Stealth macromolecular platforms for the design of MRI blood pool contrast agents
Mathurin
Grogna
a,
Rudi
Cloots
b,
André
Luxen
c,
Christine
Jérôme
a,
Catherine
Passirani
d,
Nolwenn
Lautram
d,
Jean-F.
Desreux
e,
Mike
Collodoro
f,
Marie-Claire
De Pauw-Gillet
f and
Christophe
Detrembleur
*a
aCenter for Education and Research on Macromolecules (CERM), University of Liège, B6 Sart-Tilman, B-4000, Liège, Belgium. E-mail: christophe.detrembleur@ulg.ac.be; Fax: +32-4-3663497; Tel: +32-4-3663465
bLaboratoire de chimie inorganique structurale, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
cCyclotron Research Centre, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
dINSERM U 646, 10 Rue A Boquel, 49000, Angers, France
eCoordination and Radiochemistry, University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
fLaboratory of Histology-Cytology (GIGA-R), University of Liège, B6 Sart Tilman, B-4000, Liège, Belgium
First published on 28th July 2011
Abstract
Stealth macromolecular platforms bearing alkyne groups and poly(ethylene oxide) brushes were synthesized by reversible addition fragmentation chain transfer (RAFT) polymerization. The anchoring of Gd3+-chelates bearing an azide group was then carried out by the Huisgen 1,3-dipolar cycloaddition (“click”) reaction in mild conditions, leading to macrocontrast agents for MRI applications. The gadolinium complex is hidden in the PEO shell that renders the macrocontrast agents free of any cytotoxicity and stealth to proteins of the immune system. Relaxometry measurements have evidenced an improved relaxivity of the macrocontrast agent compared to ungrafted gadolinium chelate. Moreover, this relaxivity is further enhanced when the spacer length between the Gd3+-chelate and the polymer backbone is shorter, as the result of its decreased tumbling rate. These novel products are therefore promising candidates for MRI applications.
Introduction
Magnetic resonance imaging (MRI) is a routine diagnostic tool in modern clinical medicine. MRI has many advantages as a diagnostic imaging modality. It is noninvasive, delivers no radiation burden, and has excellent (submillimetre) spatial resolution. Contrast agents (CAs) (chelating Gd3+ with suitable organic ligands such as DTPA (diethylene triamine pentaacetic acid) or DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)), that increase the relaxation rate of water molecules and improve the contrast between tissues of interest, are widely used in experimental and clinical settings. Unfortunately these CAs present two main drawbacks that lead to injection of large gadolinium doses: (i) rapid blood extraction (circulation half lifetime of about 5 min1) and (ii) poor contrast at high magnetic field.These two drawbacks can be solved by grafting CAs to biocompatible polymers. Indeed some biocompatible and water-soluble polymers have demonstrated unique pharmacokinetic properties with long blood circulation time and good tissue retention. Due to its biocompatibility, hydrophilicity, non-toxicity, good steric stabilization effect and its capacity to prevent protein adsorption,2 poly(ethylene oxide) (PEO) has emerged as a suitable candidate for that purpose. It enhances solubility of hydrophobic drugs, prolongs circulation time, minimizes non-specific uptake, and allows for specific tumor accumulation through the enhanced permeability and retention effect (EPR effect3). Moreover, according to the SBM theory4,5 (Solomon–Bloembergen–Morgan), it is recognized that slowing down molecular tumbling by grafting the contrast agent onto high macromolecular weight structures increases the relaxivity and so the contrast. In this way, recent approaches for high-relaxivity agents have involved the incorporation of Gd(III) chelates into dendrimers,6–8 micelles9–11 and linear polymers.12–16
In this paper, we aim at designing and characterizing new stealth macromolecular platforms that bind gadolinium based MRI contrast agents under very mild conditions (Scheme 1). This multifunctional platform is composed of (i) poly(ethylene oxide) grafts for ensuring water solubility and prolonged blood circulation, and (ii) alkyne groups for anchoring the gadolinium complexes by click chemistry, a well-known efficient and selective cycloaddition reaction between azide bearing molecules and alkynes.17 Furthermore, the rigid nature of the triazole linker formed during this click reaction hinders the local rotation of the Gd(III) complex that should consequently enhance the relaxivity compared to a macrocontrast agent bearing a linear and flexible linker18 (Scheme 2).
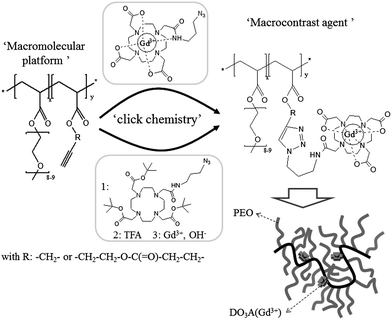 | ||
| Scheme 1 General procedure for the synthesis of the macrocontrast agent by grafting DO3A(Gd3+)–N3 (up pathway) and DO3AtBu–N3 (bottom pathway) onto alkyne bearing copolymers. | ||
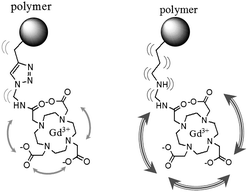 | ||
| Scheme 2 Local hindering of contrast agent due to triazole ring vs. a linear flexible linker. | ||
The combination of the stealth character of the macrocontrast agent imparted by the PEO chains with its improved relaxivity promoted by the conjugation of the gadolinium complex to the hindered macromolecule is expected to decrease the doses of injected gadolinium while maintaining satisfactory image acquisition times. Besides the design of those macrocontrast agents, their relaxivity (contrasting efficiency), cytotoxicity, and stealth character will also be evaluated in order to study their potential as MRI blood pool agents.
Experimental procedures
Materials
S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (CTA) was synthesized according to Lai et al.194-Pentynoic acid, 2,2′-azobis(isobutyronitrile) (AIBN), propargyl alcohol, triethylamine, acryloyl chloride, ethyl acetate, bromopropylamine sodium azide (NaN3), dichloromethane (CH2Cl2), heptane, dimethylsulfoxide (DMSO), poly(ethylene oxide) methyl ether acrylate (PEOMA Mn ≈ 454 g mol−1, DPPEG = 8–9, Aldrich), methyl bromoacetate, trifluoroacetic acid (TFA), dimethylformamide were purchased from Sigma-Aldrich. Magnesium sulfate (MgSO4), sodium hydroxide (NaOH), diethyl ether and potassium carbonate (K2CO3) were purchased from VWR. 1,4,7,10-Tetraazacyclododecane-1,4,7-triacetic acid, 1,4,7-tris(1,1-dimethylethyl) ester (DO3AtBu) was purchased from Chematech. Bathophenanthroline disulfonic acid disodium salt hydrate was purchased from Alfa Aesar. All components were used as received without further purification.Synthesis
1H NMR (CDCl3, TMS, 250 MHz): δ = 2.47 ppm (s, –C–H, 1H), δ = 4.72 ppm (s, –O–CH2–C, 2H), δ = 5.8 and 6.5 ppm (CH2–CH–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–, 3H)
O)–, 3H)
13C DEPT135: δ = 51.78 ppm (–O–CH2–C), δ = 75.06 ppm (–C–H), δ = 77.48 ppm (–C–H), δ = 127.34 ppm (CH2–CH–C(O)–), δ = 131.59 ppm (CH2–CH–C(O)–).
Synthesis of DO3AtBu–OMe [1-methyl ester-4,7,10-tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane]. 957 mg of DO3AtBu (1.86 mmol) was dissolved in 15 ml of acetonitrile. 0.6 g of K2CO3 and 0.212 ml of methyl bromoacetate (2.42 mmol) were then added. The resulting solution was stirred for 4 hours under reflux. After cooling to room temperature, the solvent was removed under reduced pressure and toluene (30 ml) was added to extract the product from the mixture. The organic phase was washed three times with water and was then dried with MgSO4. Solvent was removed under reduced pressure to give a yellow oil (875 mg, 80% yield). Product was analyzed by 1H NMR and ESI-MS.
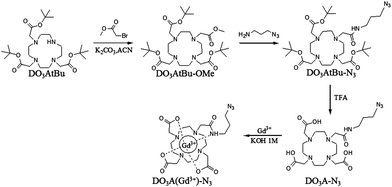 | ||
| Scheme 3 Synthetic pathway for DO3AtBu–N3 and DO3A(Gd3+)–N3. | ||
1H NMR (CDCl3, TMS, 250 MHz): δ = 1.42 ppm (s, –C–CH3, 27H), δ = 2.79 ppm (m, –N–CH2–CH2–N 16H), δ = 3.24 ppm (s, N–CH2–C(O),6H), δ = 3.38 ppm (s, N–CH2–C(O)–OMe, 2H), δ = 3.64 ppm (s, –O–CH3, 3H).
ESI-MS: m/z: 587.41 [M + H]+; 609.39 [M + Na]+.
Synthesis of DO3AtBu–N3 [1-N-3-azidopropylamine amide-4,7,10-tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane]. 875 mg of DO3AtBu–OMe (1.49 mmol) was dissolved in 3 g of N3-propylamine (APA; 30 mmol) and was stirred for 3 days at room temperature. The coupling reaction between DO3AtBu–OMe and APA was certified by mass spectrometry with the complete disappearance of the molecular peaks of DO3AtBu (m/z) 586/609 and the appearance of the molecular peak of the DO3AtBu conjugated APA (DO3AtBu–N3) (m/z) 655/677. After complete disappearance of the molecule peak of the DO3AtBu–OMe, the solvent was removed under reduced pressure. Product was purified by silica gel chromatography (100% CH2Cl2) leading to the pure compound (730 mg, 75%) as a white foam. Product was analyzed by 1H NMR, 13C NMR and ESI-MS.
1H NMR (CDCl3, TMS, 250 MHz): δ = 1.42 ppm (s, –C–CH3, 27H), δ = 1.81 ppm (quin, –CH2–CH2–N3), 2.49–3.01 ppm (very broad signals with an integration corresponding to 16H), δ = 3.19–3.35 (m, N–CH2–C(O)– and –NH–CH2–, 10H), δ = 8.72 ppm (t, –C(O)–NH–CH2–, 1H).
13C NMR (CDCl3, TMS, 63 MHz): δ = 27.76 ppm (–C–CH3), δ = 28.77 ppm (–NH–CH2–CH2–), δ = 36.18 ppm (–NH–CH2–CH2–), δ = 48.81 ppm (–CH2–N3), δ = 51.63, 52.05, 53.17, 54.55, 55.84 ppm (–N–CH2–CH2–N–), δ = 56.44 ppm (–N–CH2–C O–), δ = 57.61 ppm(–N–CH2–C(O)–), δ = 80.3 ppm (–C–CH3), δ = 170.03 ppm (–C(O)–O–tBu), δ = 171.69 ppm(–C(O)–NH–).
ESI-MS: m/z: 655.45 [M + H]+, 677.43 [M + Na]+.
Synthesis of Gd3+ complex of [1-N-3-azidopropylamine amide-4,7,10-tris(carbonylmethyl)-1,4,7,10-tetraazacyclododecane] (DO3A(Gd3+)–N3). Ligand (DO3AtBu–N3) (200 mg, 0.3 mmol) was dissolved in trifluoroacetic acid (TFA) (3 ml) and was stirred at room temperature overnight. TFA was evaporated under reduced pressure and the ligand was dissolved in water (5 ml). GdCl3·6H2O (120 mg, 0.33 mmol) was added to the solution and the pH was increased to 6 by the slow addition of a KOH solution (1 M). The solution was heated to 40 °C overnight. Finally, the uncomplexed Gd3+ ions were removed by eluting the solution through a Chelex 100 (Bio-Rad Laboratories, sodium form) resin. The resulting solution was brought to dryness under reduced pressure and the DO3A(Gd3+)–N3 complex was recovered as a pale yellow solid in almost quantitative yield (>95%).
ESI-MS: m/z: 642.16 [M + H]+.
O)–O–CH2–PEO, 64H), δ = 4.64 ppm (large, –C(O)–O–CH2–alkyne, 28H). Mn, NMR = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 g mol−1, DPn, NMR = 46, Mn, SEC = 20000 g mol−1, Mw/Mn = 1.15. Conversion is 70% for both monomers.
450 g mol−1, DPn, NMR = 46, Mn, SEC = 20000 g mol−1, Mw/Mn = 1.15. Conversion is 70% for both monomers.
The same experimental section is used for synthesizing other P[PEOMA-st-PA] with different molecular weights, excepted that the RAFT agent to monomers ratio was accordingly adapted. [PEOMA]/[PA] = 7/3, DPth = 10, [AIBN]/[CTA] = 0.1, ([PEOMA]+[PA])/[CTA] = 10.
M n, NMR = 3400 g mol−1, DPn, NMR = 7, Mn, SEC = 7500 g mol−1, Mw/Mn = 1.08. Conversion is 70% for both monomers.
1H NMR (CDCl3, 250 MHz, TMS): δ = 0.86 ppm (t, –CH2–CH3, 3H), δ = 1.16 ppm (CH3–C–, 6H), δ = 1.24 ppm (–CH2–(RAFT), 20H), δ = 1.62–1.89 ppm (large, –CH2–CH–, 120H), δ = 2.30 ppm (large, –CH2–CH–, 60H), δ = 3.36 ppm (s, CH3–O–PEO, 126H), δ = 3.62–3.72 ppm (large, –CH2–CH2–O– and –CH2–CH2–OH, 1512H), δ = 4.16 ppm (large, –CO–O–CH2–PEO and –C(O)–O–CH2–CH2–OH, 120H), Mn, NMR = 21100 g mol−1, DPn, NMR = 60, Mn, SEC = 22000 g mol−1, Mw/Mn = 1.16.
P[PEOMA-st-HEA] (DP10; 70%/30% mol) with different molecular weights was synthesized using the same experimental procedure, excepted that the RAFT agent to monomer ratio was adapted accordingly. [PEOMA]/[PA] = 7/3, DPth = 10, [AIBN]/[CTA] = 0.1, ([PEOMA]+[PA])/[CTA] = 10. Mn, NMR = 4550 g mol−1, DPn, NMR = 13, Mn, SEC = 7400 g mol−1, Mw/Mn = 1.16; conversion > 98% (complete for both monomers).
1H NMR (CDCl3, 250 MHz, TMS): δ = 0.86 ppm (t, –CH2–CH3, 3H), δ = 1.16 ppm (CH3–C–, 6H), δ = 1.24 ppm (–CH2–(RAFT), 20H), δ = 1.62–1.89 ppm (large, –CH2–CH–, 120H), δ = 2.05 ppm (–CH, 18H), δ = 2.30 ppm (large, –CH2–CH–, 60H), δ = 2.47–2.57 ppm (–C(O)–CH2–CH2–CH, 72H), δ = 3.36 ppm (s, CH3–O–PEO, 126H), δ = 3.62 ppm (large, –CH2–CH2–O–, 1428H), δ = 4.16–4.26 ppm (large, –C(O)–O–CH2–PEO and –C(O)–O–CH2–CH2–O–C(O)–, 156H), Mn, NMR = 22600 g mol−1, DPn, NMR = 60, Mn, SEC = 22500 g mol−1, Mw/Mn = 1.16.
P[PEOMA-st-AEP] (DP13; 70%/30% mol) was synthesized using the same experimental procedure by adapting the monomers to CTA molar ratio. Mn, NMR = 4900 g mol−1, DPn, NMR = 13, Mn, SEC = 7500 g mol−1, Mw/Mn = 1.17.
1H NMR (CDCl3, 250 MHz, TMS): δ = 0.86 ppm (t, –CH2–CH3, 3H), δ = 1.16 ppm (CH3–C–, 6H), δ = 1.24 ppm (–CH2–(RAFT), 20H), δ = 1.62–1.89 ppm (large, –CH2–CH–, 110H), δ = 2.30 ppm (large, –CH2–CH–, 55H), δ = 3.72 ppm (large, –CH2–CH2–OH, 110H), δ = 4.16 ppm (large, –C(O)–O–CH2–CH2–OH, 110H), Mn, NMR = 6400 g mol−1, DPn, NMR = 55, Mn, SEC = 7000 g mol−1, Mw/Mn = 1.12.
450 g mol−1) or 78 mg of P[PEOMA-st-AEP] (70 mol% PEOMA/30 mol% AEP, Mn, NMR = 22600 g mol−1 or 4900 g mol−1) (62 μmol of alkyne function) were dissolved in 2 ml of dry DMF. 1.2 mg of CuI (6.2 μmol) and azide molecule (62 μmol) were added to the copolymer solution. This mixture was stirred at room temperature overnight. Different purifications were used according to the nature of the azide molecule:
- Benzyl azide and DO3AtBu modified copolymers were purified as follows: 10 ml of CH2Cl2 and 15 ml of EDTA solution (0.5 M, pH = 7) were added to the DMF solution. The mixture was vigorously stirred for 15 min. The blue aqueous solution was removed and 15 ml of EDTA solution was again added, and the mixture was vigorously stirred for a few minutes to remove residual copper. Finally the organic phase was dried with MgSO4 and the copolymer was recovered by precipitation into a large volume of a diethyl ether/heptane mixture (50/50). Grafting yield of different copolymer was determined by 1H NMR spectroscopy and is presented in Table 3 (values around 80% for benzyl azide and between 45 and 65% for DO3AtBu–N3). Residual copper was quantified by ICP-MS analysis (<100 ppm).
- DO3A(Gd3+)–N3 modified copolymers were purified as follows: 5 ml of EDTA (0.5 M, pH = 7) was added to the DMF solution. Then the solution was dialyzed (Spectra/Por, molecular weight cut-off, 3500) against water for 48 h. Finally, the copolymers were lyophilized. Grafting yield was determined by ICP-MS spectrometry and was less than 10%. Residual copper was quantified by ICP-MS analysis (<100 ppm).
Complement consumption testing (CH50 test)
Complement consumption was assessed in normal human serum (NHS) (provided by the Etablissement Français du Sang, CHU, Angers, France) by measuring the residual haemolytic capacity of the complement system after contact with particles. The technique consisted in determining the amount of serum able to lyse 50% of a fixed number of sensitized sheep erythrocytes with rabbit antisheep erythrocyte antibodies (CH50), according to the procedure described elsewhere.22,23Complement activation was expressed as a function of the surface in order to compare particles with different mean diameters. Nanoparticle surface areas were calculated as described elsewhere,24 using the equation: S = n4πr2 and V = n(4/3)(πr3) leading to S = 3m/rρ where S is the surface area (cm2) and V the volume (cm3) of n spherical beads of average radius r (cm), weight m (μg) and volumetric mass ρ (μg cm−3).Cell culture
The human breast adenocarcinoma cell line MCF-7/BOS was kindly provided by Dr A. M. Soto (Department of Anatomy and Cellular Biology, Tufts University School of Medicine, Boston). MCF-7/BOS cells were grown at 37 °C under humidified air containing 5% CO2 in DMEM medium low glucose with 10 vol% of Foetal Bovine Serum [Origin: EU Approved (South American) FBS], 1 vol% of penicillin/streptomycin (10000 units of penicillin (base) and 10000 μg of streptomycin (base) per ml utilizing penicillin G (sodium salt) and streptomycin sulfate in 0.85% saline) and 0.5 vol% gentamycin (10 mg ml−1). The human melanoma line MEL-5 was obtained from De Giovani (University of Liege, Belgium). MEL-5 cells were grown at 37 °C in 5% CO2 in DMEM medium high glucose with 5 vol% of FBS, 1 vol% glutamine 2 mM, 1 vol% HEPES, 1 vol% of penicillin/streptomycin.
Copolymer cytotoxicity
The cytotoxicity of the copolymers was evaluated by determining the viability of the cells (MCF-7/BOS and MEL-5 [104cells per ml]) after incubation with different concentrations of copolymers (0.05 mg ml−1–30 mg ml−1) for 48 h. The percentage of viable cells was determined by the estimation of their dehydrogenase activity using the MTS tetrazolium [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)], inner salt and an electron coupling reagent (phenazine ethosulfate). The MTS tetrazolium compound is bioreduced by living cells into a formazan product.At the end of incubation period with copolymers, cells were incubated with 20 μl of a MTS solution for 4 h at 37 °C in 5% CO2. The absorbance of the solubilized formazan was measured spectrophotometrically at 490 nm with a multiplate reader (Powerwave X). Cell viability was expressed as the ratio between the amount of formazan determined for cells treated with the different copolymers and the amount for non-treated cells taken as 100%.
Characterizations
1H and 13C NMR spectra of the different polymers were recorded at 298 K with a Bruker spectrometer (250 MHz; 63 MHz for 13C) in CDCl3 or DMSO ((D1) 2 s, 16 scans, 5 wt% of polymer or 10 wt% of organic compound).000), HR 4 (5000–500000), and HR 5 (2000–4000000) (7.8 × 300 mm)]. Polystyrene standards were used for calibration.
Results and discussion
Scheme 1 shows the general structure of the statistical copolymers investigated as macromolecular platforms for the design of the MRI blood pool agents. They consist of statistical copolymers of a poly(ethylene oxide) methyl ether acrylate (PEOMA) with an acrylate bearing an alkyne group. The conjugation of the gadolinium based contrast agent with the alkyne functional macromolecule is carried out by the highly efficient and selective Huisgen cycloaddition (“click”) reaction. Two different spacers (Scheme 1) between the polymer backbone and the alkyne group are investigated in order to study the effect of their length on the properties of the corresponding macrocontrast agent (relaxivity and toxicity). The anchoring of the gadolinium complex onto the macromolecule decreases its mobility and should therefore increase its relaxivity by decreasing its tumbling rate. Moreover, hindering this complex by the surrounding PEO chains is also expected to contribute to the enhancement of its contrasting efficiency. To the best of our knowledge, there are various reports describing the controlled radical polymerization of PEO-based (meth)acrylates25–29 but not on their copolymerization with alkyne functional acrylates. This copolymerization is therefore discussed in the next section using Reversible Addition Fragmentation chain Transfer (RAFT) polymerization.26,30Synthesis of poly(poly(ethylene oxide) methyl ether acrylate)-st-poly(propargyl acrylate) (P[PEOMA-st-PA]; Scheme 4)
In order to prepare well-defined copolymers, they are synthesized by the copolymerization of poly(ethylene oxide) methyl ether acrylate (PEOMA) with propargyl acrylate (PA) by Reversible Addition Fragmentation chain Transfer (RAFT) polymerization,26,30 a powerful controlled radical polymerization technique. S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid)trithiocarbonate (CTA)19 is used as the RAFT agent in the presence of AIBN (10 mol% compared to CTA) in DMF at 80 °C. At a monomer concentration of 33 vol% in DMF, the polymerization is under 70% conversion within 300 min. A complete analysis of the kinetics of copolymerization is carried out in order to evaluate the control character of this copolymerization since, to the best of our knowledge, it has never been reported before. The experimental molecular weights are determined by 1H NMR spectroscopy by comparison of integrals corresponding to the PEO grafts at 4.16 ppm (–O–CH2–PEO) and 4.64 ppm (–O–CH2–alkyne) with the integral of the ω-chain end at 0.87 ppm (CH3–CH2–) (Fig. 1).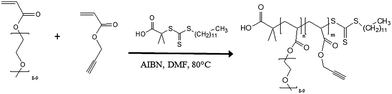 | ||
| Scheme 4 RAFT copolymerization of PEOMA with PA. | ||
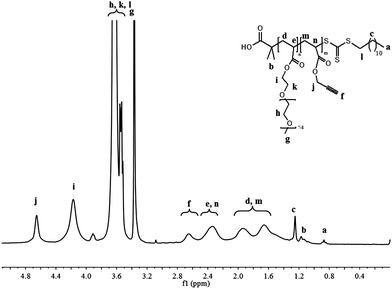 | ||
| Fig. 1 1H NMR spectrum in CDCl3 of P(PEOMA32-st-PA14) (Table 1, entry 2). | ||
As shown in Fig. 2, the experimental molecular weight increases linearly with the monomer conversion and is close to the theoretical value. The polydispersity remains low (below 1.2) all along the polymerization process. Moreover, the time dependence of ln ([M]0/[M]) is also linear (Fig. 3). All these observations are consistent with a controlled polymerization.
![Experimental molecular weights and polydispersity evolution with the monomer conversion for the PEOMA/PA copolymerization in DMF. Conditions: 80 °C; [monomers]/[CTA] = 50; [PEOMA]/[PA] = 7/3, [CTA]/[AIBN] = 10, (monomers)/DMF = 1/3 v/v. Mn exp = experimental number average molecular weight determined by 1H NMR spectroscopy using the formula.](/image/article/2011/PY/c1py00198a/c1py00198a-f2.gif) | ||
| Fig. 2 Experimental molecular weights and polydispersity evolution with the monomer conversion for the PEOMA/PA copolymerization in DMF. Conditions: 80 °C; [monomers]/[CTA] = 50; [PEOMA]/[PA] = 7/3, [CTA]/[AIBN] = 10, (monomers)/DMF = 1/3 v/v. Mn exp = experimental number average molecular weight determined by 1H NMR spectroscopy using the formula. | ||
![Time dependence of ln([M]0/[M]) for the PEOMA/PA copolymerization in DMF. Conditions: 80 °C; [monomers]/[CTA] = 50; [PEOMA]/[PA] = 7/3, [CTA]/[AIBN] = 10, (monomers)/DMF = 1/3 v/v.](/image/article/2011/PY/c1py00198a/c1py00198a-f3.gif) | ||
| Fig. 3 Time dependence of ln([M]0/[M]) for the PEOMA/PA copolymerization in DMF. Conditions: 80 °C; [monomers]/[CTA] = 50; [PEOMA]/[PA] = 7/3, [CTA]/[AIBN] = 10, (monomers)/DMF = 1/3 v/v. | ||
The composition of the copolymer, determined by 1H NMR spectroscopy, is also constant during the whole polymerization and is in line with the initial feed composition (Fig. 4), confirming the formation of a statistical copolymer.
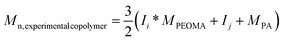
According to this optimized procedure, two copolymers of different molecular weights (Table 1, entries 1 and 2) are prepared in order to ultimately study the influence of the molecular weight of the polymer on the properties of the macrocontrast agent.
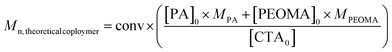
![Propargyl acrylate average composition in the copolymer during the PEOMA/PA copolymerization in DMF at 80 °C ([PEOMA]0/[PA]0 = 7/3).](/image/article/2011/PY/c1py00198a/c1py00198a-f4.gif) | ||
| Fig. 4 Propargyl acrylate average composition in the copolymer during the PEOMA/PA copolymerization in DMF at 80 °C ([PEOMA]0/[PA]0 = 7/3). | ||
| Entry | [monomer]0/[CTA]0 | Time/h | Conv.a (%) | DPPEOMa | DPPA or HEAa | M n, SECb/g mol−1 | M w/Mnb |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Determined by size exclusion chromatography (SEC) using polystyrene as standard. | |||||||
| P[PEOMA-st-PA] | |||||||
| 1 | 10 | 5 | 70% | 5 | 2 | 7500 | 1.08 |
| 2 | 50 | 5 | 70% | 32 | 14 | 42500 |
1.16 |
| P[PEOMA-st-HEA] | |||||||
| 3 | 10 | 4 | >98% | 9 | 4 | 7400 | 1.16 |
| 4 | 50 | 4 | >98% | 42 | 18 | 42000 |
1.16 |
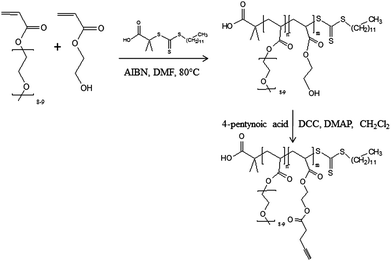
Synthesis of poly(poly(ethylene oxide) methyl ether acrylate)-st-poly(2-acryloyloxy)ethyl pent-4-ynoate) (P(PEOMA-st-AEP); Scheme 5)
The copolymer is prepared by first copolymerizing 2-hydroxyethyl acrylate (HEA) with PEOMA by RAFT, followed by the esterification of the hydroxyl groups with 4-pentynoic acid (Scheme 5). Because CTA was successfully used for the polymerization of both HEA19 and PEOMA,31 it is again used as the RAFT agent in the presence of AIBN (10 mol% compared to CTA) at 80 °C in DMF. At a monomer concentration of 33 vol% in DMF, the polymerization is quasi-complete (>98%) after 4 h with a low polydispersity (Mw/Mn = 1.16). After the polymer purification, the experimental molecular weights are determined by 1H NMR spectroscopy by comparison of integral corresponding to the polymer backbone at 4.14 ppm (–CO–O–CH2–) with the integral of the ω-chain end at 0.87 ppm (CH3–CH2) (Fig. 5). The molecular weight (Mn, NMR = 21100 g mol−1) is in rather good agreement with the theoretical value as expected for a controlled process (Mn, theor = 17500 g mol−1). By adapting the monomer to CTA ratio, a P[PEOMA-st-HEA] with a different molecular weight is prepared (Table 1, entry 4).
 | ||
| Scheme 5 RAFT copolymerization of PEOMA with AEP, followed by esterification with 4-pentynoic acid. | ||
![1H NMR spectra of P[PEOMA42-st-HEA18] (down) and P[PEOMA42-st-AEP18] (up).](/image/article/2011/PY/c1py00198a/c1py00198a-f5.gif) | ||
| Fig. 5 1H NMR spectra of P[PEOMA42-st-HEA18] (down) and P[PEOMA42-st-AEP18] (up). | ||
Esterification of the hydroxyl groups of the copolymers with 4-pentynoic acid is carried out in dry CH2Cl2 in the presence of N,N′-dicyclohexylcarbodiimide (DCC; 1.1 equivalent) and 4-(dimethylamino)pyridine (DMAP; 0.11 equivalent) at 0 °C. Under the investigated conditions, (see Experimental section), the esterification is quantitative after one night as assessed by 1H NMR analysis (Fig. 5) that evidences the complete disappearance of the signal typical of proton close to hydroxyl group –CH2–OH at 3.75 ppm and the appearance of the signal characteristic of ester group –CH2–O–C(O)– at 4.26 ppm.
Similarly to the previous copolymer, a copolymer with a different molecular weight (Table 2, entries 1 and 2) is also prepared.

Conjugation of azide functionalized molecules to alkynes bearing copolymers (P[PEOMA-st-PA] and P[PEOMA-st-AEP]) by click reaction (Scheme 1)
Benzyl azide is first used as a model molecule to optimize the conjugation conditions (with or in the absence of sodium ascorbate, bathophenanthroline disulfonic acid disodium salt hydrate, inert atmosphere,…). Optimized conditions are found to be with CuI as the catalyst (10 mol% compared to alkyne) in DMF as solvent at room temperature. As assessed by 1H NMR analysis (Fig. 6), grafting yields are between 80 and 85% for all the copolymers tested. Due to the small size of benzyl azide, there is no substantial difference between the grafting on the low and high molecular weight copolymers (Table 3, entries 1, 4 and 7, 9). A similar observation is made for the grafting on the two types of copolymers with the different spacers between the alkyne group and the polymer backbone (Table 3, entries 1, 7 and 4, 9).![1H NMR spectra of P[PEOMA32-st-PA14] (Mn = 16 450 g mol−1) and P[PEOMA32-st-PA14] conjugated with benzyl azide (Table 1, entry 5).](/image/article/2011/PY/c1py00198a/c1py00198a-f6.gif) | ||
| Fig. 6
1H NMR spectra of P[PEOMA32-st-PA14] (Mn = 16450 g mol−1) and P[PEOMA32-st-PA14] conjugated with benzyl azide (Table 1, entry 5). | ||
| Entry | Copolymer | Azide bearing molecule | Grafting yield |
|---|---|---|---|
| a Determined by ICP-MS spectrometry. b Determined by 1H NMR spectroscopy. | |||
| 1 | P[PEOMA5-st-PA2] Mn = 3400 g mol−1 | Benzyl azide | 85%b |
| 2 | DO3AtBu–N3 | 65%b | |
| 3 | DO3A(Gd3+)–N3 | <10%a | |
| 4 | P[PEOMA32-st-PA14] Mn = 16450 g mol−1 |
Benzyl azide | 80%b |
| 5 | DO3AtBu–N3 | 41%b | |
| 6 | DO3A(Gd3+)–N3 | <10%a | |
| 7 | P[PEOMA9-st-AEP4] Mn = 4900 g mol−1 | Benzyl azide | 85%b |
| 8 | DO3AtBu–N3 | 65%b | |
| 9 | P[PEOMA42-st-AEP18] Mn = 22600 g mol−1 |
Benzyl azide | 80%b |
| 10 | DO3AtBu–N3 | 47%b |
Following this reaction, the preparation of the MRI blood pool agent is then carried out by two strategies: (1) the grafting of the macroligand precursor (DO3AtBu–N3; Scheme 1), followed by the deprotection of the t-butyl groups by trifluoroacetic acid and the subsequent complexation with GdCl3 at pH = 6; or (2) by direct click reaction of the pre-formed gadolinium complex functionalized by azide group (DO3A(Gd3+)–N3; Scheme 1).
As expected, when the conjugation of more sterically hindered molecule like DO3AtBu–N3 is considered, the grafting yield is decreased to 65% when carried out on the low molecular weight copolymers (Table 3, entries 2 and 8). It drops to about 40–50% when performed on the higher molecular weight copolymers, as the result of increased steric hindrance (Table 3, entries 5 and 10).
When the grafting of DO3A(Gd3+)–N3 and DO3AtBu–N3 is considered, the final product is extracted by EDTA in order to remove the copper catalyst. ICP analysis of the final product confirms that most of the residual catalyst is removed ([Cu]0 = 5000 ppm, [Cu] after purification = 100 ppm). After reaction, the grafting yield is evaluated by 1H NMR spectroscopy (for DO3AtBu–N3) and ICP analysis (for DO3A(Gd3+)–N3). The grafting yield of DO3A(Gd3+)–N3 to P[PEOMA-st-PA] with different molecular weights is unfortunately very low (<10%) (Table 3, entries 3 and 6) compared to the DO3AtBu–N3 grafting (yield = 40–65%; Table 3, entries 2, 5, 8 and 10). This large difference with DO3AtBu–N3 is certainly the result of the folding of the ligand due to the coordination of the gadolinium by the amide and carboxylate groups, rendering DO3A(Gd3+)–N3 more sterically constrained than the uncomplexed counterpart (Scheme 6).
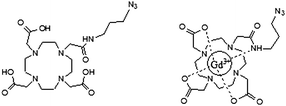 | ||
| Scheme 6 DO3AtBu–N3 (left) and DO3A(Gd3+)–N3 (right). | ||
Because of the low grafting yield of DO3A(Gd3+)–N3, only the grafting of DO3AtBu–N3 is considered in the following discussion. Macromolecules bearing DO3AtBu moieties are then reacted with trifluoroacetic acid to remove tert-butyl groups, followed by complexation of GdCl3 at pH = 6. After purifying the copolymers from excess free GdCl3, the polymers are analyzed by ICP to determine the amount of Gd3+ complexed by the macromolecule. Table 4 summarizes the structure and macromolecular parameters of the macrocontrast agents prepared by this process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | A (DP) | B (DP) | C (DP) | Gd3+a (wt%) |
| a Determined by ICP-MS spectrometry. | |||||
| 1 | –CH2– | 0–1 | 5 | 1–2 | ∼7% |
| 2 | 8–9 | 32 | 5–6 | ∼5% | |
| 3 | –CH2–CH2–O–C(O)–CH2–CH2– |
1–2 | 9 | 2–3 | ∼8% |
| 4 | 9 | 42 | 9 | ∼6% | |

Complement activation tests
Besides imparting water solubility to the macrocontrast agent, the PEO chains have to prevent the recognition of the macrocontrast agent by the immune system that is responsible for its rapid elimination from the blood circulation, restricting timing for studies.One of the macrocontrast agents (P[PEOMA32-st-(AEP8–9-DO3A(Gd3+)5–6); Table 4, entry 4) is therefore evaluated by the hemolytic CH50 test and compared to the starting copolymers (P[PEOMA32-st-AEP14] and P[PEOMA42-st-HEA18], Table 2, entry 2), and to a poly(2-hydroxyethyl acrylate) (PHEA55) as a positive control.32Fig. 7 shows that the copolymer bearing PEO grafts and the alkyne groups (P[PEOMA32-st-PAEP14]) has a very low activation of the complement. This activation slightly increases for the copolymer bearing hydroxyl groups (P[PEOMA42-st-HEA18]), known for activating the immune response.32 Importantly, the conjugation of the gadolinium complex onto P[PEOMA42-st-AEP14] (P[PEOMA42-st-(AEP9–DO3A(Gd3+)9)]) does not activate the complement, confirming that the complex is hidden in the PEO shell that renders it stealth to proteins of the immune system. This very low activation means that the macrocontrast agent is expected to have a long blood circulation time and could be ready to be evaluated by in vivo test like plasma clearance test.
![Consumption of CH50 units in the presence of PHEA55, P[PEOMA42-st-HEA18], P[PEOMA42-st-AEP18], P[PEOMA42-st-(AEP9-DO3A(Gd3+)9)] as a function of surface area.](/image/article/2011/PY/c1py00198a/c1py00198a-f7.gif) | ||
| Fig. 7 Consumption of CH50 units in the presence of PHEA55, P[PEOMA42-st-HEA18], P[PEOMA42-st-AEP18], P[PEOMA42-st-(AEP9-DO3A(Gd3+)9)] as a function of surface area. | ||
Comparison of CH50 tests realized on P[PEOMA42-st-AEP18] (Table 2, entry 2) and P[PEOMA32-st-PA14] (Table 1, entry 2) shows that the length of the spacer between the alkyne group and the polymer backbone has almost no effect on the immune response (Fig. 8). The PEO grafts are thus nicely covering them and render them stealth.
![Consumption of CH50 units in the presence of P[PEOMA32-st-PA14] and P[PEOMA42-st-AEP18] as a function of surface area.](/image/article/2011/PY/c1py00198a/c1py00198a-f8.gif) | ||
| Fig. 8 Consumption of CH50 units in the presence of P[PEOMA32-st-PA14] and P[PEOMA42-st-AEP18] as a function of surface area. | ||
Copolymer cytotoxicity
The cytotoxicity of the different copolymers is evaluated in vitro using MCF-7/BOS and MEL-5 cells. Cells (MCF-7/BOS and MEL-5) in appropriate culture media (see Experimental section) are incubated with different concentrations of polymer for 48 h.
Fig. 9 shows the ratio of survival cells population treated with 0.05 mg to 30 mg of copolymer P[PEOMA32-st-PA14] Mn = 16450 g mol−1 for MCF-7 and MEL-5 cell types. Under 10 mg ml−1cell viability is greater than 85%. These results are in good agreement with results of Pissuwan et al.33 (P[PEOMA] Mn = 10000 g mol−1) and Chang et al.34 (P[PEOMA] Mn = 20000 g mol−1). On the other hand, when the copolymer concentration of P[PEOMA32-st-PA14] is used at a higher concentration (30 mg ml−1) (higher than Pissuwan et al. and Chang et al.) cell viability decreases to a value around 50%. The half maximal inhibitory concentration (IC50, the dose to kill half of the cells after 48 h of incubation) is around 30 mg ml−1 for MEL-5 and MCF-7 cells. We have then studied the influence of the structure of the end-group on the copolymer cytotoxicity (Fig. 10). A copolymer with a thiol end group is synthesized by treatment of the copolymer with butyl amine in THF. This thiol end group copolymer has no significant influence on cell viability compared to trithiocarbonate end group (Fig. 10) in agreement with observations made by Pissuwan et al.
![Cell viability in the presence of different concentrations of copolymer P[PEOMA32-st-PA14] in a MEL-5 (○) and MCF-7(●) culture after a 48 h incubation. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Significance indicated by: *p < 0.05. On the bottom figure, polymer concentration is represented in logarithm scale conversely on the upper picture which is represented in normal scale. Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay.](/image/article/2011/PY/c1py00198a/c1py00198a-f9.gif) | ||
| Fig. 9 Cell viability in the presence of different concentrations of copolymer P[PEOMA32-st-PA14] in a MEL-5 (○) and MCF-7(●) culture after a 48 h incubation. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Significance indicated by: *p < 0.05. On the bottom figure, polymer concentration is represented in logarithm scale conversely on the upper picture which is represented in normal scale. Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay. | ||
![Cell viability of two copolymers P[PEOMA32-PA14] Mn = 16 450 g mol−1 with different end groups with same concentration 0.5 mg ml−1. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay.](/image/article/2011/PY/c1py00198a/c1py00198a-f10.gif) | ||
| Fig. 10
Cell viability of two copolymers P[PEOMA32-PA14] Mn = 16450 g mol−1 with different end groups with same concentration 0.5 mg ml−1. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay. | ||
The cytotoxicity of PEOMA-based copolymers of different functionalities (P[PEOMA42-st-HEA18], P[PEOMA42-st-AEP18], P[PEOMA32-st-PA14] and P[PEOMA42-st-(AEP9-DO3A(Gd3+)9)] has then been studied under identical conditions (0.5 mg ml−1, 48 h incubation, MEL-5 and MCF-7/BOS) and is compared in Fig. 11.
![Cell viability in the presence of different copolymers (1: P[PEOMA42-st-HEA18]; 2: P[PEOMA42-st-AEP18]; 3: P[PEOMA32-st-PA14]; 4: P[PEOMA32-st-(AEP8–9-DO3A(Gd3+)5–6)]) (0.5 mg ml−1) in MCF-7 and MEL-5 culture after 48 h. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Significance indicated by: *p < 0.05 Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay.](/image/article/2011/PY/c1py00198a/c1py00198a-f11.gif) | ||
| Fig. 11 Cell viability in the presence of different copolymers (1: P[PEOMA42-st-HEA18]; 2: P[PEOMA42-st-AEP18]; 3: P[PEOMA32-st-PA14]; 4: P[PEOMA32-st-(AEP8–9-DO3A(Gd3+)5–6)]) (0.5 mg ml−1) in MCF-7 and MEL-5 culture after 48 h. Data represent mean ± standard error of the mean (S.E.M.) (n = 4). Significance indicated by: *p < 0.05 Experiments are set up in technical replicates. Cell viability was assessed with the MTS assay. | ||
P[PEOMA42-st-HEA18] copolymer is cytotoxic (around 70% of cell viability) at 0.5 mg ml−1 on the two cell types. However esterification of hydroxyl groups with 4-pentynoic acid decreases the copolymer cytotoxicity on both cells.
The cytotoxicity of P[PEOMA32-st-PA14] and P[PEOMA42-st-(AEP9–DO3A(Gd3+)9)] is also compared. DO3(Gd3+) labeled copolymer has only a slightly higher cytotoxicity than bare copolymers. Around 80–85% viable cells are observed, indicating that the grafting of the contrast agent on the copolymer does not induce any significant cytotoxicity to the macromolecule.
Relaxometric studies of the macrocontrast agents
The four different macrocontrast agents prepared by the conjugation of DO3AtBu-azide by click chemistry on P[PEOMA-st-PA] and P[PEOMA-st-AEP], followed by the deprotection of the tert-butyl esters of the ligand by trifluoroacetic acid and the complexation of Gd3+ at pH = 6 are characterized (Table 5). Prior to analysis, free Gd3+ is removed by adding free DOTA able to strongly complex Gd3+, followed by dialysis of the copolymer against water. Freeze-drying of the final product provides the pure macrocontrast agent.| Entry | Macrocontrast agents | Relaxivity/mM−1 s−1 |
|---|---|---|
| 1 | P[PEOMA5-st-(PA0-1-DO3A(Gd3+)1-2)] Mn = 3400 g mol−1, | 11.2 (±0.1) |
| 2 | P[PEOMA32-st-(PA8–9–DO3A(Gd3+)5–6)] Mn = 16450 g mol−1 |
13.0 (±0.1) |
| 3 | P[PEOMA9-st-(AEP1–2–DO3A(Gd3+)2–3)] Mn = 4900 g mol−1 | 8.4 (±0.1) |
| 4 | P[PEOMA42-st-(AEP9–DO3A(Gd3+)9)] Mn = 22600 g mol−1 |
9.8 (±0.1) |
The contrast efficiency of the macrocontrast agents is compared in Table 5. The relaxivity (r1) of these different contrast agents is calculated after measuring longitudinal relaxation time (T1) from:
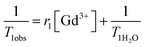
The relaxivities of the different macrocontrast agents at 20 MHz are found to range from 8.4 to 13 mM−1 s−1 and depend on their structures. For the copolymers of the same chemical nature, increasing the molecular weight of the copolymer slightly increases the relaxivity (comparison of entries 1 with 2, and entries 3 with 4, Table 5). It is a consequence of the slowing down of the rotational motion of the gadolinium complex when the molecular weight increases. Importantly, a substantial difference in relaxivity is observed between the two families of macrocontrast agents. When the length of the spacer between the gadolinium complex and the polymer backbone is decreased, the relaxivity is higher as a result of improved rigidity of the system (comparison of entries 3 and 4 with entries 1 and 2).
For the sake of comparison, the relaxivities measured for the macrocontrast agents are significantly higher than those of free and low molecular weight contrast agent DO3A(Gd3+)–N3 (5.1 mM−1 s−1), whose rotational motion is not hindered by a macromolecule. Relaxivities of our best macrocontrast agents (entries 1 and 2, Table 5) are in the same range as that measured for linear polylysine grafted by DOTA (r1 ≈ 15 mM−1 s−1).35
Full relaxometric data are then measured for two macrocontrast agent families over a large magnetic field range (from 0.01 MHz to 100 MHz) and are compared to DO3A(Gd3+)–N3 (Fig. 12). At low frequency (0.01 to 5 MHz), the relaxivity is about 1.5 higher than that of free DO3A(Gd3+)–N3. Importantly the effect of the immobilization of gadolinium on macromolecules has an even more pronounced effect on relaxivity at high frequencies (10 to 80 MHz). The maximum relaxivity for our best macrocontrast agent (P[PEOMA32-st-(PA8–9–DO3A(Gd3+)5–6]) is obtained at 30 MHz with a 250% relaxivity increase upon grafting of DO3A(Gd3+)–N3 onto the copolymer.
![Comparison of the 1H NMRD profiles of DO3A(Gd3+)–N3 (full triangles), P[PEOMA32-st-(PA8–9–DO3A(Gd3+)5–6)] (full circles) and P[PEOMA42-st-(AEP9-DO3A(Gd3+)9)] (empty circles).](/image/article/2011/PY/c1py00198a/c1py00198a-f12.gif) | ||
| Fig. 12 Comparison of the 1H NMRD profiles of DO3A(Gd3+)–N3 (full triangles), P[PEOMA32-st-(PA8–9–DO3A(Gd3+)5–6)] (full circles) and P[PEOMA42-st-(AEP9-DO3A(Gd3+)9)] (empty circles). | ||
The grafting of the gadolinium complex onto a hindered macromolecule using a spacer as short as possible is therefore beneficial to the relaxivity of the system. This relaxivity enhancement is a result of an increase of the rotational correlation lifetime of the gadolinium complex due to the bulkiness and rigidity of the whole system. Karfeld-Sulzer et al.36 has recently shown that the length between linear polymer backbone and Gd3+-complex is very important. Indeed very short spacers prevent tumbling of the complex and long spacers allow the Gd3+-complex to move freely.
Conclusion
Novel stealth functional macromolecular platforms were synthesized by RAFT copolymerization of poly(ethylene oxide) methyl ether acrylate with propargyl acrylate or with 2-hydroxyethyl acrylate followed by esterification with 4-pentynoic acid. Gadolinium complexes were grafted to these macromolecular platforms by the Huisgen 1,3-dipolar cycloaddition (“click”) reaction in mild conditions. Gadolinium based macrocontrast agents were best prepared by first grafting the gadolinium ligand precursor (DO3AtBu–N3), followed by removing the tert-butyl groups by trifluoroacetic acid and finally, by the coordination of Gd3+. When the preformed chelate (DO3A(Gd3+)–N3) was considered, grafting yields were very low due to steric hindrance.Relaxometry measurements have evidenced an improved relaxivity of the macrocontrast agent by about 250% compared to ungrafted gadolinium chelate. Moreover, this relaxivity was further enhanced when the spacer length between the Gd3+-chelate and the polymer backbone was shortened, as a result of its decreased tumbling rate. Cytotoxicity and complement activation studies have demonstrated that the macrocontrast agents were free of any cytotoxicity and that the gadolinium complex was hidden in the PEO shell, rendering the macrocontrast agents stealth to proteins of the immune system. This potential long blood circulation half-life time combined with the high relaxivity at high frequency suggests that these novel products are promising candidates for MRI applications at reasonable concentrations.
Because the gadolinium chelates are bonded to a non-degradable polyacrylate chain, the elimination behavior of these macrocontrast agents from the body has now to be studied.
Acknowledgements
C.D., M.G. and C.J. are much indebted to the “Politique Scientifique Fédérale” for financial support in the frame of the “Interuniversity Attraction Pôles Programme (IAP VI/27): Supramolecular Chemistry and Supramolecular Catalysis”, to the University of Liège and to the National Funds for Scientific Research (F.R.S.-FNRS). The authors also thank Nolwenn Lautram [Ingénierie de la Vectorisation Particulaire (Inserm)—Angers, France] for skilful assistance in the CH50 tests. JFD gratefully thanks the FNRS and the IISN of Belgium for financial support.References
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. E. Merbach and E. Toth, 2001 Search PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier and J.-P. Benoit, Biomaterials, 2006, 27, 4356–4373 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- I. Solomon, Phys. Rev., 1955, 99, 559–565 CrossRef CAS.
- N. Bloembergen, J. Chem. Phys., 1957, 27, 572–573 CrossRef CAS.
- G. Adam, J. Neuerburg, E. Spuntrup, A. Muhler, K. Scherer and R. W. Gunther, J. Magn. Reson. Imaging, 1994, 4, 462–466 CAS.
- L. H. Bryant, Jr, M. W. Brechbiel, C. Wu, J. W. Bulte, V. Herynek and J. A. Frank, J. Magn. Reson. Imaging, 1999, 9, 348–352 CrossRef CAS.
- E. C. Wiener, M. W. Brechbiel, H. Brothers, R. L. Magin, O. A. Gansow, D. A. Tomalia and P. C. Lauterbur, Magn. Reson. Med., 1994, 31, 1–8 CrossRef CAS.
- G. M. Nicolle, E. Toth, K.-P. Eisenwiener, H. R. Macke and A. E. Merbach, JBIC, J. Biol. Inorg. Chem., 2002, 7, 757–769 CrossRef CAS.
- H. Tournier, R. Hyacinthe and M. Schneider, Acad. Radiol., 2002, 9(Suppl 1), S20–S28 CrossRef.
- M. Grogna, R. Cloots, A. Luxen, C. Jérôme, C. Passirani, N. Lautram, J. F. Desreux and C. Detrembleur, Polym. Chem., 2010, 1, 1485–1490 RSC.
- G. Schuhmann-Giampieri, H. Schmitt-Willich, T. Frenzel, W. R. Press and H. J. Weinmann, Invest. Radiol., 1991, 26, 969–974 CAS.
- P. F. Sieving, A. D. Watson and S. M. Rocklage, Bioconjugate Chem., 1990, 1, 65–71 CrossRef CAS.
- D. L. Ladd, R. Hollister, X. Peng, D. Wei, G. Wu, D. Delecki, R. A. Snow, J. L. Toner, K. Kellar, J. Eck, V.C. Desai, G. Raymond, L. B. Kinter, T. S. Desser and D. L. Rubin, Bioconjugate Chem., 1999, 10, 361–370 CrossRef CAS.
- R. Rebizak, M. Schaefer and E. Dellacherie, Eur. J. Pharm. Sci., 1999, 7, 243–248 CrossRef CAS.
- S. C. Wang, M. G. Wikstrom, D. L. White, J. Klaveness, E. Holtz, P. Rongved, M. E. Moseley and R. C. Brasch, Radiology (Oak Brook, IL, U. S.), 1990, 175, 483–488 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- Y. Song, E. K. Kohlmeir and T. J. Meade, J. Am. Chem. Soc., 2008, 130, 6662–6663 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- B. Carboni, A. Benalil and M. Vaultier, J. Org. Chem., 1993, 58, 3736–3741 CrossRef CAS.
- P. Calvo, B. Gouritin, H. Chacun, D. Desmaele, J. D'Angelo, J. P. Noel, D. Georgin, E. Fattal, J. P. Andreux and P. Couvreur, Pharm. Res., 2001, 18, 1157–1166 CrossRef CAS.
- A. Aqil, S. Vasseur, E. Duguet, C. Passirani, J. P. Benoit, A. Roch, R. Muller, R. Jerome and C. Jerome, Eur. Polym. J., 2008, 44, 3191–3199 CrossRef CAS.
- A. Vonarbourg, C. Passirani, P. Saulnier, P. Simard, J. C. Leroux and J. P. Benoit, J. Biomed. Mater. Res., Part A, 2006, 78, 620–628 CrossRef CAS.
- C. Passirani, G. Barratt, J.-P. Devissaguet and D. Labarre, Life Sci., 1998, 62, 775–785 CrossRef CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M.-H. Li and B. Charleux, Chem. Commun. (Cambridge, U. K.), 2010, 46, 1950–1952 RSC.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, 2008 Search PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- M. Hurtgen, A. Debuigne, C. A. Fustin, C. Jerome and C. Detrembleur, Macromolecules (Washington, DC, U. S.), 2011, 44(12), 4623–4631 Search PubMed.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun. (Cambridge, U. K.), 2004, 2026–2027 RSC.
- C. Boyer, V. Bulmus, P. Davis Thomas, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- M. D. Rowe, C.-C. Chang, D. H. Thamm, S. L. Kraft, J. F. Harmon, Jr, A. P. Vogt, B. S. Sumerlin and S. G. Boyes, Langmuir, 2009, 25, 9487–9499 CrossRef CAS.
- C. Passirani and J.-P. Benoit, Complement Activation by Injectable Colloidal Drug Carriers, ed. Ram I. Mahato, 2005 Search PubMed.
- D. Pissuwan, C. Boyer, K. Gunasekaran, P. Davis Thomas and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun. (Cambridge, U. K.), 2009, 3580–3582 RSC.
- S. Aime, M. Botta, S. G. Crich, G. Giovenzana, G. Palmisano and M. Sisti, Bioconjugate Chem., 1999, 10, 192–199 CrossRef CAS.
- L. S. Karfeld-Sulzer, E. A. Waters, N. E. Davis, T. J. Meade and A. E. Barron, Biomacromolecules, 2010, 11, 1429–1436 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |