Controlled polymerizations for the synthesis of semiconducting conjugated polymers
Ken
Okamoto
and
Christine K.
Luscombe
*
Materials Science and Engineering Department, University of Washington, Seattle, Washington 98195-212, USA. E-mail: luscombe@u.washington.edu; Fax: + 1-206-543-3100; Tel: + 1-206-616-1220
First published on 14th June 2011
Abstract
Conjugated polymers have been under active development since the 1970s as the active material in organic field-effect transistors (OFETs), photovoltaic devices and the emissive layer in light-emitting diodes (LEDs). Extensive work has been performed to investigate the physics and chemistry of these materials, and a variety of semiconducting polymers have been synthesized using a range of polymerization techniques. One of the most important key technologies is to obtain a well-controlled polymerization, which provides polymers with narrow polydispersities and defined molecular weights. In this paper, we describe the recent progress on the synthesis of semiconducting polymers as classified as polyphenylenes, polyphenylenevinylenes, polythiophenes, polyfluorene and their block copolymers by the use of controlled polymerizations.
 Ken Okamoto | Ken Okamoto was born in Tochigi, Japan, in 1980. He received his PhD degree from Tokyo Institute of Technology in 2003 under the supervision of Professor Takaki Kanbara. After his postdoctoral research with Dr Katsuhiko Ariga at National Institute for Materials Science (NIMS) in Tsukuba for 2 years, he joined the Professor Luscombe's group as a postdoctoral fellow at University of Washington in 2010. His research interests focus on conjugated polymer design & synthesis and polymeric hybrid materials. |
 Christine K. Luscombe | Christine Luscombe was born in Kobe, Japan, in 1977. She obtained her PhD degree from the University of Cambridge under the supervision of Professor Andrew Holmes and Dr Wilhelm Huck, which was followed by postdoctoral research at UC Berkeley with Prof. Jean Fréchet (2004–2006). Luscombe has been an Assistant Professor at the Materials Science and Engineering Department, University of Washington, Seattle since September 2006 and she is the recipient of the NSF Career Award, DARPA Young Faculty Award, and Sloan Fellowship. Her research area focuses on the synthesis and design of semiconducting conjugated polymers and their applications in optoelectronic devices. |
1. Introduction
Ever since it was discovered that strong and flexible films of polyacetylene could be prepared by a Ziegler–Natta polymerization,1 numerous conjugated polymers have been developed as shown in Fig. 1.2–5 Conjugated polymers have attracted much attention due to their applications in many plastic optoelectronic devices including light-emitting diodes (LEDs),6–12 organic field-effect transistors (OFETs),13,14 photovoltaic devices,15,16 nonlinear optical devices17–19 and plastic lasers.20–23 One of the biggest advantages of semiconducting polymers is their processability derived from their solubility. However, the non-substituted semiconducting polymers shown in Fig. 1 are usually insoluble in common organic solvents and are difficult to process. Therefore, great efforts have been made to improve the solubility and processability of semiconducting polymers. The usual practice to improve the solubility of semiconducting polymers is through the introduction of long alkyl side chains onto aromatic unit.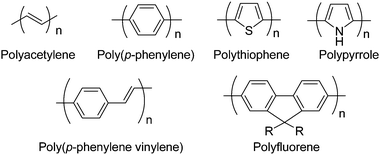 | ||
| Fig. 1 Chemical structures of a few semiconducting polymers. | ||
Many methods for the synthesis of semiconducting polymers have been developed. They have been prepared by chemical oxidation polymerization,24 electrochemical polymerization25–27 and organometallic polycondensation.28–31 Chemical and electrochemical polymerizations entail dehydrogenation: (n H–Ar–H → (Ar)n; H–Ar–H = aromatic or heterocyclic compound). Semiconducting polymers are also, in general, attainable, by using dihalo-organic compounds and several organometallics using catalytic reactions such as Suzuki–Miyaura coupling,32,33 Kumada–Tamao–Corriu coupling34–36 and Stille coupling.37 A potential drawback of the reported methods is that it is difficult to control the polydispersity, molecular weight, and the end groups of the polymers. In order to realize the goal of a controlled polymerization and to be able to create sophisticated polymer architectures, a new polymerization technique must be applied. Recent work in our group has focused on the development of controlled polymerizations to prepare semiconducting polymers. In this review, progress on the controlled synthesis of semiconducting polymers as classified as polyphenylenes, polyphenylenevinylenes, polythiophenes, polyfluorene and their block copolymers is reported.
2. Background
As we described above, one challenge in the area of semiconducting polymer synthesis is to achieve a controlled polymerization. The term “controlled polymerization” used here includes “living polymerization”. The first living polymerization was demonstrated by Michel Szwarc in 1956.38,39 He found that after the addition of a monomer to the initiator system the increase in viscosity would eventually stop but that after addition of a new amount of monomer after some time the viscosity would start to increase again. A living polymerization has been described by IUPAC as being ‘A chain polymerization from which chain transfer and chain termination are absent. In many cases, the rate of chain initiation is fast compared with the rate of chain propagation, so that the number of kinetic-chain carriers is essentially constant throughout the polymerization.’,40 and there are several features that the living polymerization allows us to achieve: (i) control over the molecular weight with narrow polydispersity (Fig. 2), (ii) allow intentional introduction of terminal groups, and (iii) introduce second monomer after the first is consumed to afford block copolymers (Fig. 3).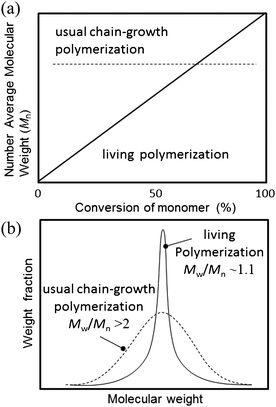 | ||
| Fig. 2 Comparison of a typical polymerization behaviour between the chain-growth polymerization and living polymerization (a) monomer/initiator ratio vs. number average molecular weight, and (b) molecular weight distribution. | ||
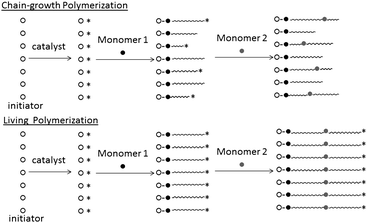 | ||
| Fig. 3 Schematic model of chain-growth polymerization and living polymerization. | ||
The two main types of semiconducting polymers are vinyl polymers and polyarylenes. The characteristic form of the vinyl polymer is that the electronically active components are incorporated into the polymer as pendant groups off an insulating polymer backbone, whereas in polyarylenes, the aromatic groups are placed within the polymer backbone (Fig. 4). Vinyl based semiconducting polymers, and a handful of polyarylenes, have been synthesized using classic living polymerization systems, such as living anionic polymerization, living ring-opening metathesis polymerization (ROMP) and atom transfer radical polymerization (ATRP). For example, polyphenylene (PP) and its derivatives,41–46 poly(9-vinylanthracene),47–51 poly(N-vinylcarbazole),52 and poly(4-diphenylaminostylene),53,54 have been prepared by living anionic polymerizations since the 1950s. Then around 1987, when the first ROMP systems were described by Grubbs and Shrock,55 ROMP was used for the preparation of polyphenylenevinylenes (PPVs).56 Meanwhile, ATRP has often been used for the synthesis of block copolymers.57 In this review, we will only focus on the synthesis of polyarylenes. With this in mind, we will first discuss the progress on the synthesis of PPs by living anion polymerization and PPVs using ROMP.
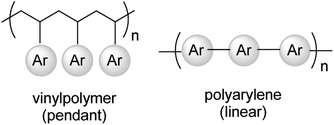 | ||
| Fig. 4 Two main types of semiconducting polymers. | ||
While vinyl polymers and a handful of polyarylenes could be synthesized in a controlled manner using existing living polymerization methods, the synthesis of the majority of polyarylenes in a controlled manner proved to be more challenging. However, there have been a growing number of studies focusing on the Ni-catalyzed dehalogenative polycondensation systems for the preparation of polythiophenes.28–31 In the usual cases of polycondensation reactions, macromolecules with a broad range of molecular weights are synthesized, because there is little difference in reactivity between monomer and polymer end groups, and so step-growth polymerization occurs. However, catalyst transfer polycondensation can convert polymerization growth mechanism from step-growth into chain-growth. The catalyst activates the polymer end group, and monomer addition keeps occurring on the same growing polymer chain. In the simplest terms, chain-growth polymerization in polyaddition systems means both non-living and living systems (see also Fig. 2 and 3), whereas chain-growth polymerization in polycondensation systems behaves as a living polymerization because chain transfer and chain termination reactions are usually absent in polycondensation reactions. For instance, Grignard metathesis (GRIM) polymerization is a synthetic method for regioregular polythiophenes recently developed by McCullough et al.58–61 and further modified by Yokozawa et al.62 One interesting feature of GRIM is its “quasi-living” chain-growth polymerization mechanism, which affords regioregular polythiophenes and the possibility of developing regioregular block copolymers with a narrow polydispersity. Following the discussion of the use of conventional living polymerization techniques for the synthesis of PPs and PPVs, we will provide an overview for the “quasi-living” polymerization systems for preparation of polythiophenes (PT), polyfluorene and polythiophene-based block copolymers.
3. π-Conjugated precursors
3.1 Polyphenylenes
Poly(p-phenylene) (PPP) has received considerable attention since the 1950s due to its high heat resistance, chemical resistance,4 electrical conductivity upon doping,63 and optoelectronic properties.64–66 Increasing the length of the conjugated p-phenylene units increases λmax of the resulting polymer to between 300 and 340 nm. However, with increasing chain length as the polymerization reaction proceeds, PPP becomes crystalline, insoluble and infusible at >6 repeat units because of enhanced interchain interactions.4,67 Therefore, there is a concomitant reduction in optoelectronic properties, due to the increasing ease in which thermal relaxation occurs rather than emissive decay. Despite the difficulty in preparing PPP, two types of PPP have been reported; one was prepared by a Ni-catalyzed dehalogenative polycondensation of p-C6H4X2 using Mg as the dehalogenating reagent according to Yamamoto et al.,2,68 and the other was obtained by an oxidative polymerization of benzene with CuCl2 according to the Kovacic method69 (Scheme 1). | ||
| Scheme 1 Synthesis of PPP by Ni-catalyzed dehalogenetive polycondensation and oxidative polymerization. | ||
As pointed out above, the syntheses of PPP are often limited by rapid formation of quasi-crystalline particles of low molecular weight segments of PPP in solution. The literature details various routes leading to PPP; however, the resulting polymers have broad molecular weight distributions, high 1,2-isomer contents, numerous structural defects, and high impurity levels.4,67 In order to obtain well-controlled and defined polymers, derivatives have been synthesized which are soluble in common organic solvents. One of the reliable routes to PPP is the dehydrogenation of poly(1,3-cyclohexadiene) (PCHD) which is easier to process compared to PPP.41,42,70,71
Before Natori reported the living anionic polymerization of 1,3-CHD in 1997,72 there had been no reports for the successful controlled polymerization of 1,3-CHD. PCHD has a structure consisting of a main chain formed by a 1,2-addition (1,2-CHD unit) and a 1,4-addition (1,4-CHD unit) (Scheme 2). The unit ratio of 1,2-CHD/1,4-CHD in the polymer chain can be controlled by the base initiator, which is typically amine or alkyllithium reagent.73–78 Natori and Sato also reported the dehydrogenation reaction of PCHD with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).79 According to their results, 1,4-phenyelene units polymer (PPP) was obtained with a controlled polymer chain length.
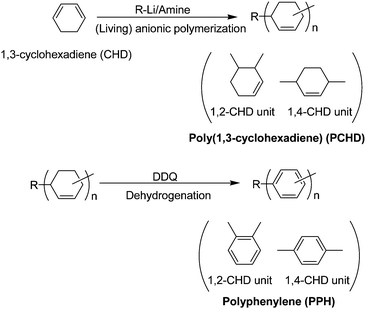 | ||
| Scheme 2 Synthesis of polyphenylene via living anionic polymerization and dehydrogenation. | ||
3.2 Polyphenylene-based block copolymers
The above PCHD route has readily permitted syntheses of binary AB and ternary ABC type copolymers of PCHD and polystyrene (PS) derivatives,80,81 where subsequent dehydrogenation gives PPP segments in the block copolymers.82–84 Because of the low solubility of PPP, the second block assists in dispersion of the PPP segments in common solvents. Furthermore, block copolymers can improve mechanical and photophysical properties, compared to individual component homopolymers by the formation of well-defined macrostructures based on stable, chemically inter-linked microstructures.85,86 For instance, PPP/PS block copolymers have been reported to form micelles in a wide range of organic solvents with PPP cores surrounded by PS shells,87 and thin films of PPP/PS/PPP self-organize into a honeycomb-like morphology86 presenting promising electroluminescence properties.88With the interesting morphology in mind, François et al. attempted the synthesis of C60 containing PPP polymers in 2002.89 The preparation of PPP/C60 copolymer was driven by the Enderlé and Mathis method,90 which described controlled grafting of up to six living anionic polymers onto C60. Two types of PPP/C60 star copolymers were synthesized as shown in Scheme 3.
 | ||
| Scheme 3 Synthesis of PPP/C60 copolymers. | ||
More recently, Natori et al. reported well-defined ternary semiconducting copolymers containing the PPP unit.91 Interestingly, the copolymers composed of three different functional parts. In the case of OLEDs, hole-transport, electron-transport, and active light emission layers are required, and so the ternary semiconducting copolymer was designed to contain poly(p-phenylene) (PP), poly(p-phenylene vinylene) (PV), and poly(styryl amine) (SA) as the hole-transport, electron-transport, and emissive layers, respectively (Scheme 4).

3.3 Polyphenylenevinylenes
A number of synthetic routes to PPV polymers have been reported. For example, (i) dehydrohalogenation of xylylene dihalides (Gilch route),92 (ii) pyrolysis of sulfonium polymer precursors (Wessling route),93 (iii) polycondensation of xylylene diphosphonates with phthalaldehydes (Wittig–Horner type condensation reaction),94 and (iv) Pd-catalyzed polycondensation of divinylbenzene with dihaloarenes (Heck type condensation reaction) (Scheme 5).95,96 The polymers obtained under these conditions had little control over the polydispersity, low molecular weight or were prepared in low yield. | ||
| Scheme 5 Synthesis of polyphenylenevinylene. | ||
Edwards and Feast first reported the synthesis of a semiconducting polymer using ROMP with their precursor route to PA in 1980.97,98 Since then, ROMP has been exploited in the synthesis of PA and PPV homopolymers and copolymers by several groups. ROMP offers the opportunity for precise control of polymer molecular weight, end groups, and narrow polydispersities. In 1992, Grubbs et al.99 demonstrated that PPV can be obtained by ROMP of substituted bicyclo[2.2.2]octadienes with excellent control of the polymer backbone structure (Scheme 6). A modification of ROMP route has also been reported (Scheme 6).100,101
 | ||
| Scheme 6 ROMP polymerization for polyphenylenevinylene. | ||
A typical strategy for increasing PPV solubility has been the incorporation of long alkyl or alkoxy side chains onto the benzene moiety. One of the most common polymers used for optoelectronic devices has been MEH-PPV (Fig. 5).102–106 Such dialkoxy-substituted PPVs have the advantage of being soluble in common organic solvents, therefore devices can be easily prepared by spin coating.
 | ||
| Fig. 5 Structure of MEH-PPV. | ||
Yu and Turner demonstrated the first controlled polymerization of substituted [2.2]paracyclophanedienes using a Grubbs catalyst to give monodisperse, soluble phenylenevinylene homopolymers.107 The ROMP of the tetra-alkoxy-substituted [2.2]paracyclophanediene derivative, dissolved in THF, was initiated by addition of a THF solution of the second-generation Grubbs catalyst (Scheme 7). This method gives polymers of controlled molecular weight, with a narrow polydispersity, very few chain defects, and an alternating cis–trans-structure. In addition, irradiation by UV light results in a photoisomerization to an all-trans-vinylene microstructure for the polymers.
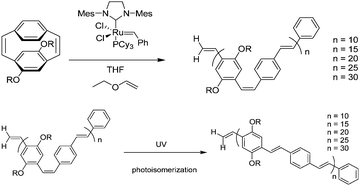 | ||
| Scheme 7 Synthesis of PPV using the second generation Grubbs catalyst to hompolymers. | ||
It would not be an exaggeration to say that the ROMP method is one of the ideal routes for synthesis of PPV polymers. Because the number-average molecular weight (Mn) increased monotonically with the monomer-to-catalyst ratio with a correlation coefficient of 0.999, and the polydispersities were all close to 1.2. Also they concluded that chain transfer, back-biting, or termination are absent and the polymerization could be regarded as “living”.
3.4 Polyphenylenevinylene-based block copolymers
A number of groups have studied the supramolecular assembly of AB or ABA type rod-coil block copolymers containing the PPV unit.108 The commonly used coil blocks, such as polystyrene derivatives or poly(ethylene) oxide, assist the self-assembling of the polymer to control the thin film morphology but do not carry any electronic function.PPV-based fully conjugated rod–rod block copolymers have been reported by the Turner group,109 where they use the second generation Grubbs catalyst for their synthesis. The polymer chain ends remain active after complete consumption of cyclophanediene monomer, and subsequently block copolymers can be obtained by additional of further cyclophanediene (Scheme 8).
 | ||
| Scheme 8 Synthesis of PPV block copolymers (R = 2-ethylhexyl). | ||
3.5 Polythiophenes
In contrast to the case of PPP, PT is considered to have close to a planar conformation which increases its charge mobility. This feature was supported by single crystal X-ray crystallography of sexithiophene.110 However, unsubstituted PTs are intractable and soluble only in solutions like mixtures of arsenic trifluoride and arsenic pentafluoride.111 Therefore, ever since Elsenbaumer et al. reported the first solution processable and environmentally stable PTs in 1987,112 a huge number of preparation methods of poly(3-alkylthiophene)s (P3AT) have been reported.58 The alkyl substituent on the thiophene ring can be incorporated into the polymer chain with two different regioregularities (rr), i.e., head-to-tail (HT) and head-to-head (HH). Sterically hindered HH linkage is considered to cause a defect in the conjugation and the electronic performance of the polymer is reduced. In the 1990s, McCullough113,114 and Rieke group115,116 independently succeeded in preparing rr HT-type poly(3-alkylthiophene-2,5-diyl)s by using a nickel-catalyzed organometallic polycondensation, and the electronic properties of rr-P3AT were found to be superior to those of their regiorandom counterparts (Scheme 9). Recently, both the Yokozawa62 and McCullough group58–60 independently developed methods for the synthesis of highly rr poly(3-hexylthiophene) (P3HT) with controlled molecular weights and narrow molecular weight distributions using the 1,3-bis(diphenylphosphino)propanenickel(II) chloride [Ni(dppp)Cl2]-catalyzed polymerization of Grignard-type monomers. | ||
| Scheme 9 Regiocontrolled synthesis of poly(3-alkylthiophene-2,5-dilyl)s. | ||
Early studies of the oxidative addition of C–X to Ni(0)Lm is well described by Yamamoto et al. The basic concepts (reductive elimination, back-donation, oxidative addition) and the basic reactions in organometallic chemistry studied by them support the organometallic polycondensation.28–31 In 2005, Yokozawa117–119 and McCullough60 independently proposed a mechanistic pathway for this GRIM polymerization in which the key step is formation of an associated zero-valent Ni–arene complex after reductive elimination (Scheme 10). The Ni complex does not diffuse to the reaction mixture, and can be inserted into the C–Br bond intramolecularly.
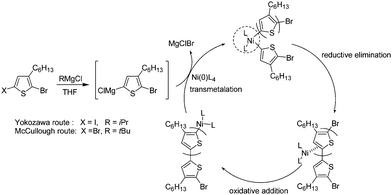 | ||
| Scheme 10 Polymerization mechanism according to McCullough and Yokozawa routes. | ||
To develop a general synthetic method, further detailed mechanistic investigations for understanding of monomers, ancillary phosphorus ligand and additives on the chain-growth pathway have been examined. The approaches for the mechanisms are classified in three groups: (i) rate and spectroscopic kinetic studies for rate-determining factor,120–122 (ii) MALDI-TOF MS, GPC and NMR studies for determination of polymerization degree and polymer end groups from obtained products,123–134 and (iii) small-molecule cross-coupling reaction via intramolecular catalyst transfer.135,136 As examples of (i), the McNeil group reported that rate studies utilizing IR spectroscopy and gas chromatography revealed that the polymerizations exhibit a first-order dependence on the catalyst (Ni(dppe)Cl2) concentration but a zeroth-order dependence on the monomer concentration. The rate and spectroscopic studies revealed that reductive elimination as the rate-determining step for 2,5-dibromo-3-hexylthiophene monomer.121 However, an alternative catalyst, Ni(dppp)Cl2, led to a different rate behaviour. Similar behaviour of the dppe ligand has previously been reported by the McCullough group in 2005,60 but the McNeil group revealed that the rate-determining steps change from reductive elimination to transmetalation when the ligand is changed from dppe to dppp.122
The following are a few recent examples where the 2nd approach was used. The Kiriy group revealed new information in polymerization mechanism using dibromobenzene as the initiator.123 Controlled experiments showed that no intermolecular catalyst-transfer occurs. They expressed the catalyst behavior as ‘random catalyst walking’ and suggested that a combination of a random catalyst walking along the chain and a sticking effect at the end groups is operating in the Kumada catalyst-transfer polycondensation (KCTP).
The Kiriy group, the Koeckelberghs group and our group have reported the synthesis of externally initiated rr-P3HT with controlled molecular weight and narrow polydispersity, respectively (Scheme 11).124–130 The external initiation is necessary for the synthesis of more hierarchical polymer architectures such as brushes and star and block copolymer. According to reaction in Scheme 11, we developed the initiated polymerization of 3-hexylthiophene monomers from an externally added cis-chloro(aryl)(dppp)nickel complex. The molecular weight of the obtained polymer increased linearly in proportion to the conversion of monomer with low polydispersity throughout the polymerization, up to a Mn of 11.2 kDa and a PDI of 1.1 versus polystyrene standards.
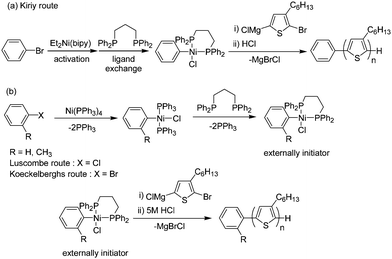 | ||
| Scheme 11 Ni-catalyzed externally initiated polymerization of (5-bromo-4-hexyl-2-thienyl)chloro magnesium: (a) 2,2′-bipyridyl (bipy)-supported initiator route and (b) tetrakis(triphenylphosphine)nickel(0) routes. | ||
We also investigated the versatility of aryl halide initiators functionalized with a phosphonate moiety.131,132 It was observed that an ortho substituent plays a role in the initiator formation and phenyl-based initiators are more stable than thiophene-based initiators (Fig. 6). The Kiriy group have reported on the steric effects of the ortho substituent on the monomer in detail133,134 (Scheme 12).
 | ||
| Scheme 12 Alkyl substitution pattern and polycondensation behaviour. | ||
 | ||
| Fig. 6 Molecular structures of initiators and their stability from the result of polymerization. | ||
Thelakkat and Lohwasser have reported key parameters influencing the kinetics of the catalyst transfer polymerization.135 In order to achieve perfect control of end groups and low polydispersity in P3HT, they examined the polymer chain ends produced when an excess of different Grignard species, quenching reagents and LiCl additives were used. The optimized procedure afforded P3HTs with 100% H/Br end groups and low PDIs.
As the 3rd approach example, van der Boom and co-workers demonstrated the intramolecular zero-valent Ni complex transfer system of Ni(PEt3)4 with brominated vinylarene.136 The resulted complex reveals that after selective η2-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C coordination reaction, subsequent intramolecular “ring-walking” oxidative addition of Ni complex was observed, even in the presence of aryl-I containing substrates (Scheme 13). On the other hand, Nakamura and co-workers reported the kinetic isotope effects and theoretical calculation study of Ni-catalyzed cross-coupling reaction.137 They indicated that because of a strong electron donor ability of Ni(0), the first irreversible step of Ni catalysis is the complexation of the Ni atom on the π-face of the haloarene. These small-molecular results are consistent with the ‘living’ behaviour of the GRIM method.
C coordination reaction, subsequent intramolecular “ring-walking” oxidative addition of Ni complex was observed, even in the presence of aryl-I containing substrates (Scheme 13). On the other hand, Nakamura and co-workers reported the kinetic isotope effects and theoretical calculation study of Ni-catalyzed cross-coupling reaction.137 They indicated that because of a strong electron donor ability of Ni(0), the first irreversible step of Ni catalysis is the complexation of the Ni atom on the π-face of the haloarene. These small-molecular results are consistent with the ‘living’ behaviour of the GRIM method.
 | ||
| Scheme 13 Selective η2-(CC) coordination of Ni(PEt3)2 to brominated vinylarene compound is followed by activation of the Ar–Br bond. | ||
The GRIM and modified GRIM methods have also been used by many groups to prepare polythiophene-based block-copolymers.60,138–150 Furthermore, although we did not describe this in the polyphenylene section, the chain-growth method enables the synthesis of substituted polyphenylenes with controlled molecular weight and narrow polydispersity (Scheme 14).121,151,152 Still each aryl monomer has required empirical development of unique reaction conditions to achieve chain-growth, e.g. ligand and additives, and the method was applied to other heterocyclic monomers including N-hexylpyrrole,153 2,3-N-octylcarbazole,154 and dihexylthieno[3,4-b]pyrazine155 (Scheme 15).
 | ||
| Scheme 14 Synthesis of PPP by the GRIM method. | ||
 | ||
| Scheme 15 Synthesis of heterocyclic homopolymers by the GRIM method. | ||
Another attractive feature of the catalyst-transfer polymerization is the development of the polymerization from organic or inorganic surfaces. Kiriy and coworkers applied this approach to grow semiconducting polymer brushes of P3HT via surface initiated chain-growth condensation polymerization (Scheme 16).124,156
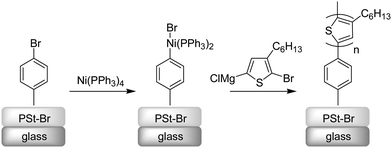 | ||
| Scheme 16 Surface-initiated grafting of P3HT. | ||
Further modifications of inorganic surface (SiO2) were reported. Gopalan and co-workers tried to modify SiO2 surface by click reaction. First, regioregular ethynyl-terminated P3HT with a molecular weight of 5900 and a polydispersity of 1.2 was synthesized by catalyst-transfer polycondensation using GRIM mediated by a Ni catalyst. The azide SAM was prepared from bifunctional molecules containing azide and siloxane as click reaction precursor and surface linker, respectively (Scheme 17).157
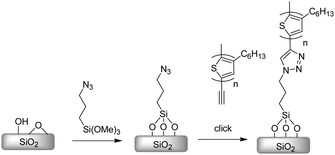 | ||
| Scheme 17 Anchoring of P3HT by click chemistry to a SAM of azide. | ||
Locklin and co-workers have reported that the surface-bound external initiator could be formed by reacting a Ni(0) complex with a thienyl bromide monolayer, and this surface-bound initiator was used to polymerize several Grignard monomers prepared from the corresponding 1,4-dihalobenzenes resulting in surface-bound with thickness up to 30 nm (Scheme 18).158,159 We have also recently investigated the “grafting from” method for the surface initiated KCTP (Scheme 19).160 As we described above, our externally initiated method was utilized to produce surface anchored poly(3-methyl)thiophene. The thickness of polymer film could be controlled by varying the monomer concentration.
 | ||
| Scheme 19 Surface-initiated grafting of P3MT from ITO. | ||
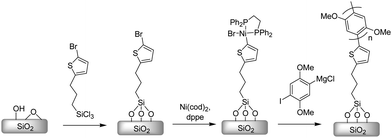 | ||
| Scheme 18 Surface-initiated grafting of PPP from SiO2. | ||
3.6 Polyfluorenes
In contrast to the GRIM method using the Ni-catalyzed polycondensation system, Yokozawa et al. reported polymerization for the synthesis of polyfluorenes via Suzuki–Miyaura coupling reaction.32,161 In this study, they focused on using arylpalladium(II) halide complexes containing a bulky phosphine ligand as the initiator complex. The polymerization reaction was carried out in the presence of tBu3Pd(Ph)Br as an externally added initiator. The phenyl ligand on the Pd(II) complex serves as an initiator unit. The polymerization of the 2-(7-bromo-9,9-dioctyl-9H-fluorene-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane proceeded via chain-growth polymerization from the initiator (Scheme 20). The molecular weight of obtained polymers was controlled with a relatively narrow polydispersity. | ||
| Scheme 20 Synthesis of polyfluorene by Suzuki–Miyaura coupling. | ||
Based on this report, Kiriy and co-workers have performed surface-initiated Suzuki polycondensation.162 Using their method (cf.Scheme 14), they succeeded to prepare poly[9,9-bis(2-ethylhexyl)fluorine] on surface-immobilized PS(Br) films, as well as on monolayers of small-molecule initiators and on patterned initiators (Scheme 21). The polymerization is fast even at room temperature and results in grafted polyfluorene films with thicknesses up to100 nm.
![Surface-initiated grafting of poly[9,9-bis(2-ethylhexyl)fluorine].](/image/article/2011/PY/c1py00171j/c1py00171j-s21.gif) | ||
| Scheme 21 Surface-initiated grafting of poly[9,9-bis(2-ethylhexyl)fluorine]. | ||
Conclusions
Numerous semiconducting polymers were prepared by living anion polymerization, ROMP and more recently, chain-growth condensation polymerization. Recent work reported using metal-catalyzed cross-coupling reactions is particularly exciting, opening new avenues for synthetic strategies which were not accessible before. The technique will contribute to the development of more elaborate semiconducting polymer architectures, including block copolymers, star polymers, and graft copolymers, which will enable us to obtain further understanding for structure–property relationships in these classes of materials.Acknowledgements
The author K.O. is grateful to Professor T. Yamamoto of Tokyo Institute of Technology and Professor T. Kanbara of University of Tsukuba for their guidance to chemistry, discussion and collaboration. Preparation of this manuscript was supported as part of the Center for Interface Science: Solar Electric Materials, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0001084 (partial support for K.O.), AFOSR SynChem program (partial support for K.O.), and NSF ((STC-MDITR DMR 0120967), MRSEC-GEMSEC DMR 0520567, CAREER Award DMR 0747489 (C.K.L.)).Notes and references
- T. Ito, H. Shirakawa and S. Ikeda, J. Polym. Sci., Part A: Polym. Chem., 1975, 13, 1943–1950 Search PubMed.
- T. Yamamoto, Y. Hayashi and A. Yamamoto, Bull. Chem. Soc. Jpn., 1978, 51, 2091–2097 CAS.
- P. Kovacic and C. Wu, J. Polym. Sci., 1960, 47, 45–54 Search PubMed.
- P. Kovacic and M. B. Jones, Chem. Rev., 1987, 87, 357–379 CrossRef CAS.
- D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, Macromolecules, 1988, 21, 294–304 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- G. Gustafsson, Y. Cao, G. M. Treacy, F. Klavetter, N. Colaneri and A. J. Heeger, Nature, 1992, 357, 477–479 CrossRef CAS.
- N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628–630 CrossRef.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121–128 CrossRef CAS.
- S. Welter, K. Brunner, J. W. Hofstraat and L. De Cola, Nature, 2003, 421, 54–57 CrossRef CAS.
- A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402–428 CrossRef.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS.
- R. C. G. Naber, C. Tanase, P. W. M. Blom, G. H. Gelinck, A. W. Marsman, F. J. Touwslager, S. Setayesh and D. M. De Leeuw, Nat. Mater., 2005, 4, 243–248 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS.
- Y. P. Cui, Y. Zhang, P. N. Prasad, J. S. Schildkraut and D. J. Williams, Appl. Phys. Lett., 1992, 61, 2132–2134 CrossRef CAS.
- K. G. Chittibabu, L. Li, M. Kamath, J. Kumar and S. K. Tripathy, Chem. Mater., 1994, 6, 475–480 Search PubMed.
- T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed. Engl., 1995, 34, 155–173 CrossRef CAS.
- F. Hide, M. A. Díaz-García, B. J. Schwartz, M. R. Andersson, Q. B. Pei and A. J. Heeger, Science, 1996, 273, 1833–1836 CAS.
- F. Hide, M. A. Díaz-García, B. J. Schwartz and A. J. Heeger, Acc. Chem. Res., 1997, 30, 430–436 CrossRef CAS.
- N. Tessler, G. J. Denton and R. H. Friend, Nature, 1996, 382, 695–697 CrossRef CAS.
- C. Kallinger, M. Hilmer, A. Haugeneder, M. Perner, W. Spirkl, U. Lemmer, J. Feldmann, U. Scherf, K. Müllen, A. Gombert and V. Wittwer, Adv. Mater., 1998, 10, 920–923 CrossRef CAS.
- M. Leclerc, F. M. Diaz and G. Wegner, Makromol. Chem., 1989, 190, 3105–3116 CAS.
- A. F. Diaz, J. I. Castillo, J. A. Logan and W. Y. Lee, J. Electroanal. Chem., 1981, 129, 115–132 CrossRef CAS.
- M. Sato, S. Tanaka and K. Kaeriyama, Makromol. Chem., 1987, 188, 1763–1771 CAS.
- L. M. Goldenberg and P. C. Lacaze, Synth. Met., 1993, 58, 271–293 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 1999, 72, 621–638 CrossRef CAS.
- T. Yamamoto, Macromol. Rapid Commun., 2002, 23, 583–606 CrossRef CAS.
- T. Yamamoto, Synlett, 2003, 425–450 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431–455 CrossRef CAS.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- K. Tamao, K. Sumitani, Y. Kiso, M. Zembayashi, A. Fujioka, S. Kodama, I. Nakajima, A. Minato and M. Kumada, Bull. Chem. Soc. Jpn., 1976, 49, 1958–1969 CAS.
- J. P. Corriu and J. P. Masse, J. Chem. Soc., Chem. Commun., 1972, 144 Search PubMed.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–523 CrossRef.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- A. D. Jenkins, P. Kratochvil, R. F. T. Stepto and U. W. Suter, Pure Appl. Chem., 1996, 68, 2287–2311 CrossRef CAS.
- C. S. Marvel and G. E. Hartzell, J. Am. Chem. Soc., 1959, 81, 448–452 Search PubMed.
- D. A. Frey, M. Hasegawa and C. S. Marvel, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 2057–2065 Search PubMed.
- F. Dawans and C. S. Marvel, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 3549–3571 Search PubMed.
- X. F. Zhong and B. François, Makromol. Chem. Rapid Commun., 1988, 9, 411–416 Search PubMed.
- B. François, S. Izzillo and P. Iratçabal, Synth. Met., 1999, 102, 1211–1212 Search PubMed.
- D. T. Williamson, T. E. Glass and T. E. Long, Abstr. Pap. Am. Chem. Soc., 2002, 224, 021 Search PubMed.
- A. Eisenberg and A. Rembaum, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 157–162 Search PubMed.
- R. H. Michel and W. P. Baker, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 163–168 Search PubMed.
- M. Stolka, J. F. Yanus and J. M. Pearson, Macromolecules, 1976, 9, 710–714 Search PubMed.
- M. Stolka, J. F. Yanus and J. M. Pearson, Macromolecules, 1976, 9, 715–719 CrossRef.
- I. Natori and S. Natori, Polym. Adv. Technol., 2010, 21, 784–788 Search PubMed.
- I. Natori, Macromolecules, 2006, 39, 6017–6024 Search PubMed.
- W. J. Feast, R. J. Peace, I. C. Sage and E. L. Wood, Polym. Bull., 1999, 42, 167–174 CrossRef.
- G. N. Tew, M. U. Pralle and S. I. Stupp, Angew. Chem., Int. Ed., 2000, 39, 517–521 CrossRef CAS.
- R. R. Schrock, J. Feldman, L. F. Cannizzo and R. H. Grubbs, Macromolecules, 1987, 20, 1169–1172 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. S. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- E. E. Sheina, J. S. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- D. M. Ivory, G. G. Miller, J. M. Sowa, L. W. Shacklette, R. R. Chance and R. H. Baughman, J. Chem. Phys., 1979, 71, 1506–1507 CrossRef CAS.
- G. Grem, G. Leditzky, B. Ullrich and G. Leising, Synth. Met., 1992, 51, 383–389 CrossRef CAS.
- G. Grem, G. Leditzky, B. Ullrich and G. Leising, Adv. Mater., 1992, 4, 36–37 CrossRef CAS.
- G. Grem and G. Leising, Synth. Met., 1993, 57, 4105–4110 CrossRef CAS.
- W. Ried and D. Freitag, Angew. Chem., Int. Ed. Engl., 1968, 7, 835–844 CrossRef.
- T. Yamamoto and A. Yamamoto, Chem. Lett., 1977, 353–356 CAS.
- P. Kovacic and A. Kyriakis, J. Am. Chem. Soc., 1963, 85, 454–458 CrossRef CAS.
- Z. Sharaby, M. Martan and J. Jagur-Grodzinski, Macromolecules, 1982, 15, 1167–1173 Search PubMed.
- G. Lefebvre and F. Dawans, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 3277–3295 Search PubMed.
- I. Natori, Macromolecules, 1997, 30, 3696–3697 CrossRef CAS.
- I. Natori, K. Imaizumi, H. Yamagishi and M. Kazunori, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 1657–1668 CrossRef CAS.
- I. Natori and S. Inoue, Macromolecules, 1998, 31, 4687–4694 CrossRef CAS.
- K. L. Hong and J. W. Mays, Macromolecules, 2001, 34, 782–786 CrossRef CAS.
- K. L. Hong and J. W. Mays, Macromolecules, 2001, 34, 3540–3547 CrossRef CAS.
- K. L. Hong, Y. N. Wan and J. W. Mays, Macromolecules, 2001, 34, 2482–2487 Search PubMed.
- D. T. Williamson, J. F. Elman, P. H. Madison, A. J. Pasquale and T. E. Long, Macromolecules, 2001, 34, 2108–2114 Search PubMed.
- I. Natori and H. Sato, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 837–845 Search PubMed.
- S. Jacobs, W. Eevers, G. Verreyt, H. J. Geise, A. De Groot and R. Dommisse, Synth. Met., 1993, 61, 189–193 CrossRef CAS.
- H. Becker, H. Spreitzer, K. Ibrom and W. Kreuder, Macromolecules, 1999, 32, 4925–4932 CrossRef CAS.
- B. François and X. F. Zhong, Makromol. Chem., 1990, 191, 2743–2753 CrossRef.
- X. F. Zhong and B. François, Makromol. Chem., 1990, 191, 2735–2741 CrossRef CAS.
- B. François and X. F. Zhong, Synth. Met., 1991, 41, 955–958 CrossRef CAS.
- B. François, G. Widawski, M. Rawiso and B. Cesar, Synth. Met., 1995, 69, 463–466 CrossRef CAS.
- G. Widawski, M. Rawiso and B. François, Nature, 1994, 369, 387–389 CrossRef CAS.
- B. François, G. Widawski and M. Rawiso, Synth. Met., 1995, 69, 491–492 Search PubMed.
- D. B. Romero, M. Schaer, J. L. Staehli, L. Zuppiroli, G. Widawski, M. Rawiso and B. Francois, Solid State Commun., 1995, 95, 185–189 Search PubMed.
- E. Mignard, R. C. Hiorns and B. Francois, Macromolecules, 2002, 35, 6132–6141 CrossRef CAS.
- Y. Ederlé and C. Mathis, Macromolecules, 1997, 30, 2546–2555 CrossRef CAS.
- I. Natori, S. Natori, K. Tsuchiya and K. Ogino, Macromolecules, 2011, 44, 256–262 Search PubMed.
- H. G. Gilch, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 1351–1357 Search PubMed.
- R. A. Wessling, J. Polym. Sci., Polym. Symp., 1985, 55–66 CAS.
- H. Rost, A. Teuschel, S. Pfeiffer and H. H. Hörhold, Synth. Met., 1997, 84, 269–270 CrossRef CAS.
- Z. N. Bao, Y. M. Chen, R. B. Cai and L. P. Yu, Macromolecules, 1993, 26, 5281–5286 CrossRef CAS.
- Z. N. Bao, Y. M. Chen and L. P. Yu, Macromolecules, 1994, 27, 4629–4631 CrossRef CAS.
- J. H. Edwards and W. J. Feast, Polymer, 1980, 21, 595–596 CrossRef CAS.
- J. H. Edwards, W. J. Feast and D. C. Bott, Polymer, 1984, 25, 395–398 CrossRef CAS.
- V. P. Conticello, D. L. Gin and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 9708–9710 CrossRef CAS.
- Y. J. Miao and G. C. Bazan, J. Am. Chem. Soc., 1994, 116, 9379–9380 CrossRef CAS.
- Y. J. Miao and G. C. Bazan, Macromolecules, 1994, 27, 1063–1064 Search PubMed.
- D. J. Braun, E. G. J. Staring, R. C. J. E. Demandt, G. L. J. Rikken, Y. A. R. R. Kessener and A. H. J. Venhuizen, Synth. Met., 1994, 66, 75–79 CAS.
- P. L. Burn, A. B. Holmes, A. Kraft, D. D. C. Bradley, A. R. Brown, R. H. Friend and R. W. Gymer, Nature, 1992, 356, 47–49 CrossRef CAS.
- P. L. Burn, A. Kraft, D. R. Baigent, D. D. C. Bradley, A. R. Brown, R. H. Friend, R. W. Gymer, A. B. Holmes and R. W. Jackson, J. Am. Chem. Soc., 1993, 115, 10117–10124 CrossRef CAS.
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, Science, 1996, 273, 884–888 CrossRef CAS.
- H. F. Wittmann, J. Gruner, R. H. Friend, G. W. C. Spencer, S. C. Moratti and A. B. Holmes, Adv. Mater., 1995, 7, 541–544 Search PubMed.
- C. Y. Yu and M. L. Turner, Angew. Chem., Int. Ed., 2006, 45, 7797–7800 CrossRef.
- S. T. Pasco, P. M. Lahti and F. E. Karasz, Macromolecules, 1999, 32, 6933–6937 CrossRef CAS.
- C. Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2010, 43, 222–232 CrossRef CAS.
- G. Horowitz, B. Bachet, A. Yassar, P. Lang, F. Demanze, J. L. Fave and F. Garnier, Chem. Mater., 1995, 7, 1337–1341 CrossRef CAS.
- J. E. Frommer, Acc. Chem. Res., 1986, 19, 2–9 CrossRef CAS.
- R. L. Elsenbaumer, K. Y. Jen and R. Oboodi, Synth. Met., 1986, 15, 169–174 CrossRef CAS.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS.
- R. D. McCullough, S. Tristram-Nagle, S. P. Williams, R. D. Lowe and M. Jayaraman, J. Am. Chem. Soc., 1993, 115, 4910–4911 CrossRef CAS.
- T. A. Chen and R. D. Rieke, J. Am. Chem. Soc., 1992, 114, 10087–10088 CrossRef CAS.
- T. A. Chen, X. M. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS.
- I. Adachi, R. Miyakoshi, A. Yokoyama and T. Yokozawa, Macromolecules, 2006, 39, 7793–7795 CrossRef CAS.
- A. Yokoyama and T. Yokozawa, Macromolecules, 2007, 40, 4093–4101 CrossRef CAS.
- J.-P. Lamps and J.-M. Catala, Macromolecules, 2009, 42, 7282–7284 Search PubMed.
- E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573–16579 CrossRef CAS.
- E. L. Lanni and A. J. McNeil, Macromolecules, 2010, 43, 8039–8044 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J. U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS.
- V. Senkovskyy, N. Khanduyeva, H. Komber, U. Oertel, M. Stamm, D. Kuckling and A. Kiriy, J. Am. Chem. Soc., 2007, 129, 6626–6632 CrossRef CAS.
- T. Beryozkina, V. Senkovskyy, E. Kaul and A. Kiriy, Macromolecules, 2008, 41, 7817–7823 CrossRef CAS.
- R. Senkovskyy, T. Tkachov, H. Beryozkina, U. Komber, M. Oertel, V. Horecha, M. Bocharova, S. A. Stamm, F. C. Gevorgyan, Krebs and A. Kiriy, J. Am. Chem. Soc., 2009, 131, 16445–16453 CrossRef CAS.
- A. Smeets, K. Van den Bergh, J. De Winter, P. Gerbaux, T. Verbiest and G. Koeckelberghs, Macromolecules, 2009, 42, 7638–7641 CrossRef CAS.
- N. Doubina, A. Ho, A. K. Y. Jen and C. K. Luscombe, Macromolecules, 2009, 42, 7670–7677 CrossRef CAS.
- N. Doubina, M. Stoddard, H. A. Bronstein, A. K. Y. Jen and C. K. Luscombe, Macromol. Chem. Phys., 2009, 210, 1966–1972 CrossRef CAS.
- H. A. Bronstein and C. K. Luscombe, J. Am. Chem. Soc., 2009, 131, 12894–12895 CrossRef CAS.
- S. D. Boyd, A. K. Y. Jen and C. K. Luscombe, Macromolecules, 2009, 42, 9387–9389 CrossRef CAS.
- N. Doubina, S. A. Paniagua, A. V. Soldatova, A. K. Y. Jen, S. R. Marder and C. K. Luscombe, Macromolecules, 2011, 44, 512–520 CrossRef CAS.
- V. Senkovskyy, M. Sommer, R. Tkachov, H. Komber, W. T. S. Huck and A. Kiriy, Macromolecules, 2010, 43, 10157–10161 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber and A. Kiriy, Macromolecules, 2011, 44, 2006–2015 Search PubMed.
- R. H. Lohwasser and M. Thelakkat, Macromolecules, 2011, 44, 3388–3397 Search PubMed.
- O. V. Zenkina, A. Karton, D. Freeman, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, Inorg. Chem., 2008, 47, 5114–5121 CrossRef CAS.
- N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15258–15259 CrossRef CAS.
- T. Yokozawa, I. Adachi, R. Miyakoshi and A. Yokoyama, High Perform. Polym., 2007, 19, 684–699 Search PubMed.
- Y. Zhang, K. Tajima, K. Hirota and K. Hashimoto, J. Am. Chem. Soc., 2008, 130, 7812–7813 CrossRef CAS.
- Q. L. Zhang, A. Cirpan, T. P. Russell and T. Emrick, Macromolecules, 2009, 42, 1079–1082 CrossRef CAS.
- Y. Zhang, K. Tajima and K. Hashimoto, Macromolecules, 2009, 42, 7008–7015 CrossRef CAS.
- S. Miyanishi, Y. Zhang, K. Tajima and K. Hashimoto, Chem. Commun., 2010, 46, 6723–6725 RSC.
- C. C. Chueh, T. Higashihara, J. H. Tsai, M. Ueda and W. C. Chen, Org. Electron., 2009, 10, 1541–1548 Search PubMed.
- T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 8794–8800 CrossRef CAS.
- T. Higashihara and M. Ueda, React. Funct. Polym., 2009, 69, 457–462 CrossRef CAS.
- A. Gutacker, S. Adamczyk, A. Helfer, L. E. Garner, R. C. Evans, S. M. Fonseca, M. Knaapila, G. C. Bazan, H. D. Burrows and U. Scherf, J. Mater. Chem., 2010, 20, 1423–1430 RSC.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS.
- K. Van den Bergh, I. Cosemans, T. Verbiest and G. Koeckelberghs, Macromolecules, 2010, 43, 3794–3800 Search PubMed.
- K. Ohshimizu and M. Ueda, Macromolecules, 2008, 41, 5289–5294 CrossRef CAS.
- T. Higashihara, K. Ohshimizu, A. Hirao and M. Ueda, Macromolecules, 2008, 41, 9505–9507 Search PubMed.
- R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012–16013 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Chem. Lett., 2008, 37, 1022–1023 Search PubMed.
- A. Yokoyama, A. Kato, R. Miyakoshi and T. Yokozawa, Macromolecules, 2008, 41, 7271–7273 CrossRef CAS.
- M. C. Stefan, A. E. Javier, I. Osaka and R. D. McCullough, Macromolecules, 2009, 42, 30–32 CrossRef CAS.
- L. Wen, B. C. Duck, P. C. Dastoor and S. C. Rasmussen, Macromolecules, 2008, 41, 4576–4578 CrossRef CAS.
- N. Khanduyeva, V. Senkovskyy, T. Beryozkina, V. Bocharova, F. Simon, M. Nitschke, M. Stamm, R. Grötzschel and A. Kiriy, Macromolecules, 2008, 41, 7383–7389 Search PubMed.
- P. Paoprasert, J. W. Spalenka, D. L. Peterson, R. E. Ruther, R. J. Hamers, P. G. Evans and P. Gopalan, J. Mater. Chem., 2010, 20, 2651–2658 RSC.
- N. Marshall, S. K. Sontag and J. Locklin, Macromolecules, 2010, 43, 2137–2144 CrossRef CAS.
- S. K. Sontag, N. Marshall and J. Locklin, Chem. Commun., 2009, 3354–3356 RSC.
- N. Doubina, J. L. Jenkins, S. A. Paniagua, K. A. Mazzio, N. A. Armstrong, S. R. Marder, A. K.-Y. Jen and C. K. Luscombe, presented in part at the MRS Spring Meeting, San Francisco, April, 2011..
- A. Yokoyama, H. Suzuki, Y. Kubota, K. Ohuchi, H. Higashimura and T. Yokozawa, J. Am. Chem. Soc., 2007, 129, 7236–7237 CrossRef CAS.
- T. Beryozkina, K. Boyko, N. Khanduyeva, V. Senkovskyy, M. Horecha, U. Oertel, F. Simon, M. Stamm and A. Kiriy, Angew. Chem., Int. Ed., 2009, 48, 2695–2698 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |