Mass transport by collisions in emulsion polymerization: why it is possible to use very hydrophobic catalysts for efficient molecular weight control
Niels M. B.
Smeets†
,
Tom G. T.
Jansen
,
Alex M.
van Herk
,
Jan
Meuldijk
and
Johan P. A.
Heuts
*
Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, Eindhoven, The Netherlands. E-mail: j.p.a.heuts@tue.nl
First published on 26th May 2011
Abstract
An alternative mass transport mechanism, based on collisions between different entities, is used to explain the performance of an extremely hydrophobic catalytic chain transfer agent (i.e.COPhBF) in emulsion polymerization. Mass transport in emulsion polymerization is generally accepted to proceed via the aqueous phase. COPhBF possesses no detectable water-solubility and would therefore be expected to be inefficient for molecular weight control in emulsion polymerization. However, proper molecular weight control using COPhBF has been demonstrated and results are presented that are consistent with the existence of an alternative mass transport mechanism in emulsion polymerization that circumvents the aqueous phase.
Introduction
Catalytic chain transfer polymerization (CCTP) has proven to be an effective technique to synthesize polymers with a predetermined molecular weight in free radical polymerization, both in homogeneous and heterogeneous systems.1–6 Molecular weight control in CCTP is attained when the radical activity of a propagating polymer chain is transferred, via an active low-spin cobalt(II) complex, to a monomeric molecule. The main advantage of using these cobalt(II) complexes as chain transfer agents is the relatively high activity combined with the catalytic nature of the chain transfer reaction. This allows for molecular weight control using minimal quantities of catalytic chain transfer agent (CCTA). Furthermore, these cobalt(II) complexes are relatively stable in aqueous environment and as such allow for implementation of CCTP in emulsion polymerization.7–15CCTP in homogeneous bulk and solution polymerization results in a uniform distribution of the CCTA and as such ensures the production of polymer with a predetermined molecular weight, a monomodal molecular weight distribution (MWD), and a polydispersity index (D) of approximately 2. In a heterogeneous emulsion polymerization, this is not necessarily the case.16Emulsion polymerization comprises a compartmentalized reaction system, which implies the necessity of all the reactants (i.e. radicals, monomer, CCTA, etc.) at the locus of polymerization, e.g. the polymer particles.17,18 This compartmentalized nature makes efficient mass transfer in emulsion polymerization a prerequisite; to ensure that the presence of any species at the locus of polymerization is not limited. The classical view on mass transport in emulsion polymerization is that it proceeds via the aqueous phase (i.e. a resistances in series model19 is applicable) and must be sufficiently fast so as not to be rate-determining.17,18,20,21 For mass transport to occur, it is suggested that the species in question must have some degree of water-solubility. For instance, in emulsion polymerization monomer diffuses from the large monomer droplets, via the aqueous phase, to the polymer particles. For most monomers used in emulsion polymerization, mass transportvia the aqueous phase is sufficiently fast despite their relatively low water-solubility; only for virtually water-insoluble monomers, such as lauryl methacrylate, this becomes problematic. Mass transfer limitations can also arise in the case where the interfacial surface area between monomer and aqueous phase is limited due to insufficient or absent agitation of the emulsion.22–26
Earlier studies have shown that mass transport of extremely hydrophobic species such as hexadecane (HD)27 and 2,5 di-tert-butyl hydroquinone (DTBHQ)28 is possible in seeded miniemulsion polymerization. The extremely low water-solubility of these species introduces a significant resistance to mass transfer and mass transport of these compounds via the aqueous phase seems to be unlikely. Consequently, it has been suggested that an alternative mass transport mechanism in emulsion polymerization must exist, and mass transport of extremely hydrophobic species in emulsion polymerization has been described by a mass transport mechanism based on collisions.29 This suggests that mass transport in emulsion polymerization does not necessarily need to proceed via the aqueous phase and hence that extremely hydrophobic species can also be transported in emulsion polymerization.
For the application of CCTP in emulsion polymerization it has been suggested that some aqueous phase solubility of the CCTA is necessary to meet the requirement of fast mass transport to ensure proper molecular weight control.7,8,10 Consequently, the majority of academic CCTA-mediated emulsion polymerization studies are performed in the presence of bis[(difluoroboryl) dimethylglyoximato]cobalt(II) (COBF) or bis[(difluoroboryl) diethylglyoximato]cobalt(II) (COEtBF) which both possess some water-solubility.10,30,31 However, some industrial examples of the use of very hydrophobic CCTAs have been reported.14,32,33 Although the number of polymer particles (Np) (i.e. the locus of polymerization) typically outnumber the number of catalyst molecules (NCCTA),7,34 mass transport is sufficiently fast to ensure uniform reaction conditions in all the polymer particles and the production of polymer with a monomodal MWD and a D of approximately 2. However, the solubility of these CCTAs in both the aqueous and organic phase of an emulsion polymerization results in partitioning, which lowers the effective CCTA concentration at the locus of polymerization and consequently a discrepancy between the target and experimental molecular weight is observed.10,31 Furthermore, the presence of the CCTA in the aqueous phase of an emulsion polymerization affects the particle formation mechanism, entry rate of radicals13,35 and renders the cobalt(II) complexes susceptible towards aqueous phase deactivation.2 These kinetic events have major consequences for the polymerization kinetics, particle size distribution (PSD) and the MWD.35
The disadvantages attributed to the effects of CCTA partitioning in emulsion polymerization can be circumvented by using bis[(difluoroboryl) diphenylglyoximato]cobalt(II) (COPhBF),35 a CCTA with no detectable water-solubility as determined by UV-Vis spectroscopy.7,8,10,31 In emulsion polymerization, this CCTA does not display any partitioning31 and consequently all the catalyst is localized in the organic phase, i.e. the monomer droplets, monomer swollen micelles and polymer particles. Consequently, as no CCTA is present in the aqueous phase of the emulsion polymerization, many of the undesired effects on the course of the polymerization, PSD and MWD are minimized or circumvented. However, COPhBF does not meet the requirement of possessing sufficient aqueous phase solubility to allow for mass transport in emulsion polymerization. Surprisingly, proper molecular weight control in emulsion polymerization with COPhBF, however, is possible.7,8,10,31,35
Studies by different authors have shown that monomodal molecular weight distributions with a D of 2 can be obtained.7,8,10,31,35 This implies that fast mass transport of the CCTA can be achieved, which is contrary to the general belief that some aqueous phase solubility is a prerequisite to allow for mass transfer in emulsion polymerization. Furthermore, these observations suggest that an alternative mass transport mechanism could be operational for COPhBF.
Here we present additional experimental proof for an alternative mass transport mechanism in emulsion polymerization, which is based on collisions and allows for mass transport of extremely hydrophobic species.
Experimental
Materials
The catalytic chain transfer agents (see Scheme 1), bis[(difluoroboryl) dimethylglyoximato]cobalt(II) (COBF), bis[(difluoroboryl) diethylglyoximato]cobalt(II) (COEtBF) and bis[(difluoroboryl) diphenylglyoximato]cobalt(II) (COPhBF) were prepared according to the method of Bakač and Espenson.7,36 For all experiments, a single batch of catalyst was used. The intrinsic activities of the different catalysts were determined in MMA emulsion polymerization at 70 °C using a modified Mayo equation;31COBF (R = methyl): CT = 21 × 103; COEtBF (R = ethyl): CT = 22 × 103 and COPhBF (R = phenyl): CT = 6 × 103. Distilled deionized water was used throughout all experiments. The monomer, methyl methacrylate (MMA, Aldrich, 99%) was distilled at reduced pressure to remove the inhibitor and stored at −24 °C prior to usage. 2,2′-Azobis(isobutyronitrile) (AIBN, Aldrich, 98%) was recrystallized from methanol (Biosolve, analytical grade) prior to usage. The initiator, 2,2′-azobis(2-methylpropionamide) dihydrochloride (V50, Aldrich, 97%), the surfactant, sodium dodecyl sulfate (SDS, Aldrich), the buffer, sodium carbonate (VWR, analysis grade) and the co-stabilizer, hexadecane (HD, Aldrich, ≥98%), were used without any further purification.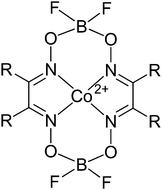 | ||
| Scheme 1 The general structure of the cobalt(II) catalytic chain transfer agents used. COBF: R = methyl, COEtBF: R = ethyl and COPhBF: R = phenyl. | ||
Emulsion polymerization
The batch experiments were performed in a 1 L glass reactor, equipped with a pitch blade impeller. A catalyst stock solution (i) was prepared by dissolving an accurate amount (0.85 mg COBF, 0.085 mg COEtBF and 1.25 or 3.13 mg COPhBF) of catalytic chain transfer agent in MMA (10 mL, 0.1 mol). An initiator solution (ii) was prepared by dissolving an accurate amount of V50 (0.54 g, 2.0 × 10−3 mol) in doubly distilled water (10 g). SDS (5.77 g, 2.0 × 10−2 mol) and sodium carbonate (0.43 g, 4.0 × 10−3 mol) were weighed and dissolved in doubly distilled water (390 g) and consequently added to the reactor. MMA (83.0 g, 0.83 mol) was added to the reactor and the resulting emulsion was purged with argon for 45 minutes. The catalyst stock solution was added and the emulsion agitated for 15 minutes prior to the addition of the initiator solution to initiate the polymerization. Samples were withdrawn periodically to monitor the conversion and molecular weight distribution.Miniemulsion polymerizations
The miniemulsion experiments were performed in a 100 mL three-necked round-bottom flask with a magnetic impeller. A catalyst stock solution (i) was prepared by dissolving an accurate amount of COPhBF (1.35 mg, 2 × 10−3 mmol) and HD (1.5 g, 6.6 × 10−3 mol) in MMA (56.6 g, 0.57 mol). SDS (0.5 g, 1.8 × 10−3 mol) and AIBN (0.1 g, 0.6 × 10−4 mol) were weighed, charged into the round-bottom flask and deoxygenated by a repeated vacuum/argon cycle. Doubly distilled water (50 mL), solution (i) (10 mL) in the case of miniemulsion A or MMA and HD (10 mL) in the case of miniemulsion B were deoxygenated with argon and added to the round-bottom flask. The resulting emulsion was stirred vigorously for 10 minutes and subsequently placed inside an ultrasound bath for 15 minutes whilst maintaining an argon atmosphere during all manipulations. After miniemulsification, miniemulsion C was prepared by mixing miniemulsions A and B in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 volume ratio. Subsequently all three miniemulsions were placed inside an oil bath at 70 °C. Samples were withdrawn periodically to monitor the conversion and molecular weight distribution.
:6 volume ratio. Subsequently all three miniemulsions were placed inside an oil bath at 70 °C. Samples were withdrawn periodically to monitor the conversion and molecular weight distribution.
Characterization
Size exclusion chromatography (SEC) was performed using a Waters 2690 separation module and a model 410 differential refractometer. A set of five Waters Styragel HR columns (HR5.0, HR4.0, HR3.0; HR1.0; HR0.5) was used in series at 40 °C. Tetrahydrofuran (THF, Aldrich) was used as the eluent at a flow rate of 1 mL·min−1, and the system was calibrated using narrow molecular weight polystyrene standards ranging from 374 to 1.1 × 106 g mol−1. Mark–Houwink parameters used for the polystyrene standards: K = 1.14 × 10−4 dL g−1, a = 0.716 and for poly(methyl methacrylate): K = 9.44 × 10−5 dL g−1, a = 0.719.Results and discussion
Partitioning of COPhBF
The partitioning behaviour of any CCTA is crucial for proper molecular weight control in emulsion polymerization. The partition coefficients (mCCTA) for different cobalt(II) complexes are expressed as the ratio of the CCTA concentrations in the organic (the monomer droplet, polymer particles and monomer swollen micelles) and (with monomer saturated) aqueous phase (mCCTA = [CCTA]org/[CCTA]aq). Typically the values of mCCTA have been determined in monomer–water systems at varying phase ratios (β), the ratio of the volumes of organic and aqueous phase (β = Vorg/Vaq).31,37 Using this method, a mCCTA of 0.72 was determined for COBF in methyl methacrylate (MMA)–water, which is in good agreement with earlier reported values ranging from 0.31 to 0.68.7,10,38 Furthermore, mCCTA for COBF and COEtBF has been determined for styrene (S)–water and butyl methacrylate (BMA)–water systems.25,37,39The previously established method for the determination of mCCTA was used to determine the mCCTA of COPhBF in an MMA–water emulsion. During the partitioning experiment a pure water phase is brought into contact with a solution of the cobalt(II) complex in MMA by vigorous agitation. As a result, both the water phase and the MMA phase will be saturated with the other solvent and the cobalt(II) complex will distribute over both phases. After phase separation, UV-Vis spectroscopy is used to determine the cobalt(II) concentrations in either phase. For COPhBF no extinction coefficient could be determined using water as a solvent, indicating that the solubility of COPhBF in water saturated with MMA is extremely low. Furthermore, in the partitioning experiments using COPhBF no partitioning occurred, i.e. the concentration of COPhBF in the MMA phase remained constant. These results are in agreement with earlier work reported by Kukulj et al.,10 wherein a comparable partitioning experiment no partitioning of COPhBF was observed.
The non-observable partitioning behaviour of COPhBF is further supported by results obtained in CCTP miniemulsion polymerization. Kukulj et al. reported CCTA mediated miniemulsion polymerization initiated by potassium persulfate (KPS).8 Dissociation of KPS results in two water-soluble oxygen-centred sulfate radicals, which are known to deactivate the active cobalt(II) complexes.2 A COBF-mediated miniemulsion polymerization initiated by KPS resulted in a major loss of the catalytic activity over the course of the polymerization. COBF possesses some aqueous phase solubility and is consequently deactivated by the sulfate radicals. As COBF is continuously replenished from the organic phase, ultimately the majority of COBF will be deactivated and control over the MWD is lost. In a similar experiment no COPhBF deactivation was observed and molecular weight control maintained throughout the course of the polymerization. This result illustrates that no measurable COPhBF partitioning occurred. COPhBF appears to be solely located in the organic phase (i.e. the monomer droplets and polymer particles) and consequently protected from aqueous phase deactivation.
In a more recent study on molecular weight control in miniemulsion polymerization using different CCTAs it was shown that no apparent loss in chain transfer activity is observed when COPhBF is used.31 Partitioning of the cobalt(II) complexes lowers the CCTA concentration at the locus of polymerization and renders a certain amount inactive in terms of molecular weight control. For COBF this results in lower apparent chain transfer activities. However, for COPhBF no apparent loss in chain transfer activity is observed. Based on predictions by a modified Mayo equation for CCTP in emulsion polymerization,31 the behaviour observed for COPhBF can only occur in the limiting case that the partition coefficient equals ∞, i.e. no partitioning of the CCTA towards the aqueous phase. The experimental results obtained in the previously reported partitioning experiments and miniemulsion polymerizations using COPhBF indeed strongly suggest that COPhBF does not partition towards the aqueous phase and is only present in the organic phase (i.e. monomer droplets, monomer-swollen micelles and polymer particles) of an emulsion polymerization.
Molecular weight control using COPhBF
COPhBF-mediated emulsion polymerizations were performed in the presence of 2.0 or 5.0 ppm (10−6 moles of COPhBF per mol of MMA) CCTA. Independent of the amount of COPhBF present nearly full conversion was obtained within 3 h of polymerization. COPhBF is known to enhance the rate of exit, which affects the course of the polymerization as well as the particle size distribution (PSD).35 The polymerizations mediated with 2.0 and 5.0 ppm display similar characteristics in terms of the PSD, volume-average particle sizes are 25.0 ± 0.2 nm with a poly of 0.14 ± 0.05. As a result of the enhanced exit and the large number of particles associated with these polymerizations (Np = 45 × 1018 dm−3) the average number of radicals per particle (![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) ) is very low ( ≈ 0.001). For a more detailed investigation of the effect of COPhBF on the emulsion polymerization kinetics the reader is referred to an earlier publication.35
) is very low ( ≈ 0.001). For a more detailed investigation of the effect of COPhBF on the emulsion polymerization kinetics the reader is referred to an earlier publication.35
Mechanistic information on the performance of COPhBF in emulsion polymerization can be extracted from an analysis of the MWD. The evolution of the MWD over the course of ab initioemulsion polymerizations mediated with 2.0 or 5.0 ppm COPhBF are shown in Fig. 1. The MWDs shown in Fig. 1A–C are monomodal and have a polydispersity index (D) of approximately 2, which is a characteristic of a MWD controlled by CCTP.
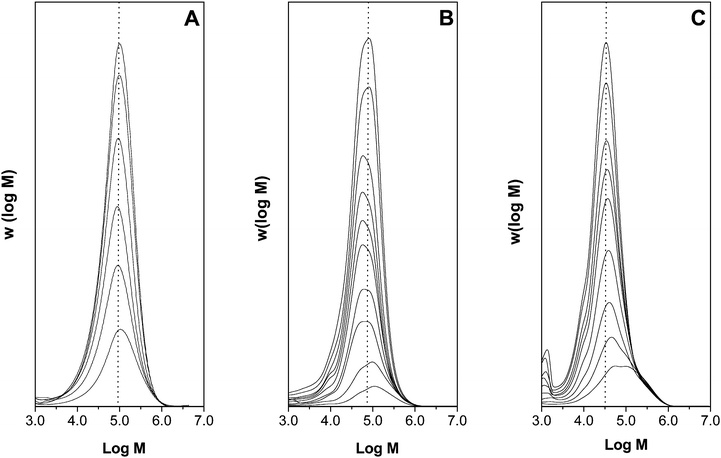 | ||
| Fig. 1 The evolution of the molecular weight distribution in the COPhBF mediated emulsion polymerization of methyl methacrylate. (A) 2.0 ppm COPhBF, (B) 2.0 ppm COPhBF and (C) 5.0 ppm COPhBF. The dotted lines indicate the locations of the peak-average molecular weights (Mp) of the molecular weight distributions at high conversion. The molecular weight distributions are scaled to conversion, for all MWDs displayed the conversion ranges from about 10% to 90%. | ||
Based on the amount of COPhBF added to the polymerization and final particle number in the polymerizations, the average number of COPhBF molecules per polymer particle (NCCTA/Np) can be calculated. For the polymerizations mediated with 2.0 and 5.0 ppm COPhBF, NCCTA/Np equals 0.070 and 0.160, respectively. Note that in the initial stage of the polymerization COPhBF also partitions towards the monomer droplets and monomer swollen micelles and that, as a consequence, NCCTA/Np will be even lower. The fact that monomodal MWDs with a D of approximately 2 are obtained for COPhBF-mediated emulsion polymerizations (Fig. 1) demonstrates that a single COPhBF molecule is able to mediate multiple polymer particles, implying that there are no mass transport limitations reducing its performance. Conversely, if any resistance to mass transport existed, multimodal MWDs would be observed with a D significantly larger than 2.16,42
Looking at Fig. 1 more closely, a shift of the MWDs towards lower average molecular weights can be observed as the polymerization progresses. This decreasing trend becomes even more obvious in the evolution of the number-average degree of polymerization (DPn) over the course of the polymerization, see Fig. 2A. The polymerizations mediated with either 2.0 or 5.0 ppm COPhBF are, based on the modified Mayo equation,31 expected to result in a DPn of 410 and 166, respectively (horizontal dotted lines in Fig. 2A). It is evident from Fig. 2A that at high conversion the DPn values indeed converge to these predicted values. However, at the initial stages of the polymerization, polymer with a higher DPn is produced. It can be seen from Fig. 2A that the decreasing trend is reproducible and present at both COPhBF loadings.
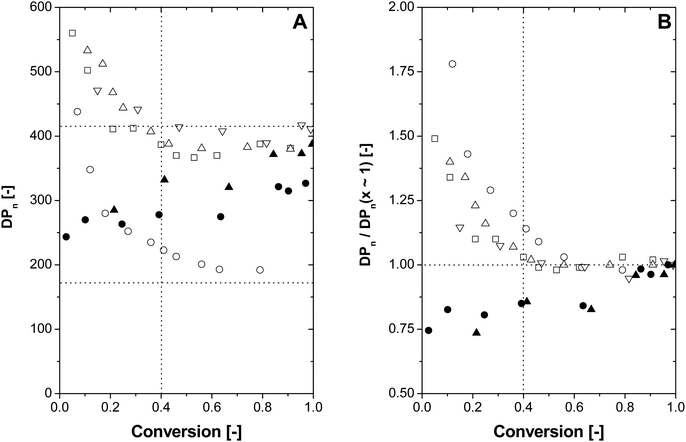 | ||
| Fig. 2 Evolution of the cummulative number-average degree of polymerization (DPn) (A) and the normalized number-average degree of polymerization (B). Normalized DPn is defined as DPn(x)/DPn(x → 1) (●) 2.0 ppm COBF, (▲) 0.2 ppm COEtBF, (△,▽,□) 2.0 ppm COPhBF and (○) 5.0 ppm COPhBF. 1 ppm is defined as 1 mol CCTA per 106 moles of monomer. Experimentally obtained number-average degree of polymerization DPn = Mw/(2 × monomer mass).40,41 The vertical dotted line indicates the conversion at which the monomer droplets have disappeared (i.e. where interval II of the emulsion polymerization finishes). The horizontal dotted lines indicate the predetermined DPn of 410 and 166 as determined from the modified Mayo equation.31 | ||
Fig. 2B shows the evolution of the DPn at a given conversion x normalized to the predetermined DPn at high conversion x → 1 (i.e., DPn(x)/DPn(x → 1)) during the course of the polymerization. Independent of the COPhBF loading, the observed decrease in DPn(x)/DPn(x → 1) in the initial stages of the polymerization occurs until a conversion of approximately 30–40% is reached (vertical dotted line in Fig. 2). Based on the Mayo equation a decrease in DPn as a function of the conversion is expected as the monomer concentration decreases throughout the course of a polymerization. However, in emulsion polymerization the monomer concentration at the locus of polymerization is assumed constant up to the disappearance of the monomer droplets (end of Interval II). Although this assumption is likely not valid for interval I, the change in the monomer concentration going from monomer swollen micelles to small polymer particles will not have a large effect on the DPn when compared to the effect of the CCTA. Therefore it can be concluded that the decrease of DPn(x)/DPn(x → 1) in the initial stages of the polymerization is due to an increase in the amount of COPhBF at the locus of polymerization.
COPhBF mass transport mechanism
The evolution of DPn and DPn(x)/DPn(x → 1) over the course of the polymerization for COBF and COEtBF is also presented in Fig. 2A and B.31COBF and COEtBF differ greatly in aqueous phase solubility when compared to COPhBF. COBF is fairly7,10,38 and COEtBF is sparingly30,38water soluble and these catalysts will readily partition, not only between monomer droplets, monomer swollen micelles and polymer particles, but also to the aqueous phase. The polymerizations were mediated using 2.0 ppm COBF and 0.2 ppm COEtBF, aiming at a predetermined final DPn of approximately 410. Based on the amount of CCTA used the NCCTA/Np for these polymerizations equaled 0.17 for COBF and 0.015 for COEtBF. For both CCTA monomodal MWDs are obtained with a D of approximately 2.35Mass transport in an emulsion polymerization is generally accepted to proceed via the aqueous phase, according to a so-called resistance in series model.17,18 As both COBF and COEtBF possess sufficient aqueous phase solubility, no mass transfer limitations are expected. Consequently, the monomer droplets, monomer swollen-micelles and polymer particles can be regarded as one continuous phase. In other words, COBF and COEtBF can readily diffuse from a monomer droplet, via the aqueous phase to a polymer particle, or vice versa. This results in a situation where the MWD is governed by a global CCTA concentration16 (CCTA = NCCTA/Np) which relates to the partitioning behavior of the CCTA between the aqueous and organic phase.
During the course of the polymerization an increase in DPn(x)/DPn(x → 1) is observed for COBF and COEtBF, see Fig. 2B. This observation follows directly from the partitioning behavior of the CCTAs. Both COBF and COEtBF partition towards the aqueous phase, which renders these complexes susceptible towards deactivation. As deactivation occurs in the aqueous phase, the aqueous phase CCTA concentration will be replenished from the organic phase. This results in a decreasing CCTA concentration at the locus of polymerization (i.e. CCTA decreases) and consequently an increase in DPn. This increase in DPn towards the end of the polymerization is typically observed in COBF and COEtBF-mediated emulsion polymerizations.
Since mass transport of COPhBF seems unlikely to occur via the aqueous phase, this result suggests the existence of an alternative mass transport mechanism in emulsion polymerization which circumvents the aqueous phase. Comparable observations have been made with regard to mass transport of extremely hydrophobic species and have been explained by a mass transport mechanism based on collisions between different entities.27–29
In what follows we will try to provide a conceivable explanation for our results using a mass transport mechanism based on collisions. First of all, the emulsion polymerization reaction system is densely packed with monomer droplets, monomer swollen micelles and/or polymer particles. Since COPhBF has no measurable water-solubility, it will reside exclusively in the organic phase. Collisions between monomer droplets, polymer particles and monomer swollen micelles occur frequently and it is not unlikely that small molecules can be exchanged between two of these entities at the moment of collision. Exchange of a COPhBF molecule between a monomer droplet and a polymer particle can only occur when the COPhBF molecule is in the proximity of the surface of the droplet or particle. In the initial stage of the polymerization a finite amount of COPhBF is localized in the bulk of the monomer droplets and does not take part in the control of the MWD. As the polymerization proceeds, the monomer droplets are shrinking in size thereby increasing the surface area to volume ratio per droplet. The probability of a COPhBF molecule being located close to the surface is increasing and consequently more COPhBF is transferred towards the polymer particles. By the time the monomer droplets disappear, COPhBF is located exclusively inside the polymer particles. Exchange of COPhBF between the polymer particles is expected to be highly effective as they possess a high surface area to volume ratio. The exchange rate of COPhBF between colliding particles is sufficiently fast to ensure proper control of the MWD, as is clearly demonstrated by the results in Fig. 1 (i.e. the polymer particles can be considered a single continuous phase with respect to CCT). Transport of COPhBF by collisions between monomer droplets and polymer particles and/or monomer swollen micelles was also briefly mentioned by Kukulj et al.10 Their conclusion was based on the performance of COPhBF in semi-batch emulsion polymerization. However, no experimental evidence was reported to support this hypothesis. Since in both our study and that by Kukulj et al. the number of catalyst molecules per particle was significantly below 1 (0.090 and 0.33, respectively), the monomodal MWDs in both studies do not originate from a “pseudo-bulk”17 situation where a single polymer particle contains a large number of COPhBF molecules.
The difference in the course of the DPn(x)/DPn(x → 1) between COBF, COEtBF on the one hand and COPhBF on the other (Fig. 2B) can be quantitatively explained with the mass transport mechanism based on collisions. The COPhBF catalyst initially resides in the monomer droplets, as the CCTA is dissolved in monomer prior to the addition to the reaction mixture. The fact that a DPn lower than that which is common for a classical emulsion polymerization is obtained already in the early stages of the polymerization indicates that prior to initiation a certain fraction of the COPhBF catalyst resides in the monomer swollen micelles. During the following 30–40% of the course of the polymerization, DPn continuously decreases as the amount of COPhBF at the locus of polymerization continuously increases. When the monomer droplets have disappeared, the total amount of COPhBF is located inside the polymer particles and a plateau DPn value of 410 is reached, which corresponds to the value expected based on the total amount of COPhBF in the system.
Exchange of COPhBF molecules between polymer particles, and as a consequence the existence of the mass transport mechanism based on the collisions between different entities, can be tested in miniemulsion polymerization. Two miniemulsions, one without COPhBF (A) and one containing 5.2 ppm (10−6 mol per mol of monomer) COPhBF (B), were mixed using a 1:6 volume ratio mixture of A and B prior to initiation (C). If there were no mass transport mechanism for COPhBF, the CCTA would be compartmentalized and a bimodal MWD would be obtained as there are two populations of polymer particles. These two populations for the separately polymerized miniemulsions are shown in Fig. 3 for distributions A and B for 0 and 5.2 ppm catalyst, respectively. However, if COPhBF mass transport is possible a (more) monomodal MWD should be obtained, governed by a global COPhBF concentration. As can be seen in Fig. 3, the MWD of the miniemulsion polymerization consisting of a mixture of two miniemulsions (C) displays a monomodal MWD, exactly positioned where expected from the overall amount of COPhBF in the system. This clearly demonstrates that the COPhBF catalyst is indeed able to mediate all polymer particles and that the CCTA is not compartmentalized. Consequently, a global COPhBF concentration governs the MWD in COPhBF-mediated (mini)emulsion polymerization which strongly supports the hypothesis for a mass transport mechanism in (mini)emulsion polymerization based on collisions.
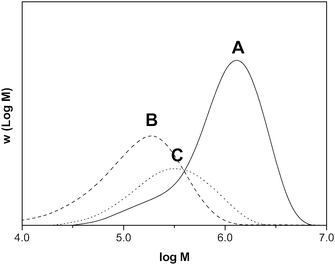 | ||
Fig. 3 Molecular weight distributions of three miniemulsion polymerizations: (A) containing 0 ppm COPhBF, (B) containing 5.2 ppm COPhBF and (C) consisting of a 1:6 volume ratio mixture of A and B. (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) 0 ppm COPhBFx = 0.96, ( ) 0 ppm COPhBFx = 0.96, (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) 5.2 ppm COPhBFx = 0.52 and (⋯) 1.2 ppm COPhBFx = 0.33. Miniemulsions A and B were mixed prior to initiation. ) 5.2 ppm COPhBFx = 0.52 and (⋯) 1.2 ppm COPhBFx = 0.33. Miniemulsions A and B were mixed prior to initiation. | ||
The main implication of a mass transport mechanism that does not depend on aqueous phase solubility is that very hydrophobic CCTAs can be readily applied in emulsion polymerization. Since these complexes do not display any detectable partitioning behavior, the aqueous phase kinetics are not affected to the same extent as in the case with e.g.COBF, which has positive effects on the rate of polymerization and the final latex properties.7–13,35
Conclusions
The results of this work strongly suggest that mass transport of COPhBF in emulsion polymerization occurs via a mass transport mechanism based on collisions. Exchange of the COPhBF catalyst occurs upon a collision between two entities, i.e., monomer droplets, monomer swollen micelles and/or polymer particles and is sufficiently fast to ensure proper molecular weight control in (mini)emulsion polymerization. Since extremely hydrophobic CCTAs are not present in the aqueous phase of an emulsion polymerization, many deleterious effects on the polymerization kinetics, molecular weight distribution and particle size distribution can be circumvented.References
- A. A. Gridnev, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1753 CrossRef CAS.
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611 CrossRef CAS.
- J. P. A. Heuts, G. E. Roberts and J. D. Biasutti, Aust. J. Chem., 2002, 55, 381 CrossRef CAS.
- L. V. Karmilova, G. V. Ponomarev, B. R. Smirnov and I. M. Belgovskii, Russ. Chem. Rev., 1984, 53, 223 Search PubMed.
- T. P. Davis, D. M. Haddleton and S. N. Richards, J. Macromol. Sci., Rev. Macromol. Chem., 1994, C34, 234 Search PubMed.
- T. P. Davis, D. Kukulj, D. M. Haddleton and D. R. Maloney, Trends Polym. Sci., 1995, 3, 365 CAS.
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Macromolecules, 1996, 29, 8083 Search PubMed.
- D. Kukulj, T. P. Davis and R. G. Gilbert, Macromolecules, 1997, 30, 7661 Search PubMed.
- S. A. F. Bon, D. R. Morsley, J. Waterson, D. M. Haddleton, M. R. Lees and T. Horne, Macromol. Symp., 2001, 165, 29 Search PubMed.
- D. Kukulj, T. P. Davis, K. G. Suddaby, D. M. Haddleton and R. G. Gilbert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 859 CrossRef CAS.
- S. C. J. Pierik, B. Smeets and A. M. van Herk, Macromolecules, 2003, 36, 9271 Search PubMed.
- D. M. Haddleton, D. R. Morsley, J. P. O'Donnell and S. N. Richards, J. Polym. Sci., Part A: Polym. Chem., 2001, 37, 3549 Search PubMed.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2009, 19, 5078 Search PubMed.
- J. Huybrechts, P. Bruylands, K. Kishenbaum, J. Vrana and J. Snuparek, Prog. Org. Coat., 2002, 45, 173 Search PubMed.
- T. Nabuurs, S. Van der Slot and A. Overbeek, Prog. Org. Coat., 2007, 58, 80 Search PubMed.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, Macromolecules, 2009, 42, 7332 Search PubMed.
- R. G. Gilbert, Emulsion Polymerization: A Mechanistic Approach, Academic Press, London, 1st edn, 1995 Search PubMed.
- A. M. van Herk, Chemistry and Technology of Emulsion Polymerization, Blackwell Publishing, Oxford, 1st edn, 2005 Search PubMed.
- J. R. Welty, C. E. Wicks, R. E. Wilson and G. Rorrer, Fundamentals of Momentum, Heat and Mass Transfer, Wiley, New York, 4th edn, 2001, ch. 29 Search PubMed.
- S. Rimmer, Macromol. Symp., 2000, 150, 149 Search PubMed.
- R. J. Leyrer and W. Mächtle, Macromol. Chem. Phys., 2000, 201, 1235 CrossRef CAS.
- B. Henninger, M. Rehfeld, W. Pauer and H.-U. Moritz, Macromol. Symp., 2004, 1, 191 Search PubMed.
- K. Tauer, H. Hernandez, S. Kozempel, O. Lazareva and P. Nazaran, Colloid Polym. Sci., 2007, 286, 499 Search PubMed.
- S. Sajjadi, Macromol. Rapid Commun., 2004, 25, 882 Search PubMed.
- M. F. Kemmere, J. Meuldijk, A. A. H. Drinkenburg and A. L. German, J. Appl. Polym. Sci., 1999, 74, 3225 Search PubMed.
- M. Zubitur, J. Mendoza, J. C. de la Cal and J. M. Asua, Macromol. Symp., 2000, 150, 13 Search PubMed.
- J. Delgado, M. S. El-Aasser, C. A. Silebi and J. W. Vanderhoff, Polym. Mater.: Sci. Eng., 1987, 57, 976 Search PubMed.
- V. S Rodriguez, M. S. El-Aasser, J. M. Asua and C. A. Silebi, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3659 Search PubMed.
- J. M. Asua, V. S. Rodriguez, C. A. Silebi and M. S. El-Aasser, Makromol. Chem., Macromol. Symp., 1990, 35–36, 59 Search PubMed.
- J. L. Waterson, D. M. Haddleton, R. J. Harrison and S. N. Richards, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1998, 39, 457 Search PubMed.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5839 Search PubMed.
- A. H. Janowicz, US Pat., 4 694 054, 1987.
- A. H. Janowicz, US Pat., 5 028 677, 1991.
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. Van Herk, Macromolecules, 2009, 42, 6422 Search PubMed.
- N. M. B. Smeets, T. G. T. Jansen, T. J. J. Sciarone, J. P. A. Heuts, J. Meuldijk and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1038 Search PubMed.
- A. Bakač, M. E. Brynildson and J. H. Espenson, Inorg. Chem., 1986, 25, 4108 CrossRef CAS.
- N. M. B. Smeets, U. S. Meda, J. P. A. Heuts, J. T. F. Keurentjes, A. M. Van Herk and J. Meuldijk, Macromol. Symp., 2007, 259, 406 Search PubMed.
- S. C. J. Pierik, Shining a Light on Catalytic Chain Transfer, PhD thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2002 Search PubMed.
- Unpublished results: for BMA, mCCTA (COBF) = 0.035 and mCCTA (COEtBF) = 8.42.
- J. P. A. Heuts, D. Kukulj, D. J. Forster and T. P. Davis, Macromolecules, 1998, 31, 2894 CrossRef CAS.
- J. P. A. Heuts, T. P. Davis and G. T. Russell, Macromolecules, 1999, 32, 6019 CrossRef CAS.
- M. E. Thomson, N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and M. F. Cunningham, Macromolecules, 2010, 43, 5467 Search PubMed.
Footnote |
| † Current address: Department of Chemical Engineering, Queen's University, Kingston, Ontario, Canada, K7L 3N6. |
| This journal is © The Royal Society of Chemistry 2011 |