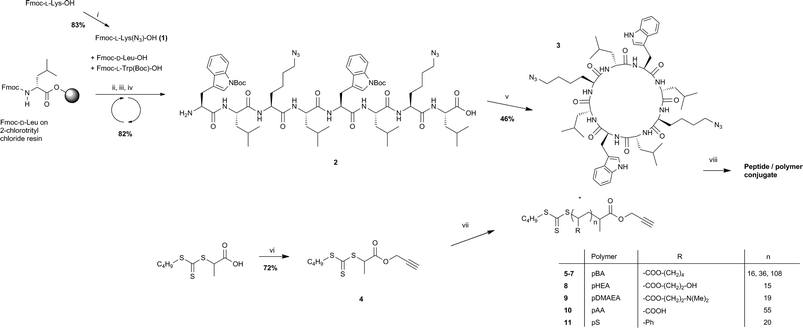 | ||
| Scheme 1 Synthesis of cyclic peptide–polymer conjugates: (i) Fmoc-L-Lys-OH, Im-SO2-N3, CuSO4, NaHCO3; (ii) Fmoc-L-Lys(N3)-OH/Fmoc-D-Leu-OH/Fmoc-L-Trp(Boc)-OH, HBTU, Hünig's base, DMF; (iii) 20% piperidine, DMF; (iv) HFIP/DCM (1:4); (v) HOBt, HBTU, Hünig's base, DMF; followed by TFA deprotection, (vi) propargyl alcohol, EDCI, DMAP, DCM, (vii) RAFT polymerization with AIBN; (viii) DMF/TFE, CuSO4, Na-ascorbate, μW. | ||
We chose to introduce the alkyne group onto the polymer by incorporating it into a chain transfer agent for reversible addition–fragmentation chain transfer (RAFT) polymerization. RAFT is a simple and effective polymerization technique, which yields well-controlled polymers exhibiting an almost infinite range of functionalities.37–40 Functionalizing the reinitiating group of a RAFT agent prior to polymerization ensures that each polymeric chain is end-capped by one alkyne group. The alkyne group is susceptible to reaction with radicals during polymerization and so is not truly orthogonal. It is, however, easily protected with moieties such as trimethylsilane (readily removed by hydrolysis after polymerization), although such protection goes against the philosophy of click chemistry. We found that for polymerizations with acetylene functionalized RAFT agents at short reaction times and low temperatures, protection is unnecessary,29,41 and so synthesized the RAFT agent 4via the esterification of the well-studied trithiocarbonate butyl-trithiocarbonate propanoic acid (BTCPA) by propargyl alcohol,31 in the presence of EDCI and DMAP. Our aims were to study the effect of polymer functionality and chain length on the formation of nanotubes and exemplify the range of functional nanotubes obtainable. 4 was therefore used to mediate the polymerization of butyl acrylate (BA), hydroxyethyl acrylate (HEA), dimethyl amino ethyl acrylate (DMAEA), acrylic acid (AA) and styrene (S) of various degrees of polymerization (Table 1).
Our initial conjugation procedure was undertaken using conventional (oil bath) heating and gave high yields although in long reaction times. In order to improve on reaction kinetics, we tested the use of microwave irradiation on the click reaction,42,43 and discovered that reaction times could be decreased from 3 days when using an oil bath to 15 minutes when using microwave irradiation, whilst keeping similar high yields. All conjugation reactions were therefore carried out in a microwave, and with a mixed solvent system of 1:1 trifluoroethanol (TFE) and dimethylformamide (DMF). The cyclic peptide was found to be remarkably insoluble, presumably due to a strong tendency to self-assemble, and so the click reaction had to be carried out in heterogeneous solution. The peptide was first sonicated with TFE to create a fine dispersion, and to this were added the polymer and CuSO4 dissolved in DMF, and the sodium ascorbate. We found that large amounts of CuSO4, well above catalytic levels, were required to drive the conjugation reactions to completion. It is likely this is due to a strong interaction between the peptide and Cu(II), which effectively inhibits the catalysis of the cycloaddition reaction until an excess of copper is present.44 The excess copper could be partially removed by passing the conjugates over a column of neutral alumina, resulting in 62% removal of the copper leaving 1.5% w/w in the conjugates.
Initial conjugation reactions were undertaken using a 50% excess of polymer, in order to drive the reaction to completion. Although the presence of free polymer is not expected to hinder self-assembly,41 we investigated the removal of the free chains after click reaction by the use of a scavenger resin. An azide modified Merrifield resin was synthesized for this purpose via a substitution reaction with NaN3.45 Loading was estimated by elemental analysis to be at 1.6 mmol g−1 of dry resin. After the conjugation reaction, the resin was added to the mixture with additional CuSO4 (2 mg) and sodium ascorbate (10 mg) and reacted in the microwave for a further 15 min at 80 °C. After removal of the resin by filtration, the amount of free polymer in the system was decreased (see Fig. S1, ESI†), although a large excess of resin was required. Indeed, despite two successive additions of 60 equiv. of resin to the reaction mixture, a small amount of free polymer remains in the sample. Given the difficulty in removing and quantifying the excess polymer, and as using large excess in one of the reactants is against the principle of a ‘click’ reaction, we investigated the conjugation reaction when reducing the amount of excess polymer. The reaction was found to proceed to completion with as little as 10% excess polymer, as evidenced by the IR spectra (Fig. 1). This small amount of excess polymer (see Fig. S1, ESI†) was deemed to be small enough in quantity so as not to require removal (see below). We note that a small excess in polymer is always required in conjugation reactions, to account for the molecular weight distribution of the sample. Table 2 summarizes the conditions for the conjugation reaction.
| # | Polymer | Equiv. polymerb | Equiv. Cub | 1H-NMR click/peptide | IR conversion | SEC Mpc |
|---|---|---|---|---|---|---|
|
a All reactions in 1:1 TFE/DMF.
b Equivalents relative to 1 mol azide sites.
c
SEC results uncorrected relative to PS standards.
d Large molecular weight made calculation of conversion by NMR too inaccurate, despite appearance of characteristic click signals.
|
||||||
| 1 | pBA16 | 1.5 | 1 | 2.5 ± 0.6 | 100% | 8800 |
| 2 | pBA16 | 1.1 | 2 | 1.9 ± 0.2 | 100% | 8700 |
| 3 | pBA36 | 1.1 | 2 | 2.0 ± 0.3 | 100% | 13500 |
| 4 | pBA108 | 1.1 | 2 | >0d | 100% | 26700 |
 | ||
| Fig. 1 IR of cyclic peptide and peptide–polymer conjugates after precipitation from water (a) in the azide region of the spectra and (b) in the amide stretching region. | ||
Full conversion was confirmed by FTIR, NMR and SEC. Total disappearance of the signal at 2100 cm−1 in the IR spectra (Fig. 1a) was observed in all cases, indicating complete reaction of the azide. Increasing carbonyl and C–H stretches were observed as expected for increasing molecular weights of polymer. 1H-NMR confirms the reaction of the azide group on the polymer by the appearance of the signal at 5.5 ppm characteristic of the RAFT R group protons adjacent to the triazole ring (see Fig. S2, ESI†). Integration of this signal relative to the signals from the peptide, while slightly inaccurate due to the presence of large amounts of polymer, suggests that the reactions indeed proceed to completion. A feature of this reaction is that the signal for these protons does not appear as a singlet, as is usually observed in such a reaction, but instead some splitting into a doublet of doublets can be observed. This is likely due to the restricted rotation caused by the conjugation of the bulky polymer to the peptide, and can be seen to a greater extent still in conjugates where each peptide ring is conjugated to 3–4 polymers. SEC analyses confirmed the molecular weight of the conjugates to be roughly double that of the unreacted polymer (see Fig. S1, ESI†).
Self-assembly
The ability of the cyclic peptide to form strong β-sheet networks, even when conjugated to the polymer, is critical if the peptide is to guide the formation of the nanotube. We confirmed β-sheet formation by FTIR, by observing the appearance of amide I bands at 1623 cm−1 and 1687 cm−1, and the amide II band at 1541 cm−1, for both unconjugated peptide and all conjugates, including those with the longest polymeric chains (Fig. 1b).46–48 Confident of the strength of these interactions, we proceeded to investigate the self-assembly properties of pBA conjugates in solution. The assembly of the conjugates was controlled by dissolving them in TFA, thus ensuring complete dissociation of potential aggregates, followed by evaporation to dryness, redissolution in DMF (1 mg ml−1) and then dropwise addition of THF (1 ml THF:100 μl DMF).23 The diluting solvent was chosen so as to be a good solvent for the polymer, but a poor solvent for the peptide, in order to encourage the aggregation of the cyclic peptides while keeping the polymer as solvated as possible.
The self-assembly process was followed by dynamic light scattering (DLS) and characterized by transmission electron microscopy (TEM). DLS showed complete dissociation of the conjugate in TFA (see Fig. S3, ESI†), and as this solution was diluted with DMF, then THF, NTs were formed. These tubes could be reversibly dissociated by heating the solution at 60 °C for 4 days with LiBr. TEM (Fig. 3a) and SEM (Fig. 2) confirm the structure of nanotubes in samples of high THF dilution of size ranging from 100 to 350 nm, and thickness ranging from 10 to 14 nm. It is important to note that the self-assembly of the conjugate is very similar with both 50% excess polymer and 10% excess polymer (see Fig. S4, ESI†), indicating that the free polymer that results from the conjugation reaction has very little effect on the structure and formation of the nanotubes. Consequently, conjugate purification prior to self-assembly is unnecessary, thus simplifying the process of tube formation. It is also remarkable that the TEM images of the nanotubes obtained from all conjugates reveal a 1 nm wide channel due to the cyclic peptide scaffold, which is not observed in the case of 4 arm conjugates, presumable because it is obscured by the higher density of the polymeric shell.29 The visibility of this channel in the case of this ‘2 arm’ peptide suggests that the polymer is physisorbed on the surface of the TEM grid, and spread relatively flat along the surface of the grid. Such an observation is consistent with the width of the observed tubes, which is much larger than what might be expected if the polymers were coiled as in solution. A final note on the self-assemblies concerns the techniques of characterisation. Although complete removal of copper is possible via washing with an EDTA solution (see Experimental section), we discovered that small amounts of copper left in the conjugates purified over neutral alumina acted as a better TEM contrast agent than stains such as RuO4 resulting in clearer images. This further highlights the usefulness of the CuAAC reaction—not only does it allow for an efficient synthesis, but it also avoids the need for additional contrast agents in the characterisation.
 | ||
| Fig. 2 Scanning electron microscopy images of pBA16 conjugates with only partial removal of copper, self-assembled in THF solution. Scale bar 200 nm. | ||
We were also interested in establishing the effect of the molecular weight of the polymer side chain on the self-assembly of the peptide. We prepared a variety of pBA conjugates, and observed that, despite the relative polydispersity in length, NTs appear to have shorter lengths (size ranging from 15 to 100 nm) and a slightly thicker shell (typically ∼15 nm), as the polymer molecular weight is increased from 2300 g mol−1 (pBA16) to 4900 g mol−1 (pBA36) and 14100 g mol−1 (pBA108). This is in agreement with the expectation that longer polymeric chains should sterically hinder the ability of conjugates to stack together, and their increased chain length would be expected to cause them to stretch further across the TEM grid (Fig. 3). Indeed, similar results have been reported for polymer nanotubes synthesized via a divergent route from a pre-assembled peptide tube.26 One of the unique advantages of the convergent approach is that it allows investigation of the effect on the self-assembly process of mixing two or more different types of conjugates. For example, mixing a fraction of conjugates prepared from longer polymers with shorter conjugates should permit the control of the length of the resulting tubes. Indeed, we observed that introducing 75% (mol) of large (pBA108) conjugate on the self-assembly of the pBA16 conjugate leads to nanotubes of length between that of the pure pBA16 and the pure pBA108 self-assembled conjugates (Fig. 3d). This approach offers a unique opportunity to control the length of nanotubes.
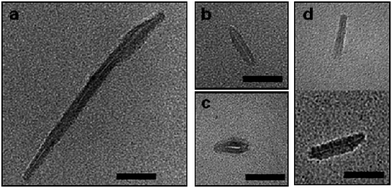 | ||
| Fig. 3 Typical TEM images self-assembled samples of (a) pBA16 conjugates; (b) pBA36 conjugates; (c) pBA108 conjugates and; (d) 75% (mol) of (pBA108) mixed with 25% (mol) of pBA16 conjugates. All samples were assembled by dilution of TFA/DMF solution with THF. Scale bar 50 nm. | ||
Functional nanotubes
The combinatorial nature of convergent synthesis also permits development of a range of functional conjugates by simply altering the nature of the polymeric chains. We exploited this opportunity by synthesizing a range of differing polymers, following the same procedure as that described above, and conjugated them to a cyclic peptide to form nanotubes exhibiting a range of functionalities in their shell. In order to illustrate the variety of functionalities accessible, we produced conjugates of poly(styrene), poly(dimethylamino ethyl acrylate), poly(acrylic acid) and poly(hydroxyethyl acrylate). Conjugates of pS20 were assembled by dilution with THF, whilst conjugates of pDMAEA19 and pAA55 were assembled by dilution with water. The resulting nanotubes offer a range of functional shells, which incorporate phenyl (hydrophobic shell), tertiary amines and carboxylic acid groups (hydrophilic and pH responsive shells), respectively (Fig. 4). Moreover, our modular approach allows the preparation of tubes with multiple functionalities on the shell, by mixing conjugates prepared from different polymers prior to self-assembly. For instance, we also prepared tubes from mixtures of pBA16/pDMAEA19 and pBA16/pS20 (1:1 w/w), as shown in Fig. 4 (see Fig. S6, ESI†, for DLS). We expect the control of shell functionality of these nanotubes offers great opportunities for applications in material, medical and nanotechnology fields.
 | ||
| Fig. 4 Typical TEM images of self-assembled conjugates of (a) pAA; (b) pDMAEA; (c) pS; (d) pHEA; (e) 50% pDMAEA, 50% pBA16 and; (f) 50% pS, 50% pBA16. Scale bar 50 nm. | ||
The self-assembly of the pHEA15 was also followed in a similar fashion. High dilutions with methanol (∼99%) led to the formation of nanotubes (Fig. 4), although in lower concentration and in a less controlled fashion (see Fig. S8, ESI†). We have previously reported that the conjugation of pHEA to amyloid-forming peptides prevents the formation of β-sheets, due to polar interaction with the polymeric chains,41 and we expect the same effect is observed here, with the pHEA inhibiting aggregation of the cyclic peptides. This effect is clearly of great interest, as pHEA conjugates could be used to further control the aggregation of cyclic peptides, thus offering another dimension to the control of the length of our nanotubes. We are currently investigating further this procedure in our laboratories.
Conclusions
We have established a new strategy to produce functional organic nanotubes of controlled structure. A convergent synthesis strategy, based on CuAAC click reaction, provides cyclic peptide/polymer conjugates of very-well defined structure. Our modular approach to self-assemble these conjugates into nanotubes is a powerful technique that allows for controlling both the size distribution of these nano-objects (by varying the size of the polymer conjugate) and the functionality of the nanotube shell (by varying the type of polymer conjugates). The preparation of functional nanotubes is a major advantage of the modular approach we have introduced here. Indeed, to date, only a small number of functional organic nanotubes have been reported. With our approach, it is now possible to generate a large number of nanotubes in a controlled and rapid process, and the structures described above are only a few examples of materials that are available. We believe this new approach will permit the design of a wide range of functional organic nanotubes of controlled structure, in a simple and efficient process.Acknowledgements
The authors thank Henry Jiang and Alison Mildenhall for their help with some of the experimental work, and Cheuk Ka Poon for useful discussions in relation to the project. We are grateful for funding through the ARC discovery program grant (DP110101608) and the ACMM for electron microscopy assistance. R.C. acknowledges the Australian government for provision of an APA research scholarship.References
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS.
- B. Eisenberg, Acc. Chem. Res., 1998, 31, 117–123 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- C.-G. Wu and T. Bein, Science, 1994, 264, 1757–1759 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- P. M. Ajayan and T. W. Ebbesen, Rep. Prog. Phys., 1997, 60, 1025 CrossRef CAS.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS.
- X. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037–2050 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- S. Scanlon and A. Aggeli, Nano Today, 2008, 3, 22–30 CrossRef CAS.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- D. W. P. M. Lowik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- H. A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- J. C. M. Van Hest,, Polym. Rev., 2007, 47, 63–92 Search PubMed.
- G. W. M. Vandermeulen and H. A. Klok, Macromol. Biosci., 2004, 4, 383–398 CrossRef CAS.
- M. A. B. Block and S. Hecht, Angew. Chem., Int. Ed., 2005, 44, 6986–6989 CrossRef CAS.
- K. Channon and C. E. MacPhee, Soft Matter, 2008, 4, 647–652 RSC.
- H. M. König and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2007, 46, 8334–8340 CrossRef.
- H. G. Börner, Prog. Polym. Sci., 2009, 34, 811–851 CrossRef.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS.
- T. D. Clark, J. M. Buriak, K. Kobayashi, M. P. Isler, D. E. McRee and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 8949–8962 CrossRef CAS.
- J. Couet, J. D. Jeyaprakash, S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297–3301 CrossRef CAS.
- M. G. J. ten Cate, N. Severin and H. G. Börner, Macromolecules, 2006, 39, 7831–7838 CrossRef CAS.
- T. Xu, N. Zhao, F. Ren, R. Hourani, M. T. Lee, J. Y. Shu, S. Mao and B. A. Helms, ACS Nano, 2011, 1376–1384 Search PubMed.
- S. Loschonsky, J. Couet and M. Biesalski, Macromol. Rapid Commun., 2008, 29, 309–315 CrossRef CAS.
- J. Couet and M. Biesalski, Small, 2008, 4, 1008–1016 Search PubMed.
- J. Couet and M. Biesalski, Macromolecules, 2006, 39, 7258–7268 CrossRef CAS.
- C. Reiriz, R. J. Brea, R. Arranz, J. L. Carrascosa, A. Garibotti, B. Manning, J. M. Valpuesta, R. Eritja, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2009, 131, 11335–11337 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Aust. J. Chem., 2010, 63, 1169–1172 Search PubMed.
- E. Vargun and A. Usanmaz, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3957–3965 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075–18077 CrossRef CAS.
- S. Dehn, R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Rev., 2011, 51, 214–234 Search PubMed.
- R. J. Griffin, Prog. Med. Chem., 1994, 131, 121–232 Search PubMed.
- J. Baskin and C. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 CrossRef CAS.
- V. Ladmiral, T. M. Legge, Y. L. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. Le, R. Mayadunne, G. Mejis, C. Moad, G. Moad, E. Rizzardo and S. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- H. Kakwere, R. J. Payne, K. A. Jolliff and S. Perrier, Soft Matter, 2011, 7, 3754–3757 RSC.
- D. T. S. Rijkers, G. W. van Esse, R. Merkx, A. J. Brouwer, H. J. F. Jacobs, R. J. Pieters and R. M. J. Liskamp, Chem. Commun., 2005, 4581–4583 RSC.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- A. J. T. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172–4174 RSC.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00202c |
| This journal is © The Royal Society of Chemistry 2011 |