Poly(norbornene) block copolymer-based shell cross-linked micelles with Co(III)–salen cores†
Yu
Liu
,
Victor
Piñón
III
and
Marcus
Weck
*
Molecular Design Institute and Department of Chemistry, New York University, New York, NY 10003-6688, USA. E-mail: marcus.weck@nyu.edu
First published on 14th June 2011
Abstract
A series of poly(norbornene) based amphiphilic triblock copolymers containing covalently attached salen ligands in the hydrophobic block, a cinnamate group-containing middle block and a poly(ethylene glycol) methyl ether-containing repeat unit as the hydrophilic block have been synthesized using living ring-opening metathesis polymerization. Micellar assemblies constructed of these copolymers were stabilized by cross-linking of the cinnamate-containing middle block using UV irradiation. The resulting shell cross-linked micelles (SCMs) have salen ligands selectively located within the hydrophobic core that are modified further by metal complexation with cobalt ions to produce SCMs core supported Co(III)-salen catalysts. The catalytic activities of these SCM catalysts were systematically investigated by hydrolytic kinetic resolution of epichlorohydrin. It was found that the composition of the copolymers and the size of SCM have substantial influences on the catalytic activity of SCMs catalysts.
Introduction
Block copolymer based micelles and nanoparticles are among the most intriguing functional nano-sized assemblies.1–6 Depending on the properties of the constituent monomers, the degree of polymerization, the block ratio, as well as structures and morphologies (size, shape, periodicity, etc.) obtained, block copolymer based micelles and nanoparticles have a broad range of applications, including nano-object assemblies,3,7drug delivery and imaging,2,8,9 metal nanoparticles synthesis and catalysis,6,10,11 surface modification,12 and stabilizers and emulsifiers.13 Owing to recent advances in living polymerization methodologies, a diverse set of copolymers with well-defined and functional building blocks are accessible providing a large library of materials that, upon self-assembly, allow for a high degree of control over morphology, dimensions and functions of the resulting micelles and nano-scale objects.14–16 Polymeric micelles assembled via noncovalent chemistry are vulnerable to external environmental stimuli, such as temperature, concentration, pH and solvents. Wooley's group pioneered an elegant strategy to overcome this limitation by stabilizing the micellar structures of block copolymers through covalent cross-links.17 These ‘shell cross-linked micelles’ (SCMs) have covalent cross-links throughout a specific domain to stabilize the micellar assemblies.18–34 The cross-linking domains are located in either the shell layer or the core–hydrophilic corona interface.18 A variety of chemistries have been employed for the crosslinking step including amide couplings, quaternizations of tertiary amines, esterifications, disulfur bond formations, 1,3-dipolar cycloadditions, radical chemistry and photo-irradiated coupling.19While SCM fabrication approaches have been optimized over the past 15 years,17 research on the practical applications of SCMs is still at an early stage. The most promising applications to date are in biomedical science, in particular drug delivery and imaging.35–40 Other applications include the synthesis of metal nanoparticles and the stabilization of metal colloids.41,42 It has been suggested in the literature that SCMs, due to the robust structure and tunability of the building blocks, might be an attractive scaffold for supported catalysis.43 In particular, the core domain of SCMs could offer a unique catalytic environment that may benefit catalytic transformations. For example, the highly dense three-dimensional arrangement of catalysts in the core could force catalysts in close proximity resulting in enhanced reactivity for catalysis that follow a bimolecular reaction mechanism. Furthermore, the confined hydrophobic environment of the core may have substrate selectivity while the hydrophilic corona and the cross-linked shell may provide a shield against catalyst poisoning and metal leaching. In addition, the stabilized core–shell structure and the gigantic size compared with small molecule-based reactants and products might allow for the recovery and recycling of SCMs catalysts. On the other hand, the permeabilities of the reactants and products through the SCMs might have a significant impact on the reaction rate. The only example of catalyst containing SCMs has been reported by O'Reilly's group who has synthesized SCM nanoparticles assembled from poly(acrylate)-b-poly(styrene) diblock copolymers.44 The core of the SCMs was functionalized with Cu(I)–terpyridine complexes that could catalyze the 1,3-dipolar cycloaddition of azido and alkynyl functionalized small molecules.
Herein, we report the synthesis of SCMs with Co(III)–salen functionalized cores assembled from poly(norbornene) based amphiphilic ABC triblock copolymers (Fig. 1). The poly(norbornene) block copolymers are synthesized viaring-opening metathesis polymerization (ROMP). The middle block (B block) is functionalized with a cinnamate group for photo-triggered crosslinking, while the terminal hydrophobic block contains a salen ligand. Selective complexation of Co with the salen in the core domain produced Co–salen functionalized SCMs catalysts that are examined as catalysts for the hydrolytic kinetic resolution (HKR) of epichlorohydrin. Our results show that the compositions of SCMs including the size and the ratio of polymerization of individual blocks have a significant influence on the catalytic activity. This study demonstrates the capability of SCMs as a catalyst support to perform catalytic reaction in aqueous solutions and shows the potential of SCMs as nanoreactors.
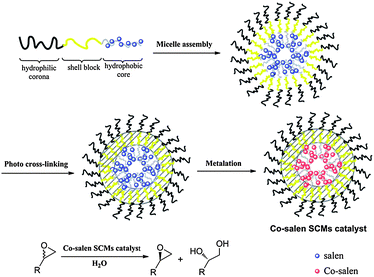 | ||
| Fig. 1 Schematic illustration of the synthesis of SCMs with Co–salen functionalized cores and their application in the HKR of epoxides. | ||
Experimental section
General methods and materials
All reagents were obtained from commercial suppliers and used without further purification unless specified below. Dichloromethane was dried by passing through columns of activated copper and alumina successively. Monomers 4,455,46,476,48 and isomerically pure exo-norbornene acid49–51 were synthesized as reported in the literature. Flash column chromatography was performed using silica gel 60 Å (230–400 mesh) from Sorbent Technologies. 1H NMR spectra were recorded at 25 °C on a Bruker AC 400 (400 MHz) spectrometer. 13C NMR spectra were obtained at 100.0 MHz at 25 °C on a Bruker AC 400 spectrometer. All chemical shifts are reported in parts per million (ppm) with reference to residual solvent peaks. Coupling constants (J) are reported in Hertz (Hz), and splitting patterns are reported as singlet (s), doublet (d), triplet (t), quartet (q) and unresolved multiplet (m). ESI-MS was conducted on an Agilent 1100 serial Capillary LCMSD Trap XCT system equipped with atmospheric pressure electrospray ionization source. Gel-permeation chromatography (GPC) analyses were performed either on a Shimadzu LC-10AD pump coupled with a UV detector with THF as the mobile phase (flow rate at 1.0 mL min−1) or on a Agilent 1200 series isocratic pump coupled with a UV detector and the Optilab ReX differential refractive index detector with CH2Cl2 as the mobile phase (flow rate at 1.0 mL min−1). For both systems the corresponding American Polymer Standards columns were used and poly(styrene)s were employed as standards for calibration. Hydrodynamic diameters of micelles and SCMs were measured at 25 °C by dynamic light scattering (DLS) using a Protein Solution DynaPro instrument with a 663 nm laser module. Size distributions (% PD: percentage of polydispersity) were determined by the DLS instrument associated software. UV-vis absorption spectra were recorded on an Agilent 8453 UV-visible Spectrophotometer. AFM images were recorded in tapping mode with either an Asylum research MFP-3D or a Digital Instruments Nanoscope IIIa using Olympus AC240TS cantilevers (Spring constant: 2 N m−1, resonant frequency: 70 kHz). The AFM samples were prepared by drop deposition of a 20 μL sample solution at a concentration of ca. 0.01 mg mL−1 onto freshly cleaved mica and allowed to dry in air. The AFM measurements were conducted under ambient conditions. The mean particle diameters were obtained from the statistical analysis of at least 40 particles in an area of 1 μm × 1 μm using Gwyddion software. The chiral GC analyses were carried out on a Shimadzu GC 14-A instrument equipped with an FID detector. A Chiraldex γ-TA column (Advanced Separation Technologies, Inc.) was used with helium as the carrier gas. Elemental analyses were performed by Quantitative Technologies, Inc. High resolution mass spectra (HRMS) were performed by the instrumentation facility of the Department of Chemistry at the University of Michigan, Ann Arbor.4-Hydroxybutyl-exo-bicyclo[2.2.1]hept-5-ene-2-carboxylate (7)
exo-Norbornene acid (0.7 g, 5 mmol), 1,4-butanediol (0.57 g, 6.25 mmol) and DMAP (67.2 mg, 0.55 mmol) were dissolved in 15 mL anhydrous CH2Cl2. The solution was cooled to 0 °C. DCC (1.13g, 5.5 mmol) was added, and the mixture was stirred at 0 °C for 1 hour and then at room temperature overnight. The precipitated urea was filtered off and the filtrate was evaporated under reduced pressure. The residue was taken up with ethyl acetate (100 mL) and filtered again. The ethyl acetate solution was washed with 0.5 M HCl solution (50 mL), saturated NaHCO3 solution (50 mL) and brine (50 mL) sequentially. The organic layer was dried over MgSO4. After filtering and removing the solvent under reduced pressure, the crude product was subjected to silica chromatography (hexane/ethyl acetate, from 100/0 to 100/30) to give the product as a colorless liquid (0.57 g, 54% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 6.08 (m, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 4.07 (t, J = 6.7 Hz, 2H, CH2-OCO-), 3.61 (t, J = 6.3 Hz, 2H, CH2-OH), 2.98 (s, 1H, CH), 2.87 (s, 1H, CH), 2.50 (s, 1H, OH), 2.17 (m, 1H, CH), 1.86 (dt, J1 = 11.8, J2 = 4.0, 1H, CH), 1.59 (m, 2H, CH2), 1.60 (m, 2H, CH2), 1.47 (m, 1H, CH), 1.32 (m, 2H, CH, CH); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.4, 138.0, 135.7, 64.3, 62.1, 46.6, 46.3, 43.2, 41.6, 30.3, 29.0, 25.2; MS (ESI-MS) calcd for C12H18O3 (m/z), 210.1; found, 211.1 (M + H+), 233.0 (M + Na+).
CH), 4.07 (t, J = 6.7 Hz, 2H, CH2-OCO-), 3.61 (t, J = 6.3 Hz, 2H, CH2-OH), 2.98 (s, 1H, CH), 2.87 (s, 1H, CH), 2.50 (s, 1H, OH), 2.17 (m, 1H, CH), 1.86 (dt, J1 = 11.8, J2 = 4.0, 1H, CH), 1.59 (m, 2H, CH2), 1.60 (m, 2H, CH2), 1.47 (m, 1H, CH), 1.32 (m, 2H, CH, CH); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.4, 138.0, 135.7, 64.3, 62.1, 46.6, 46.3, 43.2, 41.6, 30.3, 29.0, 25.2; MS (ESI-MS) calcd for C12H18O3 (m/z), 210.1; found, 211.1 (M + H+), 233.0 (M + Na+).
4-((Bicyclo[2.2.1]hept-5-ene-2-carbonyl)oxy)butanoic acid (8)
Alcohol 7 (0.47 g, 2.24 mmol) was dissolved in 4.5 mL of DMF. Pyridinium dichromate (PDC, 5.3 g, 14 mmol) was dissolved in 10.6 mL of DMF and added dropwise to the DMF solution of 7. The reaction mixture was stirred at room temperature overnight and then 50 mL ethyl acetate was added. The solution was passed through a Celite bed that was washed three times with ethyl acetate. The organic solutions were combined (200 mL total) and washed with a HCl solution (100 mL, 0.1 M) and brine (100 mL) sequentially. After the organic phase was dried over MgSO4, the solvent was removed under reduced pressure. The residue was subjected to silica chromatography (hexane/ethyl acetate, from 100/20 to 100/100) to give the product in 83% yield (413 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ (ppm) 6.11 (m, 2H, CHCH), 4.15 (t, J = 6.2 Hz, 2H, CH2-OCO-), 3.02 (s, 1H, CH), 2.91 (s, 1H, CH), 2.48 (t, J = 7.2 Hz, 2H, CH2-OH), 2.21 (m, 1H, CH), 2.00 (m, 2H, CH2), 1.86 (dt, J1 = 11.9, 1H, J2 = 4.0, CH), 1.50 (m, 1H, CH), 1.37 (m, 2H, CH, CH); 13C NMR (100 MHz, CDCl3) δ (ppm) 179.1, 176.2, 138.0, 135.7, 63.3, 46.6, 46.4, 43.1, 41.6, 30.7, 30.3, 23.8; MS (ESI-MS) calcd for C12H16O4 (m/z), 224.1; found, 225.2 (M + H+), 247.1 (M + Na+).
Salen monomer 1
Carboxylic acid 8 (200 mg, 0.89 mmol) was added to a 50 mL round bottom flask that was flushed with nitrogen before CH2Cl2 (10 mL) was added. Salen methyl alcohol52 (464 mg, 0.89 mmol), DMAP (11.9 mg, 0.098 mmol) and DCC (202 mg, 0.98 mmol) were added sequentially. The reaction mixture was heated under reflux for 12 hours. After cooling to room temperature, the urea precipitate was removed by filtration. The precipitate was washed twice with hexane/ethyl acetate (1/1) until its color became white. The organic solutions were combined and dried over MgSO4. After removal of the solvent, the residue was subjected to silica gel column chromatography using hexane/diethyl ether (100/0 to 100/30) to give the product in 78% yield (500 mg) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ (ppm) 14.10 (s, br, 1H, OH), 13.65 (s, br, 1H, OH), 8.32 (s, 1H, NCH), 8.30 (s, 1H, NCH), 7.35 (d, J = 1.3 Hz, 1H, Ar-H), 7.25 (s, 1H, Ar-H), 7.08 (s, 1H, Ar-H), 6.99 (d, J = 1.2 Hz, 1H, Ar-H), 6.13 (m, br, 2H, CHCH), 5.00 (s, 2H, CH2-Ar), 4.13 (t, J = 6.4, 2H, CH2), 3.34 (m, 2H, N-CH), 3.05 (s, 1H, CH), 2.92 (s, 1H, CH), 2.42 (t, J = 7.4, 2H, CH2), 2.21 (m, 1H, CH), 1.96 (m, 6H, 2 NCH-CH2, OCH-CH2), 1.76 (m, 1H, CHH), 1.61 (m, 1H, CHH), 1.53–1.35 (m, 6H, 2CH2, 2CHH), 1.41 (s (overlapping), 18H, 2C(CH3)3), 1.24 (s, 9H, C(CH3)3); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.1, 172.8, 165.9, 165.1, 160.7, 157.9, 140.0, 138.1, 137.6, 136.4, 135.7, 130.4, 130.1, 126.9, 126.0, 124.7, 118.4, 117.8, 72.5, 72.4, 66.6, 63.4, 46.6, 46.4, 43,1, 41.6, 35.0, 34.8, 34.7, 34.0, 33.2, 31.4, 30.9 (overlapping signals), 30.3, 29.4, 29.3, 24.3, 24.1; HRMA (ESI+) calcd for C45H62N2O6 [M + H]+, 727.4681; found, 727.4682. Elemental analysis calcd for C45H62N2O6: C, 74.35; H, 8.60; N, 3.85%; found: C, 74.33; H, 8.74; N, 3.76%.
11-Bromoundecyl-exo-bicyclo[2.2.1]hept-5-ene-2-carboxylate (9)
exo-Norbornene acid (0.83 g, 6 mmol), 11-bromo-1-undecanol (1.5 g, 6 mmol) and DMAP (80.6 mg, 0.66 mmol) were dissolved in 80 mL anhydrous CH2Cl2. The solution was cooled to 0 °C. Then, DCC (1.36g, 6.6 mmol) was added and the mixture was stirred at 0 °C for 5 minutes and then at room temperature overnight. The precipitated urea was filtered and the filtrate was evaporated under reduced pressure. The residue was taken up with 120 mL of ethyl acetate and filtered again. The ethyl acetate solution was washed with a 0.5 M HCl solution (80 mL), saturated NaHCO3 solution (80 mL) and brine (80 mL) sequentially. The organic layer was dried over MgSO4. After filtering and removing the solvent under reduced pressure, the crude product was subjected to silica gel column chromatography (hexane/ethyl acetate, from 100/0 to 100/10) to give the product as a colorless oil (2.07 g, 93% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 6.09 (m, 2H, CHCH), 4.05 (t, J = 7.0 Hz, 2H, CH2-OCO-), 3.36 (t, J = 6.9 Hz, 2H, CH2-Br), 3.00 (s, 1H, CH), 2.87 (s, 1H, CH), 2.18 (m, 1H, CH), 1.89 (m, 1H, CHH), 1.81 (m, 2H, CH2), 1.60 (m, 2H, CH2), 1.49 (m, 1H, CHH), 1.45–1.25 (m, 16H, 7 CH2, 2 CHH); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.2, 138.0, 135.8, 64.5, 46.6, 46.3, 43.2, 41.6, 33.9, 32.8, 30.3, 29.43, 29.40, 29.37, 29.2, 28.7, 28.6, 28.1, 25.9; MS (ESI-MS) calcd for C19H31BrO2 (m/z), 370.1; found, 392.9 (M + Na+).
Cinnamate monomer 2
Cinnamic acid (0.56 g, 3.8 mmol) and K2CO3 (0.74 g, 53.8 mmol) were dissolved in 25 mL of DMF. Bromide 9 (1 g, 2.69 mmol) was dissolved in 4 mL of DMF and slowly added to the reaction mixture. After stirring at 90 °C overnight, the solvent was removed and the residue was dissolved in CH2Cl2 (150 mL). The organic solution was washed with 0.5 M HCl (50 mL), saturated NaHCO3 (50 mL) and brine 50 mL sequentially. The organic layer was dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was subjected to silica gel column chromatography (hexane/ethyl acetate, from 100/0 to 100/10) to give the product as a colorless oil (1.03 g, 87% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.67 (d, J = 16.1 Hz, CH-Ar), 7.51 (m, 2H, Ar-H), 7.37 (m, 3H, Ar-H), 6.44 (d, J = 16.1 Hz, CH-Ar), 6.11 (m, 2H, CHCH), 4.19 (t, J = 6.5 Hz, 2H, CH2-O-cinnamate), 4.06 (t, J = 6.6 Hz, 2H, CH2-O-norbornene), 3.03 (s, 1H, CH), 2.89 (s, 1H, CH), 2.22 (m, 1H, CH), 1.91 (m, 1H, CHH), 1.70 (m, 2H, CH2), 1.65 (m, 2H, CH2), 1.52 (m, 1H, CHH), 1.47–1.25 (m, 16H, 7 CH2, 2CHH); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.3, 167.0, 144.5, 138.0, 135.8, 134.5, 130.2, 128.9, 128.0, 118.3, 64.7, 64.6, 46.6, 46.4, 43.2, 41.6, 30.3, 29.5 (overlapping signals), 29.3, 29.2, 28.74, 28.71, 26.0, 25.9; HRMS [ESI+] calcd for C28H38O4 [M + H]+: 439.2843; found: 439.2847. Elemental analysis calcd for C28H38O4: C, 76.68; H, 8.73%; found: C, 76.63; H, 8.77%.
Polyethylene glycol (PEG) monomer 3
exo-Norbornene acid (1.22 g, 8.8 mmol), methoxy-poly(ethylene glycol 350) (average molecular weight 350, 2.8 g, 8 mmol) and DMAP (107.5 mg, 0.88 mmol) were dissolved in 50 mL of anhydrous CH2Cl2. EDC (1.7 g, 8.8 mmol) was added and the reaction mixture was heated under reflux overnight. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was taken up with ethyl acetate (150 mL) and water (50 mL). After extraction, the organic layer was washed with saturated NaHCO3 solution (80 mL, twice) and brine (80 mL). The organic layer was dried over Na2SO4. After filtering and removing the solvent under reduced pressure, the crude product was subjected to silica gel column chromatography (CH2Cl2/methanol, from 100/0 to 100/6). Since the product is a mixture of monomers with different PEG chain lengths, the fractions from 100/2 to 100/6 (CH2Cl2/methanol) were collected. The final product was obtained as a colorless oil (3.5 g, 88% yield). 1H NMR (400 MHz, CDCl3) δ (ppm) 6.07 (m, 2H, CHCH), 4.23 (t, J = 4.9 Hz, 2H, CH2-OCO-), 3.70 (t, J = 4.9 Hz, 2H, CH2-CH2-OCO), 3.63 (m, 18H, CH2), 3.52 (m, 2H, CH2-OMe), 3.38 (s, 3H, CH3), 3.03 (s, 1H, CH), 2.89 (s, 1H, CH), 2.23 (m, 1H, CH), 1.89 (m, 1H, CHH), 1.50 (m, 1H, CHH), 1.32 (m, 2H, 2 CHH); 13C NMR (100 MHz, CDCl3) δ (ppm) 176.1, 138.0, 135.7, 71.9, 70.6, 70.5 (overlapping signals), 70.45, 69.2, 63.4, 58.9, 46.6, 46.2, 43.0, 41.6, 30.3; products were obtained as mixture with n = 5–12 ethylene glycol units. MS (ESI-MS) calcd for C19H32O7 (n = 5, m/z), 372.2; found, 395.2 (M + Na+, 15%), C21H36O8 (n = 6, m/z), 416.2; found, 439.2 (M + Na+, 67%), C23H40O9 (n = 7, m/z), 460.3; found, 483.2 (M + Na+, 97%), C25H44O10 (n = 8, m/z), 504.3; found, 527.2 (M + Na+, 100%), C27H48O11 (n = 9, m/z), 548.3; found, 571.2 (M + Na+, 96%), C29H52O12 (n = 10, m/z), 592.4; found, 615.3 (M + Na+, 83%), C31H56O13 (n = 11, m/z), 636.4; found, 659.3 (M + Na+, 55%), C33H60O14 (n = 12, m/z), 680.4; found, 703.3 (M + Na+, 33%).
General procedure for the synthesis of block copolymers a–n
The synthesis of block copolymer g is described as a representative example: Monomers 1 (14.5 mg, 20 μmol), 2 (6.6 mg, 15 μmol) and 3 (9.4 mg, 20 μmol) were weighed into three vials. The vials were covered with septa and flushed with argon. Then, a NMR tube was sealed with a septum and flushed with argon. First, 0.4 mL degassed CDCl3 was added into the vial containing 3. Once the monomer was fully dissolved, the solution was transferred into the NMR tube. A freshly prepared CDCl3 solution of Grubbs' first generation initiator (0.05 M, 20 μL) was added to the NMR tube by a syringe. The reaction solution was shaken using a vortex. The polymerization progress was monitored by 1H NMR spectroscopy. Once 3 was fully polymerized, monomer 2, dissolved in 0.2 mL CDCl3, was injected into the reaction vial. Again, 1H NMR spectroscopy was used to monitor the reaction process. When 2 was fully consumed, monomer 1 in 0.25 mL CDCl3 was added to the reaction. Once the polymerization was accomplished as determined by 1H NMR spectroscopy, ethyl vinyl ether was added to terminate the reaction. The copolymer solution was poured into a scintillation vial containing rapidly stirred pentanes (10 mL) and a yellow precipitate formed. The mixture was centrifuged at 5000 RPM for 20 min. The supernatant was decanted and the residue was redissolved in 1 mL of CH2Cl2. The precipitation process was repeated two more times until the supernatant was colorless. The precipitate was dried under vacuum. The isolated yield of block copolymer g was 94% (28.5 mg). The final block copolymer was dissolved in THF and filtered though a 0.2 μm syringe filter. This solution was used directly for micelle formation. 1H NMR (400 MHz, CDCl3) (Fig. 4) δ (ppm) 13.95 (br s, H1), 13.55 (br s, H1′), 8.21 (br m, H2/2′), 7.60 (br d, H3), 7.43 (br m, H4), 7.29 (br m, H5/6), 7.22 (br s, H7), 7.13 (br s, H8), 6.94 (br s, H7′), 6.89 (br s, H8′), 6.48 (br d, H9), 5.02–5.38 (br m, 10/10′), 4.87 (br s, H11), 4.12 (br m, H12/13), 3.96 (br m, H14/15), 3.59 (br m, H16), 3.49 (br m, H17), 3.30 (br s, H18), 3.23 (br m, H19), 2.82–3.08 (br m, H20/21), 2.38–2.70 (br m, H20′/21′/22), 2.37 (br m, 23), 1.96 (br m, H24), 1.71–1.90 (br m, H25/26), 1.45–1.71 (br m, H27/28/29), 1.41 (br m, H24), 1.35 (br m, H31/31′), 1.24 (br m, H32), 1.15 (br m, H33); 13C NMR (100 M, CDCl3) δ (ppm) 167.1, 165.9, 165.1, 160.6, 157.9, 144.6, 139.9, 137.5, 136.4, 134.5, 130.4, 130.2, 130.0, 128.9, 128.0, 126.8, 125.9, 124.8, 118.4, 118.3, 117.8, 72.4, 72.2, 72.0, 70.6 (overlapping signals), 70.5, 69.2, 66.51, 64.7, 64.5, 63.4, 59.1, 42.0 (m), 36.3 (m), 35.0, 34.7, 34.0, 33.2, 31.4, 30.7, 30.3, 29.5, 29.4, 29.3, 28.8, 26.0, 25.9, 24.3, 24.1; Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (theoretical: 46500), PDI = 1.39. DSC analyses were performed for all block copolymers (−20 °C to 150 °C at a rate of 5 °C min−1). No detectable thermal transition was observed.
000 (theoretical: 46500), PDI = 1.39. DSC analyses were performed for all block copolymers (−20 °C to 150 °C at a rate of 5 °C min−1). No detectable thermal transition was observed.
a: The incorporation ratio of monomer 4 was determined by the integration of the new signal at 2.75 ppm (CH-COOMe). Mn = 5500 (theoretical: 46500), PDI = 1.81.
b: The incorporation ratio of monomer 4 was determined by the integration of the new signal at 2.75 ppm (CH-COOMe). Mn = 5600 (theoretical: 46300), PDI = 1.98.
c: Mn = 6800 (theoretical: 51500), PDI = 1.85.
d: The incorporation ratio of monomer 4 was determined by the integration of the new signal at 2.75 ppm (CH-COOMe). Mn = 12800 (theoretical: 50000), PDI = 1.82.
e: The incorporation ratio of monomer 4 was determined by the integration of the new signal at 2.75 ppm (CH-COOMe); Mn = 14800 (theoretical: 47800), PDI = 1.66.
f: Mn = 12800 (theoretical: 56700), PDI = 2.08.
g: Mn = 15100 (theoretical: 30500), PDI = 1.39.
h: Mn = 18200 (theoretical: 31800), PDI = 1.48.
i: Mn = 14800 (theoretical: 23800), PDI = 1.62.
j: Mn = 11000 (theoretical: 17000), PDI = 1.36.
k: The incorporation ratio of monomer 6 was determined by the integration of the new signal at 0.79 ppm (CH3-CH2). Mn = 12200 (theoretical: 19500), PDI = 1.39.
l: The incorporation ratio of monomer 6 was determined by the integration of the new signal at 0.79 ppm (CH3-CH2). Mn = 13600 (theoretical: 22000), PDI = 1.43.
m: The incorporation ratio of monomer 5 was determined by the comparison of the integration change in the signal at 3.47 ppm (CH3O) before and after incorporation of the third block. Mn = 10700 (theoretical: 18400), PDI = 1.40.
n: The incorporation ratio of monomer 5 was determined by the comparison of the integration change in the signal at 3.47 ppm (CH3O) before and after incorporation of the third block. Mn = 11100 (theoretical: 19800), PDI = 1.45.
General procedure for the micelle preparation
The triblock copolymer was dissolved in THF at 5 mg mL−1 and the solution was filtered by passing through a 0.2 μm PTFE syringe filter. Micelle formation was induced by the slow addition of methanol with a syringe pump (at a rate of 7 mL h−1) to a vigorously stirred triblock copolymer solution. The final volume ratio of THF/methanol was 1:2. The micelle solution was left standing at room temperature overnight. DLS measurements were performed at 25 °C.
General procedure for the photo-irradiated cross-linking of the micelles
The micelle solution prepared as described above was thoroughly degassed by bubbling nitrogen through it before being transferred into a cylindrical quartz cell (17 mL, 1 cm path length). The solution was irradiated for 40 minutes with a 100 W xenon arc lamp from a distance of 1 cm. UV spectroscopy was used to monitor the degree of cross-linking. For some cross-linking experiments, at certain time intervals, aliquots of the micelle solution were extracted. After the removal of the solvent, the residues were dissolved in the non-selective solvent CH2Cl2 and DLS was used to check the formation of the SCMs. Once the micelles were cross-linked, the solutions were transferred to a reaction vial and degassed with nitrogen. The SCMs solutions were used directly for metal complexation.General procedure for cobalt complexation of the salen ligands within the SCMs core and SCMs catalyst activation and purification
The SCMs solution described above was transferred into a glovebox. A Co(OAc)2·4H2O solution in anhydrous methanol (0.1 M, 1.5 equiv.) was added. The solution was stirred at room temperature for two days. The color of the solution slowly changed from orange to dark red. The metalated SCMs solution was taken out of the glovebox and stirred for 5 hours with air bubbling through. The solution became dark brown indicating that the complex was converted to the catalytically active Co(III)–salen complex. The SCMs were purified by Sephadex G-50 size-exclusive column chromatography with methanol as the eluent. The collected fractions were analyzed by DLS and the fractions containing the SCMs were combined. After the solvent was removed under reduced pressure, the SCMs catalyst was redissolved in CH2Cl2/benzene (1:2). The solution was freeze-dried to generate the catalyst-containing SCMs as a brown powder. An aliquot sample was dissolved in CH2Cl2 and analyzed by DLS to ensure the metalation and oxidation process did not dissemble the micellar structures. The cobalt contents were determined by ICP-MS. Cobalt loadings ranged from 79% to 90%.
General procedure for the HKR of epichlorohydrin by SCMs catalysts
The HKR of epichlorohydrin catalyzed by SCM catalyst e is described as an example: first, 6 mg e (containing 3.14 μmol Co(III)–salen) was weighed into a 20 mL scintillation vial. Then, 5 mL degassed water was added and the mixture was stirred until the SCMs catalyst was completely dissolved. The reaction vial was immersed in a water bath and then 50 μL of epichlorohydrin was added followed by 4.5 μL of chlorobenzene. Aliquots (150 μL) were taken at certain time intervals and extracted with diethyl ether (300 μL, twice). The combined diethyl ether solutions were dried over MgSO4 and then passed through a plug of silica gel packed in a Pasteur pipette. The filtrates were concentrated to about 100 μL and subjected to chiral GC analysis. The absolute configuration of resolved epichlorohydrin was assigned by comparing it to authentic samples. After 31 hours reaction, (S)-epichlorohydrin was obtained in 44% ee (Chiraldex γ-TA, 90 °C, isothermal, tR(R, minor) = 6.16 min, tR(S, major) = 5.96 min).Results and discussions
Design and monomer synthesis
There are a number of considerations for the design of Co(III)–salen SCMs catalysts. First is the nature of the amphiphilic block copolymer. The vast majority of block copolymers used in assemblies of SCMs are synthesized via controlled radical polymerization techniques.18,19,53 Other methodologies include the use of poly(norbornene) block copolymers synthesized viaROMP.54–56 The living nature of ROMP makes it particularly valuable for the synthesis of amphiphilic block copolymers.57 In addition, ROMP offers the advantages of mild reaction conditions and high functional group tolerance allowing for the incorporation of highly diverse functionalities into polymers.58 Metal complex catalysts have been immobilized on amphiphilic poly(norbornene) block copolymers before and have been shown to catalyze organic reactions in aqueous solution.59,60 Therefore, in this study we use poly(norbornene) copolymers synthesized viaROMP to assemble the SCM catalysts.The second important issue is the choice of the cross-linking strategy. Cross-linking of the middle block should have no influence on the salen core and the corona block. Furthermore, the cross-linkable functionalities as well as the formed cross-links should not interfere with the HKR reactions. Third, the degree of cross-linking should be controllable and easy to monitor. Finally, the crosslinking strategy should not be based on the addition of a crosslinking agent and should not form byproducts. The photo-induced [2 + 2] cycloaddition between two cinnamate derivatives fulfills these requirements and has been used successfully as cross-linking strategy in SCM synthesis.28,54,61 Therefore, in this study, cinnamate functionalized exo-norbornene monomer 2 was synthesized to construct the cross-linkable block.
The hydrophilic block is based on poly(ethylene glycol) (PEG, the average number of epoxy units = 6) functionalized exo-norbornene monomer 3, while the core polymer block is based on a salen ligand-containing exo-norbornene monomer 1. Dimethyl ester 4 was copolymerized in a random fashion with 2 to space out the crosslinking units thereby reducing intramolecular cycloaddition reactions and to enhance the permeability of small molecules through the cross-linked shell.54Triethylene glycol methyl ether modified monomer 5 and alkyl chain containing monomer 6 were incorporated into the core block to tune the hydrophobicity of the SCMs. Monomers 1–3, 5,46,47 and 648 were prepared from isomerically pure exo-norbornene carboxylic acid49–51 (Scheme 1). Monomer 4 was synthesized as described in the literature.45
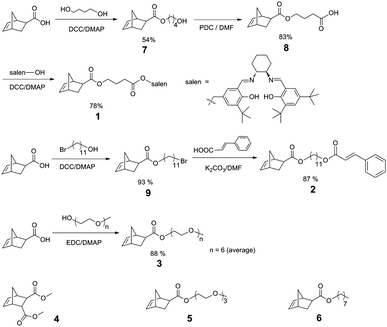 | ||
| Scheme 1 Synthetic routes towards monomers 1–3 and monomers 4–5 used in this study. | ||
Homopolymerizations
The polymerizations of monomers 4, 5 and 6 using Grubbs' first generation initiator either have been proven to be living or have been used successfully in the literature for the synthesis of block copolymers.46–48,62 The polymerization behaviors of monomers 1, 2 and 3 were investigated in chloroform using Grubbs' first generation initiator. Gel-permeation chromatography (GPC) and 1H NMR spectroscopy showed quantitative conversions of monomers 1, 2 and 3 in less than two hours depending on the monomer-to-initiator ratios. The living nature of the polymerizations was tested via a homoblock copolymerization experiment. First, a homopolymer with a monomer to initiator ratio ([M]/[I]) of 20:1 was synthesized. After completion, 200 equivalents of additional monomer were added to the reaction solution. The 20mer homopolymer and the 220mer were analyzed by GPC using THF as the eluent (Fig. 2). The GPC trace of Poly2 shows a complete shift to high molecular weight without traces of terminated low molecular weight polymer, indicating the living nature of 2. PEG polymers often have good solubilities in CH2Cl2, so we analyzed the living nature of 3viaGPC using CH2Cl2 as the eluent. Despite significant increases in the polydispersity, a complete shift of the high molecular weight trace from the low molecular weight trace was observed providing evidence of the controlled polymerization of 3. Despite complete conversion during the polymerization at low [M] to [I] ratios, 1 could not be fully converted to high molecular weight polymer. The terminated low molecular weight polymer and the unreacted monomer were always detected by GPC indicating an uncontrolled polymerization. To further probe the polymerization of 1, we monitored the carbene signal during the polymerization by 1H NMR spectroscopy. In a 20:1 [M]/[I] ratio, full initiation of Grubbs' first generation initiator was verified by the complete shift of the carbene signal from 19.9 ppm to 18.7 ppm. Upon further addition of 200 equivalents of 1, the initiated carbene signal disappeared, confirming the uncontrolled polymerization of monomer 1.
![GPC
chromatograms of the homoblock copolymer tests. (A) Poly2. Dashed line: homopolymer after complete conversion ([M] : [I] = 20 : 1, Mn = 9900, PDI = 1.27). Solid line: the same polymer after addition of additional 200 equiv. of monomer 2 ([M] : [I] = 220 : 1, Mn = 106 000, PDI = 1.68). (B) Poly3. Dashed line: homopolymer after complete conversion ([M] : [I] = 30 : 1, Mn = 12 900, PDI = 1.31). Solid line: the same polymer after the addition of additional 300 equiv. of 3 ([M] : [I] = 330 : 1, Mn = 95 000, PDI = 1.82). (C) Poly1. Dashed line: homopolymer after complete conversion ([M] : [I] = 20 : 1, Mn = 13 300, PDI = 1.23). Solid line: the same polymer after the addition of additional 200 equiv. of 1 ([M] : [I] = 220 : 1, main peak, Mn = 198 000, PDI = 1.66). All molecular weights are reported vs.poly(styrene) standards.](/image/article/2011/PY/c1py00151e/c1py00151e-f2.gif) | ||
| Fig. 2
GPC
chromatograms of the homoblock copolymer tests. (A) Poly2. Dashed line: homopolymer after complete conversion ([M]:[I] = 20:1, Mn = 9900, PDI = 1.27). Solid line: the same polymer after addition of additional 200 equiv. of monomer 2 ([M]:[I] = 220:1, Mn = 106000, PDI = 1.68). (B) Poly3. Dashed line: homopolymer after complete conversion ([M]:[I] = 30:1, Mn = 12900, PDI = 1.31). Solid line: the same polymer after the addition of additional 300 equiv. of 3 ([M]:[I] = 330:1, Mn = 95000, PDI = 1.82). (C) Poly1. Dashed line: homopolymer after complete conversion ([M]:[I] = 20:1, Mn = 13300, PDI = 1.23). Solid line: the same polymer after the addition of additional 200 equiv. of 1 ([M]:[I] = 220:1, main peak, Mn = 198000, PDI = 1.66). All molecular weights are reported vs.poly(styrene) standards. | ||
To further characterize the living nature of 2 and 3, we also conducted a series of homopolymerizations with [M]/[I] ranging from 20:1 to 100:1. Both 2 and 3 afforded a linear relationship between the [M]/[I] ratio and molecular weights (Fig. 3). These results demonstrate further the living nature of the ROMP of 2 and 3 using Grubbs' first generation initiator. It was noticed that for Poly1 and Poly2 the number-average molecular weights (Mn) deduced from GPC are close to the calculated molecular weights, but the experimental molecular weights of Poly3 obtained from GPC are significantly smaller than the calculated ones which might suggest a densely filled sphere-like conformation of Poly3 in solution.631H NMR spectroscopy was applied to analyze the degree of polymerization of Poly3 through end group analysis and a linear relationship of the degree of polymerization with the monomer to initiator feed ratio was confirmed.
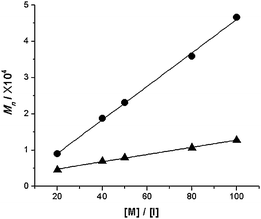 | ||
| Fig. 3 Plots of Mnvs. monomer/initiator ratios for Poly2 (circles) and Poly3 (triangles). Molecular weights are reported vs.poly(styrene) standards. | ||
Synthesis of amphiphilic triblock copolymers
The goal of this work is to construct poly(norbornene) based SCMs having Co(III)–salen functionalities within the hydrophobic core and to investigate their catalytic activities in the hydrolytic kinetic resolution of epoxides. Since monomers 2 and 3 polymerize in a living fashion, the synthesis of the triblock copolymer was executed in the following order: the polymerization was started with monomer 3 followed by monomer 2 and finally monomer 1. The block copolymerization was carried out under an inert atmosphere at ambient temperature using dry degassed CDCl3 or CHCl3 and Grubbs' first generation initiator. Once the polymerization was complete, the reaction was terminated with the addition of ethyl vinyl ether and the block copolymer precipitated from cold n-pentane. The copolymerization took between 2 and 4 hours depending on the degree of polymerization and copolymer composition. Isolated yields of all copolymers ranged from 85% to 97%.All copolymerizations were monitored by 1H NMR spectroscopy. Fig. 4 shows the 1H NMR spectroscopy characterization for the formation of copolymer g. The complete shift of the olefin signals of norbornene from 6.05 ppm to 5.0–5.4 ppm (two peaks for cis and trans isomers) indicates the complete conversion of the monomers, which was also accompanied by a broadening of all signals of the norbornene protons. From Fig. 4A–C, the newly arising signals in the 6.0 ppm to 8.5 ppm region demonstrate the sequential incorporation of the cinnamate functionality and salen ligand into the triblock copolymer. Based on the 1H NMR spectra, the ratio of repeating units of the three blocks (Poly3:Poly2:poly1) was estimated to be 1:0.8:1. By comparing the signals of each block with the carbene signal of the initiator during the polymerization, the degree of polymerization of each individual block was determined. We obtained degrees of polymerization for g of 20:16:20, which is consistent with the monomer feed ratio of 20:15:20.
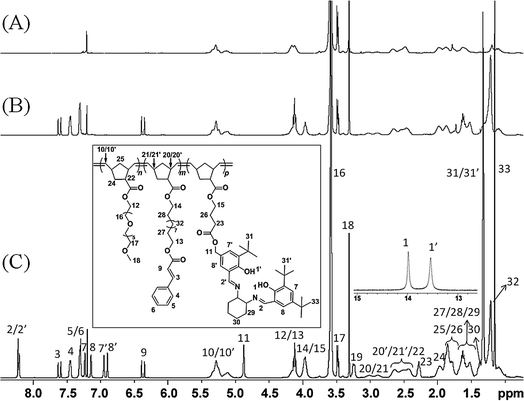 | ||
| Fig. 4 Stepwise 1H NMR characterization of the formation of triblock copolymer g (n:m:p = 20:15:20). (A) Poly3, (B) Poly3-b-Poly2 and (C) Poly3-b-Poly2-b-Poly1. | ||
The triblock copolymerization was also monitored by GPC using THF as the eluent. As a representative example, Fig. 5 shows the stepwise block polymerization characterized by GPC for copolymer e. After each step (sequential addition of monomers), a clear shift of the copolymer signal in the GPC toward higher number-average molecular weights (Mn) was observed. The polydispersity indices (PDI = Mw/Mn) ranged from 1.61 for the monoblock to 1.72 for the diblock copolymer and 1.66 for the final triblock copolymer.
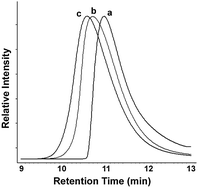 | ||
| Fig. 5 Normalized gel-permeation chromatograms of (a) monoblock polymer Poly340, (b) diblock copolymer Poly340-b-Poly215-co-420 and (c) triblock copolymer Poly340-b-Poly215-co-420-b-poly125. | ||
After establishing the copolymerization conditions for the synthesis of the amphiphilic block copolymers, we synthesized a series of triblock copolymers (Tables 1 and 2). Apart from monomers 1, 2 and 3, monomers 4, 5, and 6 were also selectively incorporated into the different blocks of the copolymers. 1H NMR spectroscopy and GPC were used to characterize all copolymers. 1H NMR spectra showed that the resulting polymer compositions closely followed the monomer feed ratios demonstrating the excellent control of the polymerization. GPC chromatograms of the final triblock copolymers all showed a monomodal distribution with polydispersities ranging from 1.36 to 2.08. The molecular weights obtained by GPC were smaller than the calculated molecular weights. In particular, increasing the portion of the hydrophilic block Poly3 led to substantial deviations from the calculated molecular weights. Poly3 has a hydrophobic backbone and hydrophilic side-chains. It has been reported that such polymeric structures have very contracted dimensions as compared to the corresponding linear homopolymers.63 Nevertheless, the combined NMR spectroscopy and GPC data proved that triblock copolymers with different polymer compositions and degrees of polymerization were obtained.
| Block copolymer |
n:m:o:p (theoretical) |
n:m:o:p (1H NMR) |
M n (calcd) | M n (GPC) | PDI |
|---|---|---|---|---|---|
| a | 75:15:5:5 |
82:16:3:5 |
46500 |
5500 | 1.81 |
| b | 60:20:10:10 |
73:21:9:8 |
46300 |
5600 | 1.98 |
| c | 60:20:0:20 |
72:20:0:22 |
51500 |
6800 | 1.85 |
| d | 40:25:10:25 |
46:27:8:24 |
50000 |
12800 |
1.82 |
| e | 40:15:20:25 |
48:16:16:25 |
47800 |
14800 |
1.66 |
| f | 40:20:0:40 |
50:20:0:40 |
56700 |
12800 |
2.08 |
| g | 20:15:0:20 |
20:16:0:20 |
30500 |
15100 |
1.39 |
| h | 15:15:0:25 |
15:15:0:25 |
31800 |
18200 |
1.48 |
| i | 15:5:0:20 |
14:5:0:17 |
23800 |
14800 |
1.62 |
| Block copolymer |
n:m:p:q:r (theoretical) |
n:m:p:q:r (1H NMR) |
M n (calcd) | M n (GPC) | PDI |
|---|---|---|---|---|---|
| j | 15:6:10:0:0 |
15:6:10:0:0 |
17000 |
11000 |
1.36 |
| k | 15:6:10:10:0 |
15:7:10:11:0 |
19500 |
12200 |
1.39 |
| l | 15:6:10:20:0 |
16:7:10:21:0 |
22000 |
13600 |
1.43 |
| m | 15:6:10:0:5 |
16:7:10:0:7 |
18400 |
10700 |
1.40 |
| n | 15:6:10:0:10 |
15:7:10:0:9 |
19800 |
11100 |
1.45 |
Micelle formation, photo-cross-linking and SCMs catalyst synthesis
The assembly of the block copolymers into micellar structures was carried out in methanol, ethanol, DMF/methanol, CH2Cl2/methanol and THF/methanol. The following procedure gave the best results: to a vigorously stirred triblock copolymer solution (5 mg mL−1) in THF, methanol was added via a syringe pump at a rate of 7 mL h−1. The final volume ratio of THF to methanol was 1:2. The resulting micelles were investigated by dynamic light scattering (DLS) (Table 3). We found that copolymers with a high ratio of the hydrophilic block were prone to micelle formation and the repeating units of the hydrophilic block need to account for at least 30% of the whole polymer. Further decrease in the ratio of the hydrophilic block led to severe aggregation (102 nm to 103 nm scale objects were detected by DLS) and poor solubility in THF/methanol solutions.
| Micelle | R/nm (% PD)a before cross-linking (MeOH) | R/nm (% PD) after cross-linking | R/nm (% PD) after metalation | ||
|---|---|---|---|---|---|
| (MeOH) | (CHCl3) | (MeOH) | (CHCl3) | ||
| a % PD: percentage of polydispersity. | |||||
| a | 13 (14%) | 12 (29%) | — | 23 (26%) | — |
| b | 15 (18%) | 15 (25%) | — | 23 (15%) | — |
| c | 18 (10%) | 15 (17%) | 15 (17%) | 22 (17%) | 22 (32%) |
| d | 15 (14%) | 13 (17%) | — | 22 (25%) | 22 (39%) |
| e | 13 (14%) | 12 (37%) | — | 31 (22%) | 33 (25%) |
| f | 22 (16%) | 20 (17%) | 23 (14%) | 29 (20%) | 26 (26%) |
| g | 18 (18%) | 14 (19%) | — | 19 (31%) | 32 (30%) |
| h | 24 (24%) | 21 (28%) | — | 26 (40%) | 31 (41%) |
| i | 18 (15%) | 17 (14%) | 22 (16%) | 23 (19%) | 21 (22%) |
| j | 14 (16%) | 14 (16%) | — | 9.2 (17%) | — |
| k | 18 (20%) | 17 (22%) | — | 20 (25%) | — |
| l | 13 (36%) | 13 (27%) | — | 12 (21%) | — |
| m | 23 (24%) | 24 (26%) | — | 23 (41%) | — |
| n | 19 (27%) | 19 (23%) | — | 15 (15%) | — |
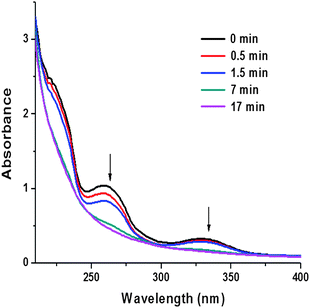
We next investigated the cross-linking of the micelles to yield the desired SCMs. The micelle solution of interest was degassed with nitrogen and then transferred to a 17 mL UV cylindrical cell with a 1 cm path length for photo-irradiation. During the reaction, aliquots were taken for either UV-vis spectroscopy analysis or DLS. The UV-vis spectra showed a decrease in the 270 nm cinnamate absorption indicating the [2 + 2] cycloaddition of the cinnamate pending groups (Fig. 6).64 After 7 minutes irradiation, the cinnamate shell was fully cross-linked as indicated by the disappearance of the cinnamate absorption. To ensure formation of the stable shell cross-linked structure, the UV irradiated cross-linking process was also monitored by DLS (ESI†). For the DLS studies, the solvent was removed roughly and the residue was quickly redissolved in dichloromethane. Dichloromethane is a good solvent for all polymer blocks of the micelle. We rationalized that if the micelle is not or only lightly cross-linked, dissociated polymers will be detected by DLS. The DLS analysis after 7 minutes reaction showed only one signal. The hydrodynamic radius was approximately 25 nm, which is close to the size of the original micelle measured in methanol. Further irradiation did not change the SCM size or polydispersity significantly and no smaller or larger species were detected. If the cross-linkable block had fewer than 5 repeating units, low molecular weight species were detected by DLS. We also found that the micelle sizes shrank after cross-linking by about 1 to 5 nm in hydrodynamic radii in methanol. This has been observed before in the literature for other SCM systems.65 Swelling of the SCMs in CH2Cl2 was detected by DLS which is analogous to literature reports that describe that SCMs swell when dissolved in non-selective solvents.54
After cross-linking, the SCMs were metalated in an inert atmosphere. The SCM solution was degassed with nitrogen and then transferred to a glovebox. Two equivalents (relative to the amount of salen ligands) of Co(OAc)2·H2O in methanol were added and the mixture was stirred for two days. The solution color gradually changed from orange to dark red. After removal from the glovebox, the Co(II)–salen functionalized SCMs were stirred for five hours while bubbling air through the solution. The final solution turned dark brown indicating that the Co–salen complexes were converted to catalytically active Co(III)–salen centers. The SCM catalysts were purified using a Sephadex G-50 column with methanol as the eluent. The SCM catalysts could be separated successfully from excess cobalt salt, other small molecules and some large sized aggregates. After removal of the solvent, the SCM catalysts were redissolved in a mixture of benzene/chloroform (1:1 ratio) and lyophilized to generate the final SCM catalyst as a brown powder. The cobalt content was determined by ICP-MS with complexation yields ranging from 79% to 90%. The cobalt loadings of the SCM catalysts were also analyzed by TGA with good agreement with the Co elemental analysis (ESI†).
The metalated SCMs were also characterized by DLS in both methanol and CH2Cl2. In most cases an increase in the hydrodynamic radius was observed in comparison to the original un-metalated SCMs. These DLS analyses proved that the metalation and oxidation process did not disassociate the micellar structures or alter significantly the SCMs. All DLS data are summarized in Table 3. Overall, the hydrodynamic radii of the metalated SCMs ranged from 10 nm to 35 nm depending on the composition of the block copolymers. Once the micelle was fully cross-linked, the micellar structures were stable in selective solvents such as methanol and non-selective solvents including CH2Cl2.
AFM was also used in the analysis of the SCM catalyst particles. Fig. 7A shows an AFM image of SCM catalyst j. Samples for the AFM measurements were prepared by dropping the SCM catalyst solution on a freshly cleaved mica wafer and drying it in air. The mean diameter of j is 24.9 nm generated by statistical analysis of at least 40 particles in an area of 1 μm × 1 μm using Gwyddion software. The particle size is narrowly distributed (±5 nm deviation), which is consistent with the low polydispersity of SCM j obtained by the DLS measurement (Fig. 7B). DLS afforded a smaller particle size (hydrodynamic radius: 9.2 nm). The average particle height in the AFM measurement is about 5 nm. One possible explanation for the size difference between AFM and DLS is the deformation of SCMs spherical structure when drying on the surface of mica.66
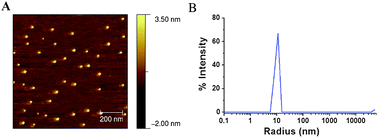 | ||
| Fig. 7 Tapping mode AFM image (A) and DLS profile (B) of SCM catalyst j. | ||
Co(III)–salen functionalized SCMs catalysis of the HKR of epoxides
One goal of this study is to develop SCM-core supported catalysts. The reaction that was chosen for the initial catalytic screening is the hydrolytic kinetic resolution (HKR) of epoxides catalyzed by chiral Co(III)–salen complexes. HKR of terminal epoxides is featured with exceptional enantioselectivity and mild reaction conditions.67,68 Since the HKR of epoxides using Co–salen complexes is based on a bimetallic mechanism,69,70catalytic centers within close proximity to each other have an enhanced cooperativity and result in a significant increase in the reaction rate.67–76 Weberskirch's group has successfully applied poly(2-oxazoline) micelle-supported Co(III) catalysts in the resolution of aromatic epoxides indicating that crosslinked micelle analogous might be an interesting target.77 We employed epichlorohydrin as the reaction substrate. Our SCM catalysts were dissolved in excess degassed water followed by the addition of epichlorohydrin. The reaction mixture was stirred in a water bath and the resolution progress was monitored by chiral gas chromatography (GC). The experimental conditions and results are listed in Table 4.| SCM catalyst | Epichlorohydrin volume/μL | Water volume/μL | Catalyst loading (mol%)b | Time/h | ee (%)c | Conversion (%)d |
|---|---|---|---|---|---|---|
| a Reactions were carried out with excess water at ambient temperature. b Catalyst loading based on cobalt. c Determined by chiral GC analysis. d Determined by GC analysis with chlorobenzene as internal standard. e Data in parentheses are the results collected at 44 hours. | ||||||
| a | 50 | 500 | 1 | 24 | <5 | 5 |
| b | 54 | 500 | 1 | 36 | <5 | <5 |
| c | 66 | 360 | 1 | 38 | 35 | 26 |
| d | 63 | 1000 | 0.5 | 42 | 50 | 35 |
| e | 50 | 500 | 1 | 31 | 44 | 31 |
| f | 45 | 250 | 1 | 38 | 55 | 37 |
| g | 43 | 250 | 1 | 32 | 79 | 46 |
| h | 46 | 250 | 1 | 32 | 82 | 47 |
| i e | 39 | 200 | 1 | 32 (44) | 91 (97) | 49 (51) |
| j | 46 | 250 | 1 | 44 | 96 | 50 |
| k | 51 | 280 | 1 | 44 | 26 | 21 |
| l | 50 | 280 | 1 | 44 | 23 | 21 |
| m | 43 | 250 | 1 | 48 | 8 | 8 |
| n | 38 | 259 | 1 | 48 | <5 | <5 |
To systemically investigate the effects of SCMs composition on the catalytic selectivity and efficiency, we initially fixed the total degree of polymerization of the triblock copolymers to 100 for all catalysis experiments, but varied the ratio between individual blocks. Comparing SCM catalysts a through f, we found that increasing the ratio of the hydrophilic block dramatically lowers catalyst efficiency. Catalysts a and b, containing the highest hydrophilic block ratio (75mer and 60mer), barely afforded any resolved products. We suggest that the PEG chains extended out from poly(norbornene) backbone forming a dense layer around the catalytic core. The longer the PEG-containing polymer block, the thicker the PEG layer imposing a barrier for the relatively hydrophobic epoxide to permeate into the catalytic core. Another possible explanation for this phenomenon is that the SCMs of a and b have a relatively low Co content, which might impede the bimetallic mechanism based HKR. Decreasing the hydrophilic block ratio led to enhanced reactivity with 44% to 55% enantiomeric excess (ee) for d, e and f, which all have 40 hydrophilic repeat units. Further decrease in the ratio of the hydrophilic block caused poor solubility of the SCM catalysts in water. Increasing the incorporation of 4 in the cross-linkable block had no positive effect on the catalytic efficiency as evidenced by the comparisons of catalytic results between d and e and also between b and c.
Next, we investigated whether changing the block copolymer length has any influence on the catalytic efficiency. We found that decreasing the total degree of polymerization from 100 to 55 increased the catalytic activity as shown by the comparison of catalytic results between f and g. With the expectation of increasing the substrate and product permeability by lowering the degree of cross-linking, we shorten the cross-linkable block to 5 or 6 repeating units. An enhancement of reaction rates was observed for i and j, which finished the resolution in 44 hours with 97% and 96% ee, respectively. Monomers 5 and 6 were incorporated into the Co(III)–salen block of polymer in order to tune the hydrophobicity of SCM core, with an attempt to further increase the permeability of water or epichlorohydrin. However, as shown by k, l, m and n, a dramatic decrease in reactivity was observed. The higher the incorporation ratio of 5 and 6, the lower the reactivity. In summary, the catalytic activity is sensitive to block copolymer composition as well as block copolymer length. However, incorporation of spacing units yielded less active catalytic materials. We suggest that the reason for this lower activity is the interference of the spacing units with the bimetallic interactions between the Co(III)–salen catalytic centers in the core of SCMs. Although a large amount of water was used in the catalytic reactions, the ring-opening reaction of the non-desired enantiomer, (S)-epichlorohydrin, was barely detected. This is proved by the strong consistency between the ee's and the conversion values, i.e. low ee values are always accompanied by low conversions. This result demonstrates the high specificity of SCMs supported Co(III)–salen catalyst towards HKR of epichlorohydrin.
Conclusions
In this manuscript we have reported the synthesis of a series of poly(norbornene) based amphiphilic triblock copolymers using ROMP. These copolymers are featured with a PEG-based hydrophilic block, a cinnamate functionalized middle block and a salen ligand modified hydrophobic block. A protocol for the formation of shell cross-linked micelles (SCMs) supported Co(III)–salen catalyst was established, in which the key steps include: micelle assemblies from the amphiphilic block copolymers, cross-linking throughout the cinnamate layer of the micelles under UV irradiation, and finally selective complexation of salen with cobalt cation followed by activation. These SCMs with Co(III)–salen functionalized core were tested for HKR of epichlorohydrin. The initial results confirmed the catalytic activity of the SCM catalysts. Systematic investigations demonstrated that the compositions of the block copolymers had a significant impact on the catalytic efficiency and selectivity. In contrast to other supported metal–salen complexes, the structure of the SCM catalysts is stabilized by the cross-linked shell, which could promote the recyclability of the catalysts. Therefore, the applications of SCM catalysts in catalytic reactions could be more environmentally friendly which will be investigated in the future.Acknowledgements
We are thankful to the Department of Energy, Office of Basic Energy Sciences through Catalysis Contract No. DEFG02-03ER15459 for financial support of this work.References
- S. Forster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- R. Shenhar, T. B. Norsten and V. M. Rotello, Adv. Mater., 2005, 17, 657–669 CrossRef CAS.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CAS.
- M. J. Monteiro, Macromolecules, 2010, 43, 1159–1168 CrossRef CAS.
- M. Lazzari and M. A. Lopez-Quintela, Adv. Mater., 2003, 15, 1583–1594 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- M. Antonietti, E. Wenz, L. Bronstein and M. Seregina, Adv. Mater., 1995, 7, 1000–1005 CAS.
- S. Klingelhofer, W. Heitz, A. Greiner, S. Oestreich, S. Forster and M. Antonietti, J. Am. Chem. Soc., 1997, 119, 10116–10120 CrossRef.
- T. Cao, W. Yin, J. L. Armstrong and S. E. Webber, Langmuir, 1994, 10, 1841–1847 Search PubMed.
- R. Pons, Polymeric Surfactants as Emulsion Stabilizers, in Amphiphilic Block Copolymers: Self-assembly and Applications, ed. P. Alexandridis and B. Lindman, Elsevier, Amsterdam, 2000, pp. 409–422 Search PubMed.
- J. F. Gohy, Block Copolymer Micelles, in Block Copolymers II, Springer-Verlag Berlin, Berlin, 2005 Search PubMed.
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS.
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295–309 CrossRef CAS.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239–7240 CrossRef.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892–16899 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776–6786 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5203–5217 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef CAS.
- V. Butun, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 12135–12136 CrossRef.
- S. Y. Liu and S. P. Armes, J. Am. Chem. Soc., 2001, 123, 9910–9911 CrossRef CAS.
- S. Fujii, Y. L. Cai, J. V. M. Weaver and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 7304–7305 CrossRef CAS.
- S. Sugihara, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2010, 49, 3500–3503 Search PubMed.
- J. F. Ding and G. J. Liu, Macromolecules, 1998, 31, 6554–6558 CrossRef CAS.
- F. T. Liu and G. J. Liu, Macromolecules, 2001, 34, 1302–1307 Search PubMed.
- M. Wang, M. Jiang, F. L. Ning, D. Y. Chen, S. Y. Liu and H. W. Duan, Macromolecules, 2002, 35, 5980–5989 CrossRef CAS.
- K. Matsumoto, T. Hirabayashi, T. Harada and H. Matsuoka, Macromolecules, 2005, 38, 9957–9962 CrossRef CAS.
- Y. T. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- Y. T. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81–89 CrossRef CAS.
- S. Y. Zhang and Y. Zhao, J. Am. Chem. Soc., 2010, 132, 10642–10644 Search PubMed.
- A. N. Koo, H. J. Lee, S. E. Kim, J. H. Chang, C. Park, C. Kim, J. H. Park and S. C. Lee, Chem. Commun., 2008, 6570–6572 RSC.
- L.-Y. Li, W.-D. He, J. Li, B.-Y. Zhang, T.-T. Pan, X.-L. Sun and Z.-L. Ding, Biomacromolecules, 2010, 11, 1882–1890 CrossRef CAS.
- Y. C. Wang, Y. Li, T. M. Sun, M. H. Xiong, J. A. Wu, Y. Y. Yang and J. Wang, Macromol. Rapid Commun., 2010, 31, 1201–1206 Search PubMed.
- A. Roesler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95–108 CrossRef CAS.
- Y. Li, K. Hindi, K. M. Watts, J. B. Taylor, K. Zhang, Z. Li, D. A. Hunstad, C. L. Cannon, W. J. Youngs and K. L. Wooley, Chem. Commun., 2010, 46, 121–123 RSC.
- A. M. Nystrom, Z. Q. Xu, J. Q. Xu, S. Taylor, T. Nittis, S. A. Stewart, J. Leonard and K. L. Wooley, Chem. Commun., 2008, 3579–3581 RSC.
- S. Y. Liu, J. V. M. Weaver, M. Save and S. P. Armes, Langmuir, 2002, 18, 8350–8357 CrossRef CAS.
- T. Sanji, Y. Ogawa, Y. Nakatsuka, M. Tanaka and H. Sakurai, Chem. Lett., 2003, 980–981 Search PubMed.
- R. K. O'Reilly, Philos. Trans. R. Soc., A, 2007, 365, 2863–2878 Search PubMed.
- A. D. Ievins, X. F. Wang, A. O. Moughton, J. Skey and R. K. O'Reilly, Macromolecules, 2008, 41, 2998–3006 CrossRef CAS.
- M. S. Morgan, R. S. Tipson, A. Lowy and W. E. Baldwin, J. Am. Chem. Soc., 1944, 66, 404–407 CrossRef CAS.
- J. R. Carlise, R. M. Kriegel, W. S. Rees and M. Weck, J. Org. Chem., 2005, 70, 5550–5560 Search PubMed.
- S. K. Yang, A. V. Ambade and M. Weck, Chem.–Eur. J., 2009, 15, 6605–6611 CrossRef CAS.
- J. M. Pollino, K. P. Nair, L. P. Stubbs, J. Adams and M. Weck, Tetrahedron, 2004, 60, 7205–7215 CrossRef CAS.
- D. D. Manning, L. E. Strong, X. Hu, P. J. Beck and L. L. Kiessling, Tetrahedron, 1997, 53, 11937–11952 CrossRef CAS.
- J. D. Roberts, E. R. Trumbull, W. Bennett and R. Armstrong, J. Am. Chem. Soc., 1950, 72, 3116–3124 CrossRef CAS.
- C. D. Vernooy and C. S. Rondestvedt, J. Am. Chem. Soc., 1955, 77, 3583–3586 CrossRef CAS.
- M. Holbach, X. L. Zheng, C. Burd, C. W. Jones and M. Weck, J. Org. Chem., 2006, 71, 2903–2906 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- M. Sandholzer, S. Bichler, F. Stelzer and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2402–2413 Search PubMed.
- B. Rupp, T. Bauer and C. Slugovc, Proc. SPIE, 2009, 7393, 73930Y/73931–73930Y/73939 Search PubMed , (Nanophotonic Materials VI).
- J. Liu, Y. Liao, X. He, J. Yu, L. Ding and M. Xie, Macromol. Chem. Phys., 2011, 212, 55–63 Search PubMed.
- D. Smith, E. B. Pentzer and S. T. Nguyen, Polym. Rev., 2007, 47, 419–459 Search PubMed.
- R. H. Grubbs, Handbook of Metathesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- G. M. Pawar, B. Bantu, J. Weckesser, S. Blechert, K. Wurst and M. R. Buchmeiser, Dalton Trans., 2009, 9043–9051 RSC.
- G. M. Pawar, J. Weckesser, S. Blechert and M. R. Buchmeiser, Beilstein J. Org. Chem., 2010, 6, 28 Search PubMed.
- X. Z. Jiang, S. Z. Luo, S. P. Armes, W. F. Shi and S. Y. Liu, Macromolecules, 2006, 39, 5987–5994 CrossRef CAS.
- K. Stubenrauch, C. Moitzi, G. Fritz, O. Glatter, G. Trimmel and F. Stelzer, Macromolecules, 2006, 39, 5865–5874 CrossRef CAS.
- K. Ito, Y. Tomi and S. Kawaguchi, Macromolecules, 1992, 25, 1534–1538 Search PubMed.
- J. X. Chen, A. R. Vaino, R. L. Smith and S. C. Collins, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3482–3487 Search PubMed.
- J. Skey and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3690–3702 Search PubMed.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS.
- D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS.
- R. Breinbauer and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 3604–3607 CrossRef CAS.
- X. L. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105–1112 CrossRef CAS.
- N. Madhavan, C. W. Jones and M. Weck, Acc. Chem. Res., 2008, 41, 1153–1165 CrossRef CAS.
- P. Goyal, X. L. Zheng and M. Weck, Adv. Synth. Catal., 2008, 350, 1816–1822 CrossRef CAS.
- X. L. Zheng, C. W. Jones and M. Weck, Adv. Synth. Catal., 2008, 350, 255–261 CrossRef CAS.
- X. Zhu, K. Venkatasubbaiah, M. Weck and C. W. Jones, ChemCatChem, 2010, 2, 1252–1259 Search PubMed.
- B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309–1312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum of homopolymer Poly140, thermogravimetric analysis (TGA) of the SMCs and profiles of DLS monitoring UV irradiated cross-linking of micelles. See DOI: 10.1039/c1py00151e |
| This journal is © The Royal Society of Chemistry 2011 |