Reversible cross-linking of hydrophilic dynamic covalent polymers with radically exchangeable alkoxyamines in aqueous media
Jing
Su
a,
Yoshifumi
Amamoto
a,
Masamichi
Nishihara
b,
Atsushi
Takahara
*abc and
Hideyuki
Otsuka
*abc
aGraduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
bInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: takahara@cstf.kyushu-u.ac.jp; otsuka@ms.ifoc.kyushu-u.ac.jp; Fax: +81-92-802-2518; Tel: +81-92-802-2515
cInternational Research Center for Molecular Systems, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan
First published on 29th June 2011
Abstract
Reversible cross-linking of alkoxyamine-containing hydrophilic dynamic covalent polymers in aqueous media has been accomplished. Water-soluble polymers with alkoxyamine units in the side chains were synthesized by radical copolymerization of 2-(dimethylamino)ethyl methacrylate and methacrylic esters with alkoxyamine units, and subsequent protonation of dimethylaminoethyl groups by hydrochloric acid. By heating the polymers in water at 100 °C in a closed system, hydrogels were formed by radical exchange reactions. Because the radical reactions are tolerant of water, the exchange reactions of alkoxyamine units in the side chain caused the cross-linking reaction even in aqueous media. Cross-linking behaviors depended on the amounts of alkoxyamine units in the polymers and the reaction concentrations. A de-cross-linking reaction was also accomplished by radical exchange reaction between the cross-linked polymer and added hydrophilic alkoxyamine compound. This dynamic cross-linking nature in aqueous media provides a foundation for a wide range of polymer reactions, and it can be applied to environmentally benign dynamic materials.
Introduction
Dynamic covalent chemistry combines the robustness of covalent bonds with the reversibility of non-covalent interactions,1–3 which makes it a powerful tool for the synthesis of impressively complex structures under thermodynamic controls.4 Reversible covalent reactions such as disulfide exchange,2c,d,5olefin metathesis,6 and thermoreversible Diels–Alder reaction7 have been utilized for dynamic covalent chemistry and applied to molecular systems, including self-assembly8 and structural transformation on environmental change.9–13 In response to equilibrium perturbations, such as changes in concentration, temperature, ionic factors, electric or magnetic fields, chemical or biological agents, and mechanical stress, constitutional alterations may result from re-equilibration of the product.14–17 Considerable attention has also been attracted in terms of applying dynamic covalent chemistry to developing new polymers such as dynamic covalent polymers,2b,14,18–20 which could be considered as reorganizable polymers or stimuli-responsive polymers, consisting of reversible covalent bonds. We have previously reported formations of multiblock copolymers21 and graft copolymers22 from dynamic covalent polymers containing alkoxyamine.Cross-linking systems have also been prompted by dynamic covalent chemistry. For example, sequence-selective cross-linking of oligoamide in aqueous media to afford the corresponding macrocyclic oligoamide has been reported.23 In polymer chemistry, reversible covalent bonds have provided covalently cross-linked polymers with particular abilities such as sol–gel transition,9,24,25 network structure variation,26 and self-healing properties27–31 with no decrease in their high mechanical strengths and stabilities. So far, we have reported a reversible cross-linking system in organic solvents using alkoxyamine compounds, which are adducts of styryl radicals and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), as reversible covalent bonds. The alkoxyamine compounds are widely known as initiators for living radical polymerizations, reaching equilibrium between the alkoxyamines (dormant species) and radicals (active species) over 60 °C. Additionally, in homolysis, each unit is exchanged by crossover of the radicals to form different alkoxyamine compounds.22,32,33 Therefore, the cross-linking reactions occur by the radical exchange of the polymers with alkoxyamine units in their side chains.25
In comparison to polymer reactions in organic solvents, those in aqueous media can be applied as environmentally benign polymeric materials such as coatings and paints. Water is evidently the most abundant and environmentally benign solvent and has the industrial advantage that it can be used in large amounts without producing hazardous waste.34 Because the radical reactions are tolerant of many functional groups and solvents, similar to olefin metathesis polymerization,35 the exchange reaction of alkoxyamine units is a candidate for cross-linking units in aqueous media. In this paper, reversible cross-linking in aqueous media using water-soluble dynamic covalent polymers with alkoxyamine units is discussed, as represented in Scheme 1. The cross-linking reactions are traced from the perspective of the amounts of alkoxyamine in the polymer and reaction concentrations. In addition, de-cross-linking reaction is attempted via radical exchange reaction of the cross-linked polymers with hydrophilic alkoxyamine compound.
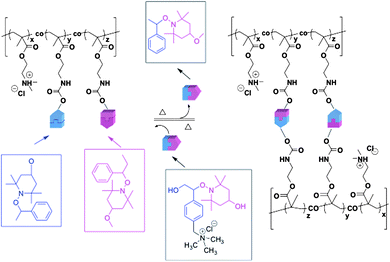 | ||
| Scheme 1 Cross-linking and de-cross-linking reactions of water-soluble dynamic covalent polymersvia radical exchange reaction of alkoxyamine units. | ||
Experimental
Materials
4-Acetyl-TEMPO was synthesized by reference to previous reports.10,25,33 Methacrylic esters with alkoxyamine units (1 and 2) were synthesized as previously reported.94-Chloromethyl styrene was kindly supplied by AGC Seimi Chemical Co. Ltd. and used without further purification. Benzoyl peroxide (BPO) was purchased from Nacalai Tesque Inc. and used without further purification. L(+)-Ascorbic acid and 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) were purchased from Wako Pure Chemical Industries and used without further purification. Trimethylamine in ethanol solution was purchased from Sigma-Aldrich Co. and used without further purification. 2-(Dimethylamino)ethyl methacrylate (DMAEMA) was purchased from Wako Pure Chemical Industries and purified by distillation under reduced pressure over calcium hydride.Measurement
1H (400 MHz) NMR spectroscopic measurements were carried out at 25 °C with a Bruker AV-400 spectrometer using tetramethylsilane as an internal standard in chloroform-d (CDCl3) and DMSO-d6. The number-average molecular weights (Mn) and molecular weight distribution of soluble polymers were determined by gel permeation chromatography (GPC) using N,N-dimethylformamide (DMF) or water as eluents. DMF-eluent GPC was recorded on a JASCO instrument equipped with a JASCO 2031plus refractive index (RI) detector and two poly(hydroxyethyl methacrylate) gel columns (Shodex OHpak SB-804HQ, 0.5 mL min−1). DMF containing 0.01 M LiBr was used as an eluent. A calibration curve was constructed from a series of well-defined polystyrene standard samples. Aqueous GPC measurement was performed with a Shimadzu high-performance liquid chromatography (HPLC) system connected to three polystyrene gel columns of Tosoh G3000PWXL-CP (pore size 20 nm, bead size 7 μm) and G5000PWXL-CP (pore size 100 nm, bead size 10 μm) × 2 and equipped with a RI detector (Shimadzu RID-10A) using an acetic acid aqueous solution (500 mM) containing sodium nitrate (200 mM) as an eluent at a rate of 0.6 mL min−1. A calibration curve was constructed from a series of well-defined poly[{2-(methacryloyloxy)ethyl}trimethyl ammonium chloride] standard samples. The dynamic viscoelastic measurements were carried out with a Physica MCR101 rheometer with a parallel circular plate 12 mm and 20 mm in diameter in water (10 wt%) at 25 °C. The storage modulus (G′) was evaluated based on the average values of numerous measurements.Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900, Mw/Mn = 2.06.
900, Mw/Mn = 2.06.
Results and discussion
Synthesis of water-soluble polymer
Water-soluble polymers consisting of alkoxyamine and hydrophilic side chains were synthesized, as shown in Scheme 2, in two steps: radical copolymerization of DMAEMA and methacrylic esters with alkoxyamine units (1 and 2), and protonation of dimethylaminoethyl groups by hydrochloric acid. Poly(DMAEMA-co-1-co-2) (3) was synthesized by radical copolymerization of DMAEMA, 1, and 2 using V-70 as a low-temperature radical polymerization initiator (decomposition half-time: 10 h at 30 °C). The polymerization was carried out at 40 °C because the alkoxyamine units are stable at this temperature.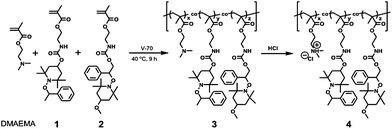 | ||
| Scheme 2 Preparation of water-soluble polymer by radical copolymerization of DMAEMA, 1, and 2, and subsequent protonation of dimethylaminoethyl groups. | ||
As shown in Table 1, four copolymerization experiments with different feed ratios of DMAEMA, 1, and 2 were performed. After purification, the composition of each copolymer was estimated by 1H-NMR measurements (Fig. 1), and Mn and Mw/Mn were evaluated by GPC using DMF as an eluent. In all cases, the compositions of 1 and 2 in the copolymers ((y + z)/(x + y + z)) were very close to the monomer feed ratios of DMAEMA, 1, and 2. This result indicates that four types of random copolymers (3a–d) with different alkoxyamine compositions were successfully obtained by radical copolymerization of DMAEMA, 1, and 2. The molecular weights of the polymers estimated by GPC increased with the ratio of 1 and 2, because of the difference in molecular weight between DMAEMA and alkoxyamine-containing monomers.

The dimethylaminoethyl groups in polymers 3a–d were subsequently converted into ammonium groups by treating the polymers with 2 N of hydrochloric acid, as shown in Scheme 2. After the purification, the polymers were dissolved in water. It was confirmed that protonation of the dimethylaminoethyl groups proceeded quantitatively. Furthermore, the molecular weights of the polymers estimated by aqueous GPC increased with the ratio of 1 and 2, with these results corresponding to the polymers 3a–d. This result verifies that the water-soluble polymers 4a–d were obtained (0.51 g, yield 79% for 4a; 0.62 g, yield 93% for 4b; 0.62 g, yield 91% for 4c; 0.62 g, yield 90% for 4d).
Cross-linking reaction
Since polymers 4a–d have radically exchangeable alkoxyamine units in the side chain, it was expected that the cross-linking reaction could proceed even in aqueous media. Aqueous solutions of polymers 4a–d were heated at 100 °C in closed systems under different concentrations. Fig. 2 shows a photograph of 10 wt% aqueous solutions of polymer 4b before and after heating for 24 h. The gelation of the system was confirmed after heating with no change in color observed. At 100 °C, styryl and nitroxide radicals derived from the alkoxyamine units in the side chain were generated, and their radical exchange and formation of different alkoxyamines caused cross-linking points to form. If carbon–carbon coupling such as styryl–styryl coupling takes place, the formation of nitroxide radicals changes the colorless solution to red. Because of the persistent radical effect,36carbon–carbon coupling reactions hardly occurred. | ||
| Fig. 2 Photograph of 10 wt% aqueous solutions of polymer 4b before heating (left) and after heating (right) at 100 °C in a closed system for 24 h. | ||
Dynamic viscoelasticity measurement was performed using a rheometer with a parallel circular plate in water (10 wt%) at 25 °C. The relationships between storage modulus (G′) and angular frequency (ω) of 10 wt% aqueous solutions of polymer 4b before cross-linking, and after cross-linking for 12 h and 24 h, were measured at 25 °C, as shown in Fig. 3. The storage modulus (G′) prior to the cross-linking reaction suggests that characteristic angular frequency dependence exhibits a typical tendency of linear polymers. As the reaction proceeded, almost no frequency dependence of storage modulus (G′) was observed, and the average G′ was 2.55 × 103 Pa, which suggests that the prepared polymer samples have gel characteristics because of their network structure.37 These data confirm that the network structure of the polymer chains was formed after heating.
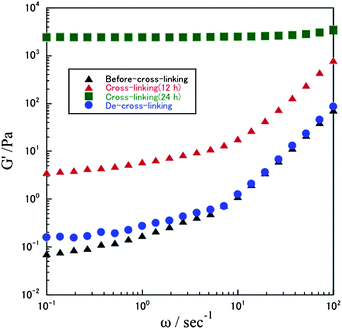 | ||
| Fig. 3 Storage modulus (G′) vs. angular frequency (ω) of 10 wt% aqueous solutions of polymer 4b before cross-linking, after cross-linking for 12 h and 24 h, and after de-cross-linking at 25 °C. | ||
The cross-linking behavior depends notably upon the composition of the copolymers and the polymer concentrations. Fig. 4 shows the yield of the gels prepared from 4a–d in various initial polymer concentrations. In the cases of 4a and 4b, gelation of the systems was confirmed particularly in high concentrations, and higher yields of the gel prepared from 4a were obtained in all initial concentrations compared with those for 4b. Furthermore, in both cases, higher yields of the gel were formed with an increase in initial polymer concentration. In the cases of 4c and 4d, however, gelation of the system was barely observed, and the yield was almost zero even in high concentrations. This result indicates that the gelation occurred only when increasing the ratio of alkoxyamine units in the side chains and is related to overlap concentration of polymer chains.
 | ||
| Fig. 4 Yields of water-insoluble products after heating water-soluble polymers 4a–d in water at 100 °C in closed systems under various concentrations. | ||
A simplified reaction behavior describing the cross-linking is discussed from the viewpoint of intramolecular and intermolecular cross-linking reactions. Fig. 5 shows GPC curves of the reacted polymers after heating 4c in various concentrations. As mentioned above, macroscopic gelation of the system was not confirmed after heating 4c. In the case of 1 wt% condition, a peak was observed in the region of lower molecular weight compared with that for 4c before heating. It is considered that an intramolecular cross-linking reaction proceeded preferentially because of the low concentrations, and hydrodynamic size became lower than that before heating. Meanwhile, under the high-concentration conditions, peaks in regions of higher molecular weight were observed. Because of the close distances between polymer chains, intermolecular cross-linking occurred in part, which is not sufficient for gelation of the system. As a result, the polymers that reacted in the different concentrations display different macromolecular structures brought about by intramolecular and intermolecular cross-linking (Scheme 3). The tendency was clearly observed, however, the quantitative discussion on intra/interefficiency is not possible in the present system.
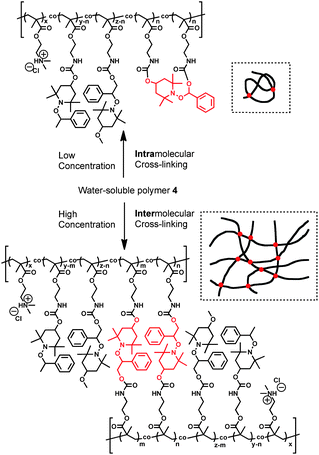 | ||
| Scheme 3 Intramolecular and intermolecular cross-linking reactions of polymer 4. | ||
 | ||
| Fig. 5 GPC curves of the reacted polymers before and after heating aqueous solution of 4c (8 wt%, 6 wt%, 4 wt%, 2 wt%, and 1 wt% concentrations) at 100 °C in closed systems for 24 h. | ||
De-cross-linking reaction
The gels cross-linked by reversible covalent bonds have the potential for reverse de-cross-linking reaction that could turn macroscopic gels into solutions. Here, water-soluble alkoxyamine (7) was designed as a de-cross-linking agent and was prepared according to Scheme 4. Although water-soluble alkoxyamines attached by PEG have been reported,38 they tend to form aggregates under heating. The de-cross-linking reaction was carried out by heating the cross-linked polymer swollen in water with an excess amount of 7 (20 equiv. per alkoxyamine unit) at 100 °C in a closed system for 24 h. After heating, the gel turned into solution. This result proves that the cross-linking points and 7 are thermally dissociated, and radical exchange reaction proceeds between the generated radicals. De-cross-linking reaction progresses as a result. | ||
| Scheme 4 Preparation of water-soluble alkoxyamine 7. | ||
Fig. 6 shows aqueous GPC curves of polymer 4b and the de-cross-linked polymer, which was generated by treatment with the gel prepared from 4b with water-soluble alkoxyamine. Before cross-linking, the Mn of polymer 4b was 39700, which was determined by the aqueous GPC system. After de-cross-linking reaction of the gel prepared from 4b, the Mn of the de-cross-linked polymer was 66000, indicating that most polymer chains are de-cross-linked.
 | ||
| Fig. 6
GPC curves of polymer 4b (Mn = 39700, Mw/Mn = 3.04) and de-cross-linked polymer by treatment with the gel prepared from 4b with water-soluble alkoxyamine (Mn = 66000, Mw/Mn = 2.38). | ||
Dynamic viscoelasticity measurement also supports the progress of the de-cross-linking reaction. Fig. 3 shows the relationships between storage modulus (G′) and angular frequency (ω) of 10 wt% aqueous solutions of polymer 4b before cross-linking and after de-cross-linking of the gel prepared from 4b. The plots for the two samples are very similar, indicating that the network structure is broken down by treatment with the gel prepared from 4b with water-soluble alkoxyamine.
Conclusions
Reversible cross-linking reactions of alkoxyamine-containing dynamic covalent polymers in an aqueous system have been demonstrated. Water-soluble polymers with dynamic covalent bonds in the side chain were synthesized by radical copolymerization of DMAEMA, 1, and 2, and subsequent protonation of dimethylaminoethyl groups. Cross-linking reactions of water-soluble polymers in aqueous media were achieved by heating the polymer to 100 °C in closed systems, and it was found that gel structures clearly depended on the amounts of cross-linkable alkoxyamine units in the polymer chains and concentrations of the polymer. De-cross-linking reaction was also accomplished in aqueous media by radical exchange reaction between the cross-linked polymers and the water-soluble alkoxyamine compound. Since radical reactions are tolerant of many functional groups and solvents, the present system may be applied to systems that are more complicated, such as organic/aqueous two-phase systems.Acknowledgements
The authors gratefully acknowledge the financial support of a Grant-in-Aid for Scientific Research (20350057) from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan. The present work is also supported by a Grant-in-Aid of the Global COE Program, “Science for Future Molecular Systems” under MEXT, Japan. H.O. gratefully acknowledges the financial support of Funding Program (Green Innovation GR077) for Next Generation World-Leading Researchers from the Cabinet Office, Government of Japan.Notes and references
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousin, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- (a) P. T. Glink, A. I. Oliva, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 1870–1875 CrossRef CAS; (b) T. Nishinaga, A. Tanatani, K. Oh and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 5934–5935 CrossRef CAS; (c) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS; (d) Y. Furusho, T. Oku, T. Hasegawa, A. Tsuboi, N. Kihara and T. Takata, Chem.–Eur. J., 2003, 9, 2895–2903 CrossRef CAS; (e) R. Cacciapaglia, S. D. Stefano and L. Mandolini, J. Am. Chem. Soc., 2005, 127, 13666–13671 CrossRef CAS; (f) H. Kudo, R. Hayashi, K. Mitani, T. Yokozawa, N. C. Kasuga and T. Nishikubo, Angew. Chem., Int. Ed., 2006, 45, 7948–7952 CrossRef CAS.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- (a) C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC; (b) N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS; (c) J. R. Nitschke and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11970–11974 CrossRef CAS.
- (a) W. J. Lees and M. W. George, J. Org. Chem., 1993, 58, 642–647 CrossRef CAS; (b) H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1050–1152 RSC.
- (a) M. J. Marsella, H. D. Maynard and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 1101–1103 CrossRef CAS; (b) H. Otsuka, T. Muta, M. Sakada, T. Maeda and A. Takahara, Chem. Commun., 2009, 1073–1075 RSC.
- (a) Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636–2641 CrossRef CAS; (b) X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 1997, 278, 1601–1604 CrossRef; (c) R. Gheneim, C. P. Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- (a) J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS; (b) R. F. Service, Science, 2005, 309, 95 CrossRef CAS.
- Y. Amamoto, Y. Higaki, Y. Matsuda, H. Otsuka and A. Takahara, J. Am. Chem. Soc., 2007, 129, 13298–13304 CrossRef CAS.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2009, 42, 8733–8738 CrossRef CAS.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- R. Nicolay, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361 CrossRef CAS.
- X. X. Chen, M. A. Dam, K. Ono, A. Mal, H. B. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- (a) T. Ono, T. Nobori and J.-M. Lehn, Chem. Commun., 2005, 1522–1524 RSC; (b) Y. Ruff and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 3556–3559 CrossRef CAS; (c) Y. Ruff, E. Buhler, S.-J. Candau, E. Kesselman, Y. Talmon and J.-M. Lehn, J. Am. Chem. Soc., 2010, 132, 2573–2584 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS.
- J.-M. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- W. G. Skene and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- (a) Y. Higaki, H. Otsuka, T. Endo and A. Takahara, Macromolecules, 2003, 36, 1494–1499 CrossRef CAS; (b) Y. Higaki, H. Otsuka and A. Takahara, Polymer, 2003, 44, 7095–7101 CrossRef CAS; (c) Y. Higaki, H. Otsuka and A. Takahara, Polymer, 2006, 47, 3784–3791 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 1696–1701 CrossRef CAS.
- M. F. Li, K. Yamato, J. S. Ferguson, K. K. Singarapu, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2008, 130, 491–500 CrossRef CAS.
- G. H. Deng, C. M. Tang, F. Y. Li, H. F. Jiang and Y. M. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- A. Izuka and H. H. Winter, Macromolecules, 1997, 30, 6158–6165 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- M. W. Urban, Prog. Polym. Sci., 2009, 34, 679–687 CrossRef CAS.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- G. V. Kolmakov, K. Matyjaszewski and A. C. Balazs, ACS Nano, 2009, 3, 885–892 CrossRef CAS.
- (a) C. J. Hawker, G. G. Barclay and J. Dao, J. Am. Chem. Soc., 1996, 118, 11467–11471 CrossRef CAS; (b) B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. D. Cola, Angew. Chem., Int. Ed., 2010, 49, 6881–6884 CrossRef CAS.
- (a) H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, Chem. Commun., 2002, 2838–2839 RSC; (b) H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064–4065 CrossRef CAS; (c) G. Yamaguchi, Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2005, 38, 6316–6320 CrossRef CAS; (d) H. Otsuka, K. Aotani, Y. Higaki, Y. Amamoto and A. Takahara, Macromolecules, 2007, 40, 1429–1434 CrossRef CAS.
- (a) K. Sato, M. Kamigaito and M. Sawamoto, Macromolecules, 2000, 33, 5836–5840 CrossRef; (b) K. Sato, S. Sato and M. Kamigaito, J. Am. Chem. Soc., 2007, 129, 9586–9587 CrossRef; (c) J. Huber and S. Mecking, Macromolecules, 2010, 43, 8718–8723 CrossRef CAS.
- K. Zhang, J. Cui, M. Lackey and G. N. Tew, Macromolecules, 2010, 43, 10246–10252 CrossRef CAS.
- (a) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS; (c) A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC; (d) A. Studer and T. Schulte, Chem. Rec., 2005, 5, 27–35 CrossRef CAS; (e) W. Tang, T. Fukuda and K. Matyjaszewski, Macromolecules, 2006, 39, 4332–4337 CrossRef CAS.
- A. Izuka and H. H. Winter, Macromolecules, 1997, 30, 6158–6165 CrossRef CAS.
- Y. J. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228–5234 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |