Recent progress on photochromic diarylethene polymers
Qianfu
Luo
,
Hui
Cheng
and
He
Tian
*
Key Labs for Advanced Materials and Institute of Fine Chemicals, East China University of Science & Technology, Shanghai, 200237, P. R. China. E-mail: tianhe@ecust.edu.cn; Fax: (+86)-21-64252288
First published on 24th June 2011
Abstract
Organic photochromic materials based on diarylethenes and their derivatives have been intensively investigated from fundamental research to technological applications. This review briefly presents the latest advances in photochromic polymers containing diarylethenes and their derivatives. Polymer matrices doped with diarylethene derivatives and diarylethene electroactive polymers will also be included. The new and significant research in the field of photochromic diarylethenes polymers is highlighted. Their design and preparation methods, various properties and possible applications are briefly discussed.
 Qianfu Luo | Qianfu Luo received his B.S. and M.S. degrees in organic chemistry from Henan normal University in 1998 and 2001, respectively. In 2004, he received his PhD degree at East China University of Science & Technology (ECUST) supervised by Prof. He Tian. After postdoctoral research at the Institute of Advanced Materials, Fudan University, he joined the faculty of the ECUST in 2006. His current research interests are focused on the photochromic materials, organic synthesis and luminescent materials. |
 Hui Cheng | Ms. Hui Cheng is a first-year graduate student supervised by Dr. Luo at East China University of Science & Technology. |
 He Tian | Prof. He Tian received his PhD degree in 1989 from the East China University of Science & Technology (ECUST, Shanghai). In 1999, he was appointed Cheung Kong Distinguished Professor by the Education Ministry of China. His current research interests include the syntheses of novel functional organic dyes or copolymers and the development of inter-disciplinary materials science that determines the electronic and optical properties of materials. Prof. Tian has published over 280 papers in international journals and 5 academic books (in Chinese). He has also been awarded 48 Chinese patents. Now he is Co-Editor of Dyes and Pigments (Elsevier) and Advisory Editorial Board Member of Polymer Chemistry (RSC) and Chemical Science (RSC). |
1. Introduction
Organic photochromic materials based on diarylethenes and their derivatives have attracted more and more attention in recent years.1–8 In fact, they are considered as one of the most promising photochromic families because of their unique thermal stability and fatigue resistance compared with other organic photochromic materials.2,3 Generally, the photochromic process of diarylethene involves a reversible light-triggered transformation between a thermodynamically stable molecule and its isomer.2 Accompanying their reversible structural changes, various physical and chemical properties change accordingly.9–17 Namely, their internal structures and macroscopic properties can be controlled or tuned by external stimulus or triggers. Their controllable characteristics offer vast possibilities for practical applications, such as optical memory and switches, data storage and retrieval, optical computers and display systems, all-optical modulation and holography, waveguide and actuator.18–26Recently, new developments based on photochromic diarylethenes (DTE) are coming forward, photochromic organometallics, nanostructures, biomolecules, supra-molecules and chiral diarylethenes have been widely prepared and investigated.27–37 These reported photochromic diarylethene systems could be operated in solutions, polymeric matrices, and amorphous forms and even in the crystalline state.38–41 In fact, practical common applications usually require materials in solid films. Therefore, a polymeric photochromic material is preferred rather than a low-molecular-weight compound for the practical application. So more and more attention has been focused on the study of photochromic polymeric systems containing diarylethenes.42–49
Obviously, there were some reviews about low-molecular-weight photochromic diarylethenes,50–56 but much fewer reviews on photochromic polymer containing diarylethenes.57 During the past 5 years, a lot of new photochromic polymer containing diarylethenes emerged and motivated us to write this review. Here, we will briefly review some of the latest advances in the field of photochromic polymers based on diarylethenes, mainly involving diarylethene copolymers and homopolymers. Polymer matrices doped with diarylethene derivatives and diarylethene electroactive polymers will also be mentioned. Some of the latest examples of the research in the field of photochromic polymeric diarylethenes are highlighted. Their design and preparation methods, excellent properties and potential application are summarized briefly.
2. Photochromic polymers doped with diarylethene derivatives
There are two main ways that the photochromic units were introduced into a polymer matrix, as dopants dispersed in a matrix or as part of macromolecule covalently grafted onto the polymer. Dispersing the photochromic units into a polymer matrix is an easy and efficient strategy for preparing photochromic films.58–64 So far, many photochromic diarylethene derivatives were embedded in various polymer matrices to investigate their performance and functions, for instance, bisthienylethene-bridged naphthalimide dimer65 (shown in Scheme 1) dispersed in PMMA to demonstrate two-dimensional recording, bisbenzo-thienylethene-cored dendrimers66 (Scheme 2) placed in polycarbonate of bisphenol A (PC) films to increase photo-cyclization conversion ratio, and diarylethene derivatives with perylene bisimide67 (Scheme 3) dispersed in various rigid and soft polymer matrices to measure and analyze single-molecule photochromic reactions. These systems exhibit notable performances in solid state polymeric matrices. On the one hand, most organic small molecules could be conveniently doped in suitable polymeric matrices to form solid films. On the other hand, although we expect that photochromic small-molecules doped in matrices retain their individual reactivity just as in solution, they are more or less affected by the restriction of molecular mobility, inhomogenity of the matrices, existence of various relaxations and limitation of concentration, etc. In order to avoid these drawbacks photochromic polymers with photochromic diarylethenes as part of the macromolecules were developed.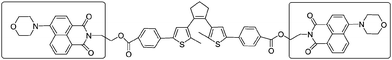 | ||
| Scheme 1 Bisthienylethene-bridged naphthalimide dimer. | ||
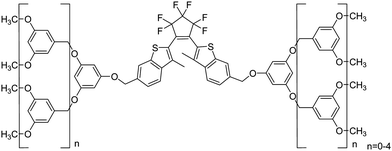 | ||
| Scheme 2 Bisbenzothienylethene-cored dendrimers. | ||
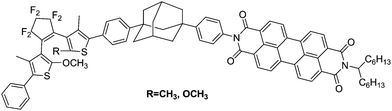 | ||
| Scheme 3 Diarylethene derivatives with perylene bisimide. | ||
3. Photochromic polymers covalently bonded with diarylethenes
Although small-molecule diarylethene derivatives dispersed in a polymeric matrix is an easy and efficient way to obtain photochromic solid film, the performance of the small-molecule will be inevitably affected by the external environment mentioned above. Moreover, polymeric matrix in these systems is treated only as a substrate or solution for the functional small-molecules. They do not have any significant additive effect on their functions in contrast to the photochromic polymers connected covalently with diarylethenes. Photochromic diarylethenes covalently bonded to polymers have many advantages, such as high concentration, homogeneous system, tailored structure and additive effects in function.57 Therefore, diarylethene-bonded photochromic polymers, including diarylethene copolymers and homopolymers have been extensively studied. The main intention of this research is combining and optimizing various properties in a polymeric system through elaborate structure design and architecture.3.1 Photochromic copolymers based on diarylethenes
Because multiple components and different functions can be strategically combined to form a homogeneous polymer, design and synthesis of photosensitive copolymer materials that combined photochromic diarylethenes with various functional molecules have attracted considerable interest.In 2005, Bobrovsky and co-workers68 synthesized a ternary comb-shaped photochromic copolymer containing diarylethene, cholesteric and phenylbenzoate side groups by the radical copolymerization of the corresponding acrylic monomers (Scheme 4). The photochromic diarylethene was chosen as an acceptor and another fluorescent dopant, coumarin 152 (2 wt.-%), was selected as a donor for the fluorescence-resonance energy transfer (FRET). In addition, in order to fine-tune the intensity of the selective light reflection, a chiral photosensitive cholesteric was introduced into the ternary copolymer too.
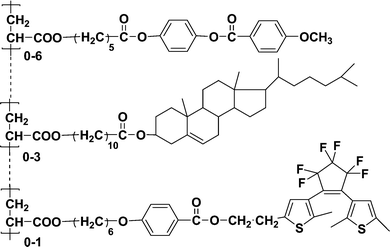 | ||
| Scheme 4 Ternary copolymer. | ||
Because the peak of the fluorescence emission (coumarin 152) partly overlapped with the absorption band of the diarylethene (closed form), the emission intensity of the fluorescence could be manipulated by the FRET process from the excited molecular fluorophore (donor) to the photochromic diarylethene (acceptor), illustrated in Scheme 5. Although the selective excitation band of the fluorophore might be slightly overlapped with the absorption band associated with the photo-cyclization of diarylethene, it makes possible to enhance the cycloreversion reactions and to photo-regulate the fluorescence intensity. This photosensitive system provides a potential application in reversible optical data recording and storage.
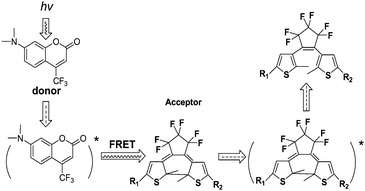 | ||
| Scheme 5 The fluorescence-resonance energy transfer (FRET) between excited molecular fluorophore to the photochromic diarylethene. | ||
It is also the combination of the fluorescent and photochromic properties, but Branda and Wolf69 chose the covalent attachment of the photochromic dithienylethene (DTE) to the fluorescent polythiophene polymer, as shown in Scheme 6. The diarylethene-functionalized polythiophene is prepared by a click reaction between the copolymer azide-polythiophene and the substituted dithienylethene derivative. Due to the energy overlap between the emission from the polythiophene and the absorbance of the ring-closed dithienylethene, the polythiophene emission was almost fully quenching by the ring-closed form of the dithienylethene. Compared with the mixture of the dithienylethene and polythiophene, the covalent attachment of the DTE component to the fluorescent polymer was more efficient for the fluorescence quenching because the latter could afford more effective quencher concentration. Based on this system, reactivity-gated chemosensors were developed.
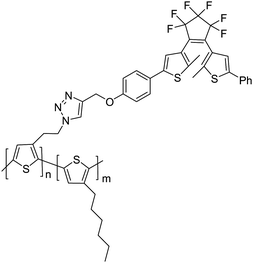 | ||
| Scheme 6 Diarylethene-functionalized polythiophenes. | ||
In addition, a series of multicolored copolymers and homopolymers containing different photochromic dithienylethenes were also designed and prepared,70 as illustrated in Scheme 7. These polymers were constructed by the mild ring opening metathesis polymerization (ROMP) process. This approach could be used to prepare a variety of high-content homopolymers under different conditions.
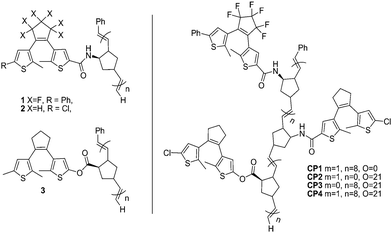 | ||
| Scheme 7 Multi-colored homo-polymers and the structures of copolymers. | ||
Irradiating them with the appropriate wavelengths of light, the homo- and co-polymers showed a full spectrum of colors in their ring-closed states. Each photochromic copolymer containing several different dithienylethene components could display four to eight possible states. Although each state could not be directly addressed, the multi-addressable, multicolored materials of this type have potential applications in multi-frequency optical data storage, and display technologies.
Multifunction is one of the most significant advantages for copolymer systems. In order to develop multifunctional copolymers possessing the capability of fluorescence, liquid crystallinity, and photoswitching, Akagi and co-workers71 synthesized novel photoresponsive liquid crystalline (LC) conjugated copolymers by adding dithienylethene moieties into the side chains of poly(phenylene-vinylene) [P1] and poly(bithienylene-phenylene) [P2] (Scheme 8). The main preparation processes of the polymers were the copolymerizations between their corresponding monomers via Still polycondensation reaction. Upon irradiation with UV and visible light, their absorption and fluorescence exhibited reversible changes. Especially, the macroscopically aligned P2 film demonstrated reversible photoswitching properties in linearly polarized fluorescence by liquid crystalline and photoresponsive diarylethene moieties in the side chain. These copolymers are proposed for the application of advanced optical memory devices with multi-read-write functions.
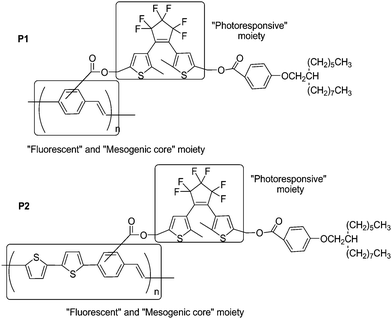 | ||
| Scheme 8 Photoresponsive liquid crystalline conjugated copolymers. | ||
Although the former macroscopically aligned film of the copolymer successfully realized photochemical control of linearly polarized fluorescence, it was difficult and time consuming in the fabrication of a smaller size device. In order to avoid this difficulty in processing, the chiral alkyl groups ((S)-2-nonanol) was introduced into the polymer on the side chains, as described in Scheme 9.72 Upon irradiation with ultraviolet and visible light, these helically π-stacked conjugated polymers showed a reversible quenching and emitting behavior in right- or left-handed circularly polarized fluorescence (CPF) through the photoisomerization of the dithienylethene moiety in the side chain. Because CPF is free from the macroscopic alignment of the polymer, polymers could conveniently exhibit CPF spectra in cast films. These properties are useful for chiroptical materials with photochemical switchable capability.
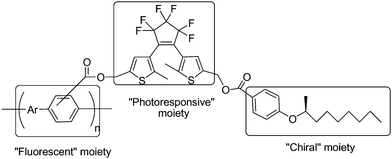 | ||
| Scheme 9 Helically π-stacked conjugated polymers appended by chiral alkyl groups. | ||
Nowadays, poor solubility and low content of photochromic units in photochromic polymers still call for solutions. Li and Tian73 designed and synthesized a high-content pendant photochromic copolymer with a dithienylethene dimer bridged by a fluorene and an alkyl fluorene unit (Scheme 10). This molecular design could obtain more freedom through functionalization of the bridge unit. The polymer was obtained by the well-known palladium-catalyzed Suzuki coupling reaction. Because each of its monomers contains two perpendicular dithienylethene units, the content of active photochromic dithienylethene component in the polymer could be improved effectively (53 wt.-%). The polymer showed good solubility in common organic solvents, such as chloroform and tetrahydrofuran, from which optically transparent and high content polymer films could be obtained easily by coasting. The polymer exhibited photochromic reaction in both solid film and solution. Moreover, the fluorescence intensity could be modulated by the photochromic reaction. Its high-content and photoresponsive characteristics will possibly be applied as molecular memory and switching devices.
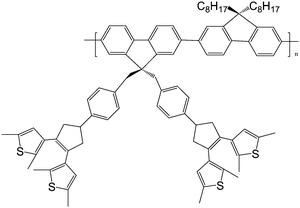 | ||
| Scheme 10 Photochromic copolymer with the dithienylethene dimer bridged by fluorenes. | ||
Besides desired fluorescence quenching by photocyclization mentioned above, “Concentration quenching effect” of fluorescence emission has been the challenge for the practical high-density medium of fluorescent photochromic diarylethene systems. In order to solve this problem, Park et al.74 developed an unconventional fluorescent photochromic polymer (DCS-BTE) by Knoevenagel condensation of phenylene diacetonitrile and dithienylethenes appended by benzoyl groups (shown in Scheme 11). This copolymer showed enhanced fluorescence emission with increasing concentration (so-called “aggregation-induced enhanced emission”). Namely, the fluorescence quantum efficiency of the polymer (open-ring form) in solid film was larger than that in solution. The molecular planarity and J-type aggregation in the 1,4-bis(β-cyano-4-methylstyryl) benzene units were gated by the specific intermolecular interactions in the solid state. Moreover, modulated alternatively by UV and visible light, the strongly fluorescent poly-(DCS-BTE) film demonstrated high contrast bistable photochromic switching. These characteristics suggest their viable application in the erasable optical information storage and retrieval. This report provides a new idea to deal with the challenge of non-destructive detection methods.
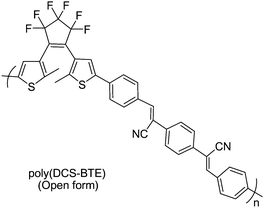 | ||
| Scheme 11 Unconventional fluorescent photochromic polymer. | ||
Due to the fact that photochromic diarylethenes show a persistent and reversible photochemical conversion, and their π-electron delocalization is extended more in the closed-isomers than in the open-isomers, photochromic diarylethenes are also expected to be used in photo-electrical switching. Kim and co-workers75,76 synthesized a series of copolymers containing photochromic diarylethenes and other large conjugated systems (Scheme 12), such as phenylquinoline and substituted p-phenylene vinylene, to investigate their photo-electric properties. These main chain copolymers were prepared by the Wittig polycondensation and the Friedlander condensation reaction from their relevant monomers. It was found that the polymer containing diarylethene and quinoline units showed a high quantum yield (due to the enforced antiparallel conformation of the dithienylethene) and highly efficient electric transport properties in the closed form because of the extension of π-electron delocalization. When modulated by UV and visible light, the copolymer incorporating diarylethene and modified p-phenylenevinylene units performed a reversible photo-electrical switching effect.
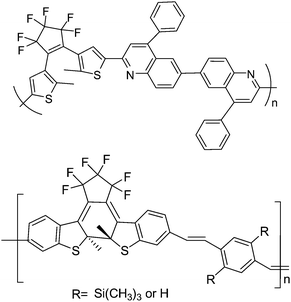 | ||
| Scheme 12 The copolymer incorporating diarylethene and modified p-phenylenevinylene units. | ||
Irie and co-workers77 also designed and synthesized an extended π-conjugated main chain copolymer by introducing large and twisted oligo-phenylene-fluorene units between diarylethene sites, as shown in Scheme 13. The polymer was prepared by the palladium-catalyzed cross-coupling reaction between the substituted diarylethene and the modified 2,7-bis-biphenylfluorene monomers. The polymer film demonstrated photochromic properties and photo-conducting behaviour. Alternating irradiation with UV and visible light, the electrical conductivity of the polymer could be switched many times. The electrical-conductivity was also investigated in solid films. The polymer showed a large conductivity modulation due to the high photochromic reactivity. The authors believed this photoresponsive polymer with photochromic diarylethenes had potential for photo-mode molecular memory and switching devices.
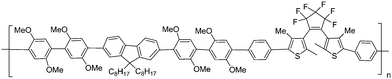
For most photochromic diarylethene derivatives, the colored isomers (closed-forms) return to the initial colorless isomers (open-forms) under visible light. However, Kobatake and Yamashita78 designed and synthesized photochromic diarylethene polymers whose colored state has a high photostability under room light (Scheme 14). Because the ring-opening quantum yield was strongly suppressed by the introduction of the cyclohexyloxy groups at the 2- and 2′-positions of the diarylethenes, the polymers showed good photostability under visible room light in solution and in the net film. Moreover, the polymers demonstrated excellent photocoloration and rapid thermal bleaching of the closed-ring forms above 150 °C as well as the performance of the diarylethene monomers. The polymer and copolymer were obtained by a radical polymerization and copolymerization of the appropriate monomers initiated with 2,2′-azobisisobutyronitrile. Although the photostability and fatigue-resistance of the closed-isomers have yet to be improve for practical application, the photochromic polymers are expected to be used for rewritable display materials and image recordings in a write-by-light/erase-by-heat system.
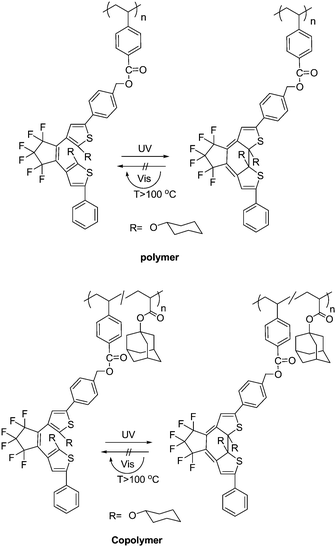 | ||
| Scheme 14 Photocoloration and thermal bleaching of the diarylethenes derivatives. | ||
Recently, gold nanoparticles (AuNPs) covered or protected with various photochromic compounds have been widely reported. In order to evaluate the electric field interaction far from the gold surface, Nishi and Kobatake79 developed a new type of photochromic diarylethene polymer-gold nanoparticle complex (Au-poly(DE) and Au-Poly(St)-block-poly(DE)), in which the photochromic polymers were introduced around the gold nanoparticle in a high density (Scheme 15). The low-polydispersity and –SH terminated diarylethene polymers and block copolymers were synthesized by reversible addition fragmentation chain transfer (RAFT) radical polymerization, and then the polymer-gold nanoparticle complexes were prepared according to Brust's method using these photochromic polymers. Upon alternate irradiation with ultraviolet and visible light, these nanoparticle complexes exhibited reversible photochromism just as the Au-free polymer diarylethenes did. Due to the local electric field interaction from the surface plasmon resonance of the gold nanoparticle to the diarylethene far from the gold nanoparticles, the absorption maximum of the diarylethene closed-ring isomer showed a bathochromic shift, especially in the solid state. In addition, the study also found that the influence of the local electric field on the photochromic diarylethene depended on the distance between the diarylethene chromophore and the gold surface. This makes the nanoparticle useful for a sensor of the medium around the particle.
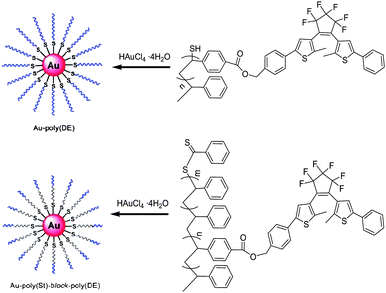 | ||
| Scheme 15 Photochromic diarylethene polymer-gold nanoparticle complexes. | ||
Furthermore, Kobatake and coworkers synthesized a series of water soluble gold nanoparticles by citrate reduction and a seeding growth method before exchanging the water-soluble covering agent with polymer diarylethnenes.80 It was found that the larger size Au-poly(DE) exhibited a reversible spectral change in the local surface plasmon resonance (LSPR) band, but the smaller size Au-poly(DE) prepared by Brust's method mentioned above hardly showed any such spectral shift. This phenomenon indicated that the sensitivity of the LSPR band strongly depended on the particle size of the gold nanoparticle, which could be potentially used for plasmonic materials with a light-controllable LSPR band.
3.2 Homopolymer containing photochromic diarylethenes
Due to the restriction of structural design and synthetic method, homopolymer based on photochromic diarylethenes have been reported much less often than copolymers. Ahn and co-workers81,82 reported two photochromic homopolymers based on diarylethenes with benzothiophene group (poly-BTF6) and the oxides of sulphur (poly-BTFO4) in high yields (Scheme 16). They were initially synthesized using Friedel–Crafts alkylation via the respective monomers. This new polymerization method was convenient and efficient in the polymerization of many functional aromatic compounds. The high density diarylethene main-chain homopolymers exhibited efficient photochromic properties in solution and in solid film. Moreover, the fatigue-resistance characteristics of the polymers to UV and visible light were significantly enhanced compared with their monomers. The fluorescence quantum yield of the poly-BTFO4 was much higher than that of the monomers in solution, even in solid film, although the fluorescence quantum yields of the closed-ring isomer of poly-BTFO4 is a little low. These properties are potentially useful for photoinduced fluorescence switching.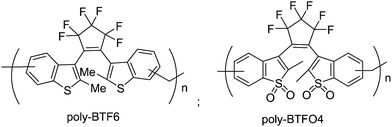 | ||
| Scheme 16 Photochromic homopolymers based on diarylethenes with benzothiophene group and the oxides of sulphur. | ||
In order to study the photochromic reaction process of polymer film, Uchida and co-workers synthesized a network polymer film by oxidation polymerization of a diarylethene derivative (Scheme 17).83 Moreover, time-resolved transient absorption spectra and picosecond transient absorption spectroscopy of the polymer were investigated during the photochromic process. It was found that the cyclization and cycloreversion reaction of the film took place in less than 30 ps, which were almost the same rapid as those of diarylethene monomers in solution. The rapid response is probably attributable to the fact that the photochromic diarylethene molecular parts are fixed in the photo-reactive anti-parallel conformation by oxidation polymerization from the closed-ring form. This rapid response photochromic polymer film may be used for rewritable optical memory and display.
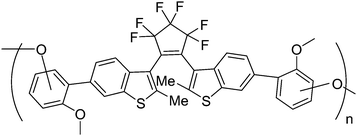 | ||
| Scheme 17 Network polymer based diarylethene derivatives. | ||
4. Diarylethene electroactive polymers
Practical applications usually require materials as solid films. Electropolymerization is a simple but efficient method for producing polymer films, especially for conducting polymer films. It has a few advantages such as the controllable film thickness and the monitored polymerization process, which will be suitable for immobilization of some photochromic diarylethenes systems. Recently, several electroactive polymers based on diarylethene have been developed. Their spectroscopic, electrochemical and photochromic properties have been investigated.For example, Kim and co-workers84 designed and synthesized a diarylethene derivative substituted with redoxactive 3,4-ethylenedioxythiophene to induce electrochemical anodic polymerization (Scheme 18). Upon electrochemical oxidation of the compound, a red-purple polymeric film was deposited on the working electrode. The electrochemical oxidation of the modified diarylethene (open state) resulted in the formation of its polymeric film (closed state) via electrocyclic ring cyclization induced by electrochemical reduction or oxidation, as depicted in Scheme 18. The film growth and thickness could be controlled conveniently by cyclic voltammetry and the electrolysis time. Although the process of the electropolymerization was not completely clear, this paper offered a convenient deposition method of a diarylethene polymer film.
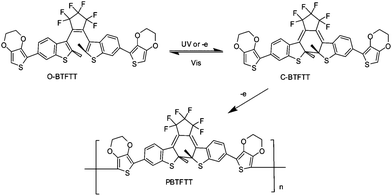 | ||
| Scheme 18 Electrocyclization and electrochemical polymerization of electroactive diarylethene derivative. | ||
Recently, electroactive polymer thin films containing dithienylcyclopentene were fabricated via electro-polymerization of electropolymerisable methoxystyryl- substituted diarylethenes (Scheme 19).85 The photochromic dithienylethene and the electropolymerizable methoxystyryl units in the polymer thin film retain their individual functions and properties because they are spaced by a phenyl. It was demonstrated that the polymer thin film could be conveniently switched by light and electrons between the open and closed form of the photochromic dithienylethene moiety. The thickness and stability of the polymer films were also investigated. It was found that their mechanical stability adhering to the electrode depended on the solvent and the electrode materials, and the thickness of the films was also limited to several monolayer equivalents, which may limit its application.
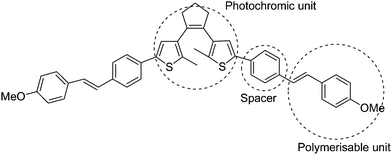 | ||
| Scheme 19 Electropolymerisable methoxystyryl- substituted diarylethenes. | ||
In addition, Feringa et al.86 also designed and synthesized an alkene bridged terthiophene system, which combined a conjugated terthiophene unit and a photochromic dithienylethene unit (Scheme 20). Interestingly, cyclic voltammetry of the open form (1Fo) produced an alkene bridged sexithiophene based polymer film (poly-1Fo) through the oxidative α,α-terthiophene coupling, but in the closed form (1Fc) the oxidation polymerization did not occur because of the formation of a stable dication, as illustrated in Scheme 20. It showed that the electropolymerizability of the terthiophenes could be controlled by photoswitching of the dithienylethene. The polymeric material may have a basic use in controlling electropolymer deposition.
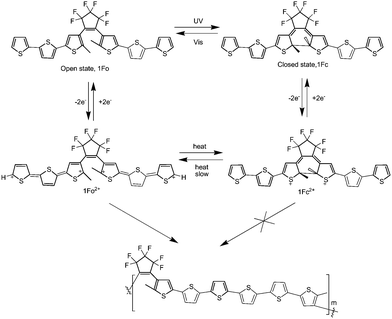 | ||
| Scheme 20 Photochemical switching between polymerizable (1Fo) and non-polymerizable (1Fc) states. | ||
5. Conclusion
Photochromic materials based on diarylethenes are expected to have more widespread applications. Considering the needs of practical applications and manufacturing processes, photochromic diarylethene polymers are more appealing. Photochromic polymers doped with diarylethene derivatives and diarylethene electroactive polymers are a simple and effective selection for forming functional films. Although they have respective strong points, the reactivity is strongly affected by the matrices and undesirable side reactions induced by electrolysis. Polymers covalently bonded with photochromic diarylethenes, by contrast, have many advantages such as high photochromic content, optical homogeneity, better stability and compatibility, especially tailorable structure and additive effect. Therefore, many multi-functionalized polymer systems coupling photochromic diarylethenes have been strategically designed and investigated. They are expected to have wide application in multi-frequency optical data storage and retrieval, rewritable optical memory and display, light-controllable plasmonic materials and chiroptical materials, various sensors and switches, etc. And a series of polymerization methods including Friedlander condensation, Knoevenagel condensation, Wittig polycondensation, Suzuki coupling, Friedel–Crafts alkylation, oxidation polymerization, ring opening metathesis polymerization and silesquinoxane and siloxane chemistry have been employed and developed.68–83 However, the multi-functional diarylethenes and incorporating multiple components into a polymer system often result in multi-step reactions and then decrease the overall yields greatly, which are disadvantageous to mass production. Moreover, how to keep the individual properties and reactivity in a solid polymeric system is also a challenge for further potential applications.Acknowledgements
Q-F. Luo thanks NSFC/China (No. 20702014) and the open project from Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education (2009YJG04) for financial support. The National Basic Research 973 Program (2011CB808400) is acknowledged for financial support.References
- B. L. Feringa, Molecular Switches, Wiley–VCH, Weinheim, 2001 Search PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS.
- H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- T. B. Norsten and N. R. Branda, J. Am. Chem. Soc., 2001, 123, 1784–1785 CrossRef CAS.
- C. T. Poon, W. H. Lam, H. L. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2010, 132, 13992–13993 CrossRef CAS.
- J. Areephong, H. Logtenberg, W. R. Browne and B. L. Feringa, Org. Lett., 2010, 12, 2132–2135 CrossRef CAS.
- S. Delbaere, G. Vermeersch, M. Frigoli and G. H. Mehl, Org. Lett., 2010, 12, 4090–4093 CrossRef CAS.
- V. W. W. Yam, J. K. W. Lee, C. C. Ko and N. Y. Zhu, J. Am. Chem. Soc., 2009, 131, 912–913 CrossRef CAS.
- G. Y. Jiang, Y. L. Song, X. F. Guo, D. Q. Zhang and D. B. Zhu, Adv. Mater., 2008, 20, 2888–2898 CrossRef CAS.
- M. Herder, M. Pätzel, L. Grubert and S. Hecht, Chem. Commun., 2011, 47, 460–462 RSC.
- S. J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS.
- H. Tian, B. Z. Chen, H. Y. Tu and K. Müllen, Adv. Mater., 2002, 14, 918–923 CrossRef CAS.
- Q. Chen, Y. Feng, D. Q. Zhang, G. X. Zhang, Q. H. Fan, S. N. Sun and D. B. Zhu, Adv. Funct. Mater., 2010, 20, 36–42 CrossRef CAS.
- C. Bertarelli, A. Bianco, F. D'Amore, M. C. Gallazzi and G. Zerbi, Adv. Funct. Mater., 2004, 14, 357–363 CrossRef CAS.
- X. L. Meng, W. H. Zhu, Q. Zhang, Y. L. Feng, W. G. Tan and H. Tian, J. Phys. Chem. B, 2008, 112, 15636–15645 CrossRef CAS.
- A. Spangenberg, R. Métivier, J. Gonzalez, K. Nakatani, P. Yu, M. Giraud, A. Léaustic, R. Guillot, T. Uwada and T. Asahi, Adv. Mater., 2009, 21, 309–313 CrossRef CAS.
- H. Tian, B. Qin, R. X. Yao, X. L. Zhao and S. J. Yang, Adv. Mater., 2003, 15, 2104–2107 CrossRef CAS.
- M. Frigoli and G. H. Mehl, Chem.–Eur. J., 2004, 10, 5243–5250 CrossRef CAS.
- R. Li, C. S. Santos, T. B. Norsten, K. Morimits and C. Bohne, Chem. Commun., 2010, 46, 1941–1943 RSC.
- Y. C. Jeong, D. G. Park, I. S. Lee, S. I. Yang and K. H. Ahn, J. Mater. Chem., 2009, 19, 97–103 RSC.
- L. N. Lucas, J. de. Jong, J. van. Esch, R. M. Kellogg and B. L. Feringa, Eur. J. Org. Chem., 2003, 155–166 CrossRef CAS.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS.
- S. Z. Pu, G. Liu, L. Shen and J. K. Xu, Org. Lett., 2007, 9, 2139–2142 CrossRef CAS.
- J. A. Gan, L. Sanguinet and J. L. Pozzo, Mol. Cryst. Liq. Cryst., 2005, 430, 37–43 CrossRef CAS.
- J. W. Kang, J. J. Kim and E. Kim, Opt. Mater., 2002, 21, 543–548 CrossRef.
- Y. Chen, D. X. Zeng, N. Xie and Y. Z. Dang, J. Org. Chem., 2005, 70, 5001–5005 CrossRef CAS.
- (a) M. N. Roberts, C. J. Carling, J. K. Nagle, N. R. Branda and M. O. Wolf, J. Am. Chem. Soc., 2009, 131, 16644–16645 CrossRef CAS; (b) J. C. Boyer, C. J. Carling, B. D. Gates and N. R. Branda, J. Am. Chem. Soc., 2010, 132, 15766–15772 CrossRef CAS.
- T. Nakagawa, Y. Hasegawa and T. Kawai, Chem. Commun., 2009, 5630–5632 RSC.
- H. Kai, S. Nara, K. Kinbara and T. Aida, J. Am. Chem. Soc., 2008, 130, 6725–6727 CrossRef CAS.
- T. Tsujioka, R. Takagi and T. Shiozawa, J. Mater. Chem., 2010, 20, 9623–9627 RSC.
- (a) K. Uchida, S. Sukata, Y. Matsuzawa, M. Akazawa, J. J. D. Jong, N. Katsonis, Y. Kojima, S. Nakamura, J. Areephong, A. Meetsma and B. L. Feringa, Chem. Commun., 2008, 326–328 RSC; (b) J. J. D. Jong, T. B. Tiemersma–Wegman, J. H. Esch and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 13804–13805 CrossRef.
- K. M. Yeo, C. J. Gao, K. H. Ahn and I. S. Lee, Chem. Commun., 2008, 4622–4624 CAS.
- Y. Tanaka, T. Ishisaka, A. Inagaki, T. Koike, C. Lapinte and M. Akita, Chem.–Eur. J., 2010, 16, 4762–4776 CrossRef CAS.
- M. Singer and A. Jäschke, J. Am. Chem. Soc., 2010, 132, 8372–8377 CrossRef CAS.
- J. Kärnbratt, M. Hammarson, S. Li, H. L. Anderson, B. Albinsson and J. Andréasson, Angew. Chem., Int. Ed., 2010, 49, 1854–1857 CrossRef.
- T. Shiozawa, M. K. Hossain, T. Ubukata and Y. Yokoyama, Chem. Commun., 2010, 46, 4785–4787 RSC.
- X. J. Piao, Y. Zou, J. C. Wu, C. Y. Li and T. Yi, Org. Lett., 2009, 11, 3818–3821 CrossRef CAS.
- A. J. Myles, B. Gorodetsky and N. R. Branda, Adv. Mater., 2004, 16, 922–925 CrossRef CAS.
- M. Irie, Bull. Chem. Soc. Jpn., 2008, 81, 917–926 CrossRef CAS.
- T. Tsujioka, R. Takagi and T. Shiozawa, J. Mater. Chem., 2010, 20, 9623–9627 RSC.
- H. Furukawa, M. Misu, K. Ando and H. Kawaguchi, Macromol. Rapid Commun., 2008, 29, 547–551 CrossRef CAS.
- (a) H. Nakashima and M. Irie, Polym. J., 1998, 30, 985–989 CrossRef CAS; (b) H. Nakashima and M. Irie, Macromol. Chem. Phys., 1999, 200, 683–692 CrossRef CAS; (c) K. Uchida, M. Saito, A. Murakami, S. Nakamura and M. Irie, ChemPhysChem, 2003, 4, 1124–1127 CrossRef CAS.
- J. J. D. Jong, P. R. Hania, A. Pugzlys, L. N. Lucas, M. Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef.
- S. Ryo, Y. Ishibashi, M. Murakami, H. Miyasaka, S. Kobatake and M. Irie, J. Phys. Org. Chem., 2007, 20, 953–959 CrossRef CAS.
- Y. Tsuboi, R. Shimizu, T. Shoji and N. Kitamura, J. Am. Chem. Soc., 2009, 131, 12623–12627 CrossRef CAS.
- C. C. Corredor, Z. L. Huang, K. D. Belfield, A. R. Morales and M. V. Bondar, Chem. Mater., 2007, 19, 5165–5173 CrossRef CAS.
- T. L. Andrew, H. Y. Tsai and R. Menon, Science, 2009, 304, 917–921 CrossRef.
- S. Wang, X. C. Li, B. Z. Chen, Q. F. Luo and H. Tian, Macromol. Chem. Phys., 2004, 205, 1497–1507 CrossRef CAS.
- F. Stellacci, C. Bertarelli, F. Toscano, M. C. Gallazzi, G. zotti and G. Zerbi, Adv. Mater., 1999, 11, 292–295 CrossRef CAS.
- (a) H. Tian and S. Wang, Chem. Commun., 2007, 781–792 RSC; (b) H. Tian and Y. L. Feng, J. Mater. Chem., 2008, 18, 1617–1622 RSC.
- M. Morimoto, S. Kobatake and M. Irie, Chem. Rec., 2004, 4, 23–38 CrossRef CAS.
- V. Guerchais, L. Ordronneau and H. L. Bozec, Coord. Chem. Rev., 2010, 254, 2533–2545 CrossRef CAS.
- S. Kobatake and M. Irie, Annu. Rep. Prog. Chem., Sect. C, 2003, 99, 277–313 RSC.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129 CrossRef CAS.
- S. Nakamura, S. Yokojima, K. Uchida, T. Tsujioka, A. Gordberg, A. Murakami, K. Shinoda, M. Mikami, T. Kobayashi, S. Kobatake, K. Matsuda and M. Irie, J. Photochem. Photobiol., A, 2008, 200, 10–18 CrossRef CAS.
- M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS.
- T. J. Wigglesworth, A. J. Myles and N. R. Branda, Eur. J. Org. Chem., 2005, 1233–1238 CrossRef CAS.
- D. Kitagawa, I. Yamashita and S. Kobatake, Chem. Commun., 2010, 46, 3723–3725 RSC.
- S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS.
- D. Medvedeva, A. Bobrovsky, N. Boiko, V. Shirinyan and M. Krayushkin, Macromol. Chem. Phys., 2006, 207, 770–778 CrossRef CAS.
- A. Bianco, G. Lardino, A. Manuelli, C. Bertarelli and G. Zerbi, ChemPhysChem, 2007, 8, 510–514 CrossRef CAS.
- S. Kobatake, H. Imagawa, H. Nakatani and S. Nakashima, New J. Chem., 2009, 33, 1362–1367 RSC.
- K. Uchida, M. Saito, A. Murakami, T. Kobayashi, S. Nakamura and M. Irie, Chem.–Eur. J., 2005, 11, 534–542 CrossRef CAS.
- C. C. Corredor, Z. L. Huang and K. D. Belfield, Adv. Mater., 2006, 18, 2910–2914 CrossRef CAS.
- G. Jiang, S. Wang, W. F. Yuan, L. Jiang, Y. Song, H. Tian and D. B. Zhu, Chem. Mater., 2006, 18, 235–237 CrossRef CAS.
- Y. Fujimoto, T. Ubukata and Y. Yokoyama, Chem. Commun., 2008, 5755–5757 RSC.
- T. Fukaminato, T. Umemoto, Y. Iwata, S. Yokojima, M. Yoneyama, S. Nakamura and M. Irie, J. Am. Chem. Soc., 2007, 129, 5932–5938 CrossRef CAS.
- D. Medvedeva, A. Bobrovsky, N. Boiko, V. Shibaev, I. Zavarzin, M. Kalik and M. Krayushkin, Macromol. Rapid Commun., 2005, 26, 177–182 CrossRef CAS.
- J. Finden, T. K. Kunz, N. R. Branda and M. O. Wolf, Adv. Mater., 2008, 20, 1988–2002 CrossRef.
- T. J. Wigglesworth and N. R. Branda, Chem. Mater., 2005, 17, 5473–5480 CrossRef CAS.
- H. Hayasaka, T. Miyashita, K. Tamura and K. Akagi, Adv. Funct. Mater., 2010, 20, 1243–1250 CrossRef CAS.
- H. Hayasaka, K. Tamura and K. Akagi, Macromolecules, 2008, 41, 2341–2346 CrossRef CAS.
- X. C. Li and H. Tian, Macromol. Chem. Phys., 2005, 206, 1769–1777 CrossRef CAS.
- S. J. Lim, B. K. An and S. Y. Park, Macromolecules, 2005, 38, 6236–6239 CrossRef CAS.
- H. Choi, H. Lee, Y. Kang, E. Kim, S. O. Kang and J. Ko, J. Org. Chem., 2005, 70, 8291–8297 CrossRef CAS.
- E. Kim and H. W. Lee, J. Mater. Chem., 2006, 16, 1384–1389 RSC.
- T. Kawai, Y. Nakashima and M. Irie, Adv. Mater., 2005, 17, 309–314 CrossRef CAS.
- S. Kobatake and I. Yamashita, Tetrahedron, 2008, 64, 7611–7618 CrossRef CAS.
- H. Nishi and S. Kobatake, Macromolecules, 2008, 41, 3995–4002 CrossRef CAS.
- H. Nishi, T. Asahi and S. Kobatake, J. Phys. Chem. C, 2009, 113, 17359–17366 CAS.
- Y. C. Jeong, D. G. Park, E. Kim, S. I. Yang and K. H. Ahn, Macromolecules, 2006, 39, 3106–3109 CrossRef CAS.
- Y. C. Jeong, S. I. Yang, E. Kim and K. H. Ahn, Macromol. Rapid Commun., 2006, 27, 1769–1773 CrossRef CAS.
- K. Uchida, A. Takata, S. Ryo, M. Saito, M. Murakami, Y. Ishibashi, H. Miyasaka and M. Irie, J. Mater. Chem., 2005, 15, 2128–2133 RSC.
- J. Lee, T. Kwon and E. Kim, Tetrahedron Lett., 2007, 48, 249–254 CrossRef CAS.
- P. Wesenhagen, J. Areephong, T. F. Landaluce, N. Heureux, N. Katsonis, J. Hielm, P. Rudolf, W. R. Browne and B. L. Feringa, Langmuir, 2008, 24, 6334–6342 CrossRef CAS.
- J. Areephong, T. Kudenrnac, J. J. D. Jong, G. T. Carroll, D. Pantorott, J. Hjelm, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 12850–12851 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |