A specific inhibitory effect of multivalent trehalose toward Aβ(1-40) aggregation†
Masaya
Wada
a,
Yuta
Miyazawa
b and
Yoshiko
Miura
*a
aDepartment of Chemical Engineering, Graduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka, 819-0395, Japan. E-mail: miuray@chem-eng.kyushu-u.ac.jp; Fax: +81-92-802-2800; Tel: +81-92-802-2749
bSchool of Materials Science, Japan Advanced Institute of Science and Technology, 1-1 Asahidai, Nomi, Ishikawa, 923-1292, Japan
First published on 21st May 2011
Abstract
The inhibitory effect of disaccharides and their polyvalent compounds toward aggregation of Aβ(1-40) was investigated. Polyvalent trehalose, maltose and lactose were synthesized, and their inhibitory effects were investigated together with those of the corresponding monomeric disaccharides. The inhibitory effects of the monomeric disaccharides were weak, but were amplified by multivalency. Trehalose and polyvalent trehalose induced formation of aggregates with specific pseudo-spherical morphology, which did not occur in the presence of the other disaccharides or in the absence of additives. Addition of polyvalent trehalose neutralized the cytotoxicity of Aβ, while monomeric trehalose induced weak cytotoxicity (toward HeLa cells). The specific inhibitory effects of trehalose and polyvalent trehalose on Aβ aggregation are discussed in the terms of the conformation of those molecules.
Introduction
The major pathological hallmarks of Alzheimer disease (AD) are amyloid deposits, neurofibrillary tangles and selective neuronal loss in the patient's brain.1 The principal component of amyloid deposits is amyloid β peptide (Aβ) composed of 39–42 peptide residues, of which the major components are Aβ40 and Aβ42. It has been reported that neuronal cell decrease is strongly related to accumulation of Aβ, and in particular to toxic oligomeric aggregates 7–10 nm in size. The detailed mechanism of AD, and the development of inhibitors toward Aβ aggregation have received continuous attention. There have been reports of a variety of compounds that inhibit Aβ aggregation, including peptides,2 dopamine,3 dendrimers4 and sugars.5 Most importantly, there are strong correlations with sugars such as proteoglycan and the protein in vivo,6 thus it is highly possible that sugars interact with Aβ.Trehalose (α-D-glucopyranosyl-(1-1)-α-D-glucopyranoside), in particular, has received much attention because remarkable biological properties of trehalose have been reported, including stabilization of lipid bilayers7 and DNA duplex,8 moisture-retention capability9 and inhibition of protein aggregation.10 Sierks et al.11 and Hoshi et al.12 have reported inhibition of Aβ aggregation by addition of trehalose. Inhibition by trehalose of protein aggregation in insulin amyloid13 and mice with Huntington disease14 has also been reported.
However, the effect of trehalose on protein aggregation in amyloidosis is far from being understood in detail. In previous studies the dose of trehalose was large,11–14 so the efficacy of trehalose toward protein inhibition was low compared with that of other protein inhibitors, because of weak affinities. Trehalose interacts with and stabilizes proteins by hydrogen bonding to proteins15,16 and the hydration shell around proteins.17 In spite of the weakness of the inhibitory effect, trehalose might be a good inhibitor for aggregation of various proteins, because all proteins form both hydrogen bonds and hydration shells.
It has been reported that the weak interaction of sugar with protein can be enhanced by the multivalent effect,18,19 and various polymeric systems have been reported such as glycopolymers,20,21 glycodendrimers22,23 and glycopeptides.24 We have previously reported various glycopolymers that amplified the interaction between protein and saccharide.25,26 Although trehalose is not a specific “ligand” for protein, the interaction of trehalose with protein is considered to be enhanced by multivalency. Consequently, we synthesized glycopolymer-carrying trehalose, and investigated its inhibitory effect toward Aβ aggregation. Considering the biological activity of trehalose, the novel multivalent trehalose is an attractive compound due to its inhibition of protein aggregation. In addition, it expands the range of compounds with specific trehalose activity.
In the present study we investigated the biological activities of multivalent trehalose and reference disaccharides (lactose and maltose) (Fig. 1), in terms of their inhibitory effect on Aβ aggregation. The protein aggregation behavior was monitored by the fluorescence of thioflavin T (ThT) in the presence of glycopolymers, and the morphological changes induced by addition of glycopolymers were investigated by atomic force microscopy (AFM). The cytotoxicity of Aβ was investigated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using HeLa cells. We have reported a polyvalent trehalose with an alkyl chain.27 But the inhibitory effect of the compound was affected by its amphiphilicity.28 In the present study, we designed polyvalent saccharides with shorter linkers and copolymerized with acrylamide to eliminate the effect of amphiphilicity.
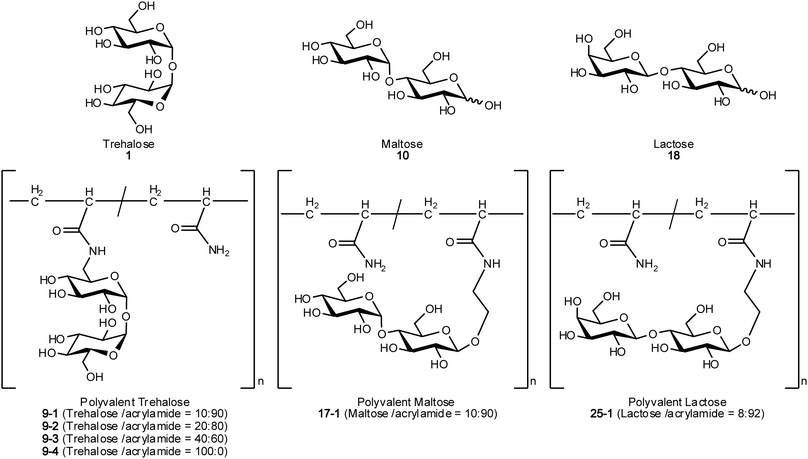 | ||
| Fig. 1 Chemical structures of compounds used in this research. | ||
Experimental section
Materials
The following reagents were used as received: amyloid β protein (human, 1-40) (Peptide Institute, Inc., Osaka, Japan); Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and penicillin–streptomycin solution (Invitrogen Co. GIBCO, Carlsbad, CA, USA); D-trehalose monohydrate, lactose monohydrate, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), thioflavin T (ThT) and sodium chloride (NaCl) (Kanto Chemical Co., Inc., Tokyo, Japan); maltose monohydrate (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan); ammonium hydroxide (28–30%) (Sigma-Aldrich, USA); 2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulfonic acid (HEPES) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Dojindo, Japan); and 2, 2′-azobis-(2-amidinopropane)dihydrochloride (AAPD) (Wako Chemical, Osaka, Japan). Other chemicals such as multivalent disaccharides were synthesized. The detailed synthetic schemes and characterization are described in the ESI†.Measurements
1H (300 MHz, 500 MHz) NMR spectra were recorded in D2O, CDCl3 or MeOD at room temperature with Varian Inova 300 and 500 spectrometers (Agilent Technologies, Inc., Santa Clara, CA, USA), and mass spectra were obtained with an ESI-MS LCQ-DECA XP instrument (Thermo Scientific, Waltham, MA, USA). Gel permeation chromatography (GPC) was conducted with a JASCO 800 high-performance liquid chromatograph (Jasco, Tokyo, Japan), using a Shodex SB-804HQ column (Shodex, Tokyo, Japan) with phosphate buffered saline (PBS) as eluent and pullulan as molecular weight standard. Fluorescence intensity was measured with an LS55 instrument (Perkin-Elmer, Waltham, MA, USA), and an SPA400-SPI3800N instrument (Seiko Instruments Inc., Tokyo, Japan) with DF40P (40 N cantilever) was used for atomic force microscopy (AFM). A microplate reader, Multiskan JX, purchased from ThermoFisher Scientific Inc. (Waltham, MA, USA), was used for MTT assay.Glycopolymers
1-O-(α-D-Glucopyranosyl)-6-(2-propene amido)-6-deoxy-α-D-glucopyranoside, 4-O-(α-D-glucopyranosyl)-1-O-{2-(2-propene amido)ethyl}-β-D-glucopyranoside and 4-O-(β-D-galactopyranosyl)-1-O-{2-(2-propene amido)ethyl}-β-D-glucopyranoside were synthesized by conventional procedures (see ESI†). Acrylamide derivatives of trehalose, maltose and lactose were copolymerized with acrylamide using AAPD as radical initiator (Scheme 1).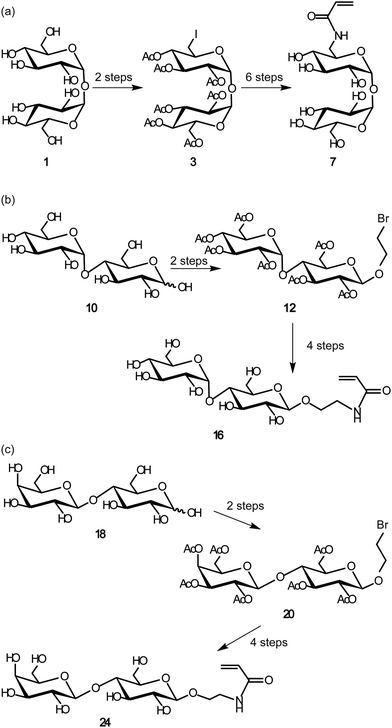 | ||
| Scheme 1 Syntheses of acrylamide derivatives of disaccharides of: (a) trehalose monomer, (b) maltose monomer, and (c) lactose monomer. | ||
Preparation of Aβ(1-40)
0.12 μmol Aβ40 was monomerized with 240 μl of ice-cold HFIP, and 11 μl of the solution dispensed into each of 21 tubes. The Aβ40 solution in the tubes, cooled in ice, was evaporated to dryness with a stream of nitrogen, and the Aβ40 residues preserved at −80 °C. For ThT assay, the Aβ40 stocks were dissolved in 22 μl of 0.03% NH3 solution to give 500 μM concentrations.ThT assay29
ThT assay samples were made up in 50 mM phosphate buffer with 100 mM NaCl (pH 7.5), with the following concentrations of solutes: ThT, 20 μM; Aβ40, 23 μM; sugar and glycopolymer 10 mM (the concentration of glycopolymer indicates the concentration of the sugar unit). The volume of each test sample was 100 μl, which was added to a 96 well plate and the wells covered with film. The plate was incubated and shaken at 400 rpm (Incubator FMS, EYELA, Tokyo) at 37 °C. Fluorescence intensity was measured at 485 nm at 1 h intervals, with excitation at 450 nm.Kinetic analysis of ThT assay results
Kinetic analysis was applied to Aβ40 aggregation on the basis of the reported two step reaction mechanism.30 Autocatalytic nucleation takes place in the first step, followed by fibril elongation in the second step. The mechanism is represented by eqn (1) and (2), with associated rate constants kn and ke, respectively.| nM → Pn (kn, nucleation process) | (1) |
| M + Pn → Pn+1 (ke, fibril elongation process) | (2) |
Experimental data were fitted using eqn (3):
| f = (ρ{exp [(1 + ρ)kt] − 1})/(1 + ρ exp [(1 + ρ)kt]) | (3) |
Morphology analysis
The sample preparation procedures for AFM were as for ThT assay. The samples (23 μM Aβ40, 10 mM sugar and glycopolymer, 100 μl in 50 mM phosphate buffer with 100 mM NaCl and pH 7.5) were incubated at 37 °C and shaken at 400 rpm. After 48 h 5 μl drops of sample solution were placed on black mica, dried for 24 h at room temperature, rinsed (100 μl × 6) with ultra-pure water, dried for 10 min under vacuum, then allowed to dry at room temperature prior to examination with AFM.Cell culture31
HeLa cells were suspended in DMEM with 10% FBS and 1% penicillin–streptomycin. The cells were plated at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells well−1 on a tissue culture treated 24-well plate, in a humidified atmosphere (5% CO2/95% room air) at 37 °C. One day after plating, the culture medium was changed to 450 μl DMEM supplemented with 5% FBS. To generate aggregation, a sample that was 23 μM in Aβ40 and 10 mM in sugar and glycopolymer in 50 mM phosphate buffer with 100 mM NaCl and pH 7.5, was used. The total volume (100 μl) of each sample was added to the wells of a 96 well plate (IWAKI) together with 500 mM NaCl and 200 mM HEPES at pH 7.4, incubated at 37 °C, and shaken at 400 rpm for 24 h. After the change of medium, 50 μl of pre-incubated sample was applied to the medium. The HeLa cells were further cultured for 24 h and assayed for MTT reduction.32 MTT was dissolved in PBS at 5 mg ml−1 and the cell culture medium was mixed with MTT solution with DMEM supplemented with 5% FBS (in the ratio 1:9). Then 24-well plates of cell culture were charged with 300 μl of the medium and the cells cultured for 4 h, and the absorbance of solubilized MTT formazan product was measured at 570 nm (reference at 650 nm).
000 cells well−1 on a tissue culture treated 24-well plate, in a humidified atmosphere (5% CO2/95% room air) at 37 °C. One day after plating, the culture medium was changed to 450 μl DMEM supplemented with 5% FBS. To generate aggregation, a sample that was 23 μM in Aβ40 and 10 mM in sugar and glycopolymer in 50 mM phosphate buffer with 100 mM NaCl and pH 7.5, was used. The total volume (100 μl) of each sample was added to the wells of a 96 well plate (IWAKI) together with 500 mM NaCl and 200 mM HEPES at pH 7.4, incubated at 37 °C, and shaken at 400 rpm for 24 h. After the change of medium, 50 μl of pre-incubated sample was applied to the medium. The HeLa cells were further cultured for 24 h and assayed for MTT reduction.32 MTT was dissolved in PBS at 5 mg ml−1 and the cell culture medium was mixed with MTT solution with DMEM supplemented with 5% FBS (in the ratio 1:9). Then 24-well plates of cell culture were charged with 300 μl of the medium and the cells cultured for 4 h, and the absorbance of solubilized MTT formazan product was measured at 570 nm (reference at 650 nm).
Results
Synthesis of glycopolymers
The acrylamide derivatives of disaccharides of trehalose (1), maltose (10) and lactose (18) were synthesized (Scheme 1). 1 was selectively iodinated at the 6′ position, and the other hydroxy groups were acetylated to give 3. 10 and 18 were acetylated and glycosylated with 2-bromoethanol in the presence of BF3·OEt2 to give 12 and 20, respectively. The compounds obtained were subjected to azidation, amination and condensation with acryloyl chloride, affording the acrylamide derivatives of the three disaccharides (7, 16 and 24). Polyacrylamide derivatives with disaccharides were polymerized by free radical copolymerization with acrylamide using AAPD as initiator. In the case of polyvalent trehalose, the sugar contents in the polymers were varied from 10 to 100% (9-1, 9-2, 9-3 and 9-4). In the other polyvalent disaccharides, the sugar content was about 10% (17-1 and 25-1) based on our previous reports.24 The polymers obtained were purified by dialysis and used for subsequent studies. The synthesized glycopolymers (9-1, 9-2, 9-3, 9-4, 17-1 and 25-1) and the original disaccharides (1, 10 and 18) were subjected to the assay for inhibition of protein aggregation (Fig. 1 and Table 1).| No. | M n (×105) | M w (×105) | M w/Mnb | Yield (%) | Sugar content ratesc (%) | n |
|---|---|---|---|---|---|---|
| a Solvent (H2O) 500 μl, AAPD 3 mol%. b By pullulan standard. c By 1H NMR. | ||||||
| 9-1 | 1.1 | 1.6 | 1.6 | 80 | 9.7 | 98 |
| 9-2 | 1.0 | 1.6 | 1.6 | 67 | 20 | 147 |
| 9-3 | 1.0 | 1.5 | 1.4 | 80 | 63 | 235 |
| 9-4 | 1.1 | 1.6 | 1.4 | 41 | 100 | 285 |
| 17-1 | 0.67 | 1.3 | 1.9 | 72 | 9.7 | 60 |
| 25-1 | 0.53 | 1.1 | 2.2 | 77 | 7.8 | 41 |
Inhibitory effects of trehalose and polyvalent trehalose
Aβ without disaccharides and polyvalent disaccharide additives showed a characteristic sigmoidal curve, indicating aggregation with increasing incubation time (Fig. 2).29 The time-course of fluorescence change with a trehalose (1) additive was scarcely different from that without additives. Moreover, the addition of 1 shortened the lag phase from ∼6 h to ∼4 h. The final fluorescence intensity after 12–24 h was almost the same as that without additives. In addition, 1 did not inhibit Aβ aggregation even with addition of more concentrated (>100 mM) trehalose solution (Fig. S1†). The result with trehalose was different from previous results, and is discussed later.11,12,27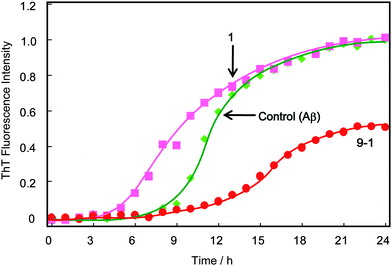 | ||
| Fig. 2 Time course of the fluorescence change of ThT with Aβ(1-40) in the presence and absence of 1 and 9-1, at pH 7.4 and 37 °C with 400 rpm shaking. The concentrations of Aβ and 1 or 9-1 were 23 μM and 10 mM, respectively. These concentrations of polyvalent disaccharides indicate the sugar unit concentrations. | ||
In contrast, addition of the polyvalent trehalose (9-1) strongly inhibited aggregation of Aβ: the lag phase was 4 h longer than in the case without additives. The final fluorescence intensity with 9-1 was 47% smaller than the value without additives, or in the presence of 1.
Inhibitory effect of other disaccharides and polyvalent disaccharides
The inhibitory effects of the disaccharides of maltose (10) and lactose (18) were investigated (Fig. 3a). Monomeric maltose (10) elongated the lag phase, and decreased by 20% the final fluorescence intensity compared with that without additives. The ThT trace in the presence of lactose (18) was similar to that for trehalose, in which the lag phase was a little shorter than the original value (∼5 h), and the final fluorescence intensity was almost the same.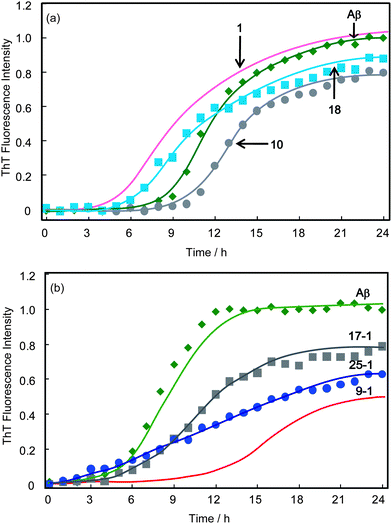 | ||
| Fig. 3 Time course of the fluorescence change of ThT with Aβ(1-40) in the presence and absence of disaccharides and polyvalent disaccharides: (a) 1, 10 and 18 and (b) 9-1, 17-1 and 25-1; at pH 7.4 and 37 °C with 400 rpm shaking. The concentrations of Aβ and 1, 9-1, 10, 17-1, 18 or 25-1 were 23 μM and 10 mM, respectively. These concentrations of polyvalent disaccharides indicate the sugar unit concentrations. | ||
Polyvalent maltose (17-1) and lactose (25-1) showed an inhibitory effect on Aβ aggregation based on the multivalent effect, and the inhibitory effect was much stronger than with the monomeric disaccharides (Fig. 3b). 17-1 and 25-1 decreased the final fluorescence intensity to 16 and 33%, respectively, of the intensity in the absence of additives. Of the three types of glycopolymers, 9-1 (polyvalent trehalose) showed the strongest inhibitory effect.
Kinetic analyses of ThT assay
ThT fluorescence traces were fitted in terms of amyloid nucleation (kn) and fibril elongation (ke) rate constants using eqn (3) (Table 2). Monovalent trehalose (1) increased kn and decreased ke relative to the values without additives (Aβ40 control), while polyvalent trehalose (9-1) decreased both kn and ke, indicating the strong inhibitory effect of polyvalent trehalose. Polyvalent trehalose decreased kn by one-third and ke by five-ninths, relative to the respective values without additives, confirming a strong inhibitory effect due to multivalency. The most noticeable difference between trehalose and polyvalent trehalose was in kn which for polyvalent trehalose was just 6% of the value for trehalose. In the case of maltose and lactose, and polyvalent disaccharides (17-1 and 25-1) the magnitude of ke decreased.| No. | k n/s−1 | k e/l mol−1 s−1 |
|---|---|---|
| Aβ40 (Control) | 6.2 × 10−7 | 8.8 |
| 1 | 3.2 × 10−6 | 3.9 |
| 10 | 1.1 × 10−7 | 6.4 |
| 18 | 2.9 × 10−6 | 3.7 |
| 9-1 | 2.1 × 10−7 | 5.1 |
| 17-1 | 3.1 × 10−6 | 3.9 |
| 25-1 | 8.7 × 10−6 | 2.0 |
Sugar content effect of polyvalent trehalose
The inhibitory effect of polyvalent trehalose was investigated with sugar contents 9.7% (9-1), 20% (9-2), 63% (9-3) and 100% (9-4) by ThT fluorescence assay (Fig. 4). The time-course of ThT was dependent on the sugar content of the additive, but surprisingly the inhibitory effect of polyvalent trehaloses with higher sugar content (9-3 and 9-4) was weak. Addition of polyvalent trehalose with the highest sugar content (9-4) did not inhibit Aβ aggregation but resulted in a 128% increase of ThT fluorescence, and 9-3 only slightly inhibited Aβ aggregation. In contrast the polyvalent trehaloses with moderate sugar content (9-1 and 9-2) showed strong inhibition effects from the ThT time-course and the final fluorescence intensity. The results confirmed that 9-2 had the largest inhibitory effect.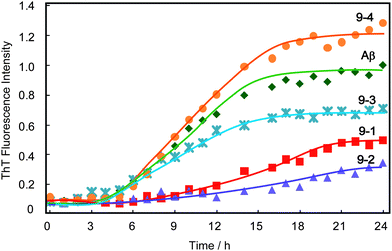 | ||
| Fig. 4 Time course of the fluorescence change of ThT with Aβ(1-40) in the presence and absence of polyvalent disaccharide additives 9-1, 9-2, 9-3, and 9-4, at pH 7.4 and 37 °C with 400 rpm shaking. The concentrations of Aβ and polyvalent disaccharides were 23 μM and 10 mM, respectively. These concentrations of polyvalent disaccharides indicate the sugar unit concentrations. | ||
Morphology of Aβ in the presence of disaccharide and polyvalent disaccharide additives
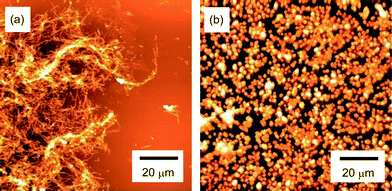
The morphology of the Aβ aggregates was investigated by AFM (Fig. 5 and 6). Aβ without additives formed fibrillar aggregates 1.1–3.3 μm in length, 62–83 nm in width and 4.2–10 nm high (Fig. 5a). Addition of polyvalent trehalose (9-1) changed the morphology of Aβ, and a mixture of pseudo-spherical aggregates and small fibrils was formed (Fig. 5b). The pseudo-spherical aggregates were 25–56 nm in height and 500–690 nm in diameter, and the small fibrillar aggregates were 250–310 nm long, 71 nm wide, and 0.88 nm high. Monomeric trehalose (1) also changed the morphology, and produced fibrillar and pseudo-spherical aggregates (Fig. 6a and b)). The pseudo-spherical aggregates (39–55 nm in diameter and 205–207 nm high) were smaller than those formed with polyvalent trehalose (9-1). | ||
| Fig. 5 AFM images of the aggregates of Aβ(1-40) obtained by incubation of Aβ (23 μM) at pH 7.4 and 37 °C with 400 rpm shaking for 48 h. The images correspond to (a) without additives (Aβ only), (b) addition of 9-1 (10 mM), (c) addition of 17-1 (10 mM), and (d) addition of 25-1 (10 mM). | ||
 | ||
| Fig. 6 AFM images of the aggregates of Aβ(1-40) with 1 (10 mM) obtained by incubation of Aβ (23 μM) at pH 7.4 and 37 °C with 400 rpm shaking for 48 h. The images show (a) fibrils and (b) pseudo-spherical objects. | ||
Polyvalent disaccharides 17-1 and 25-1 reduced the size of Aβ fibrils (Fig. 5c and d), but only fibrillar aggregates were found. The fibrils were smaller (length × height × width = 0.25–2.0 μm × 81–138 nm × 8.5–11 nm for 17-1, and 0.31–2.1 μm × 56–82 nm × 3.3–8.6 nm for 25-1) than those for the control sample.
In vitro neutralization of Aβ toxicity with polyvalent disaccharide additives
Addition of Aβ reduced HeLa cell viability to 62% due to the cytotoxicity of Aβ (Fig. 7). Addition of trehalose (1) either did not reduce the cytotoxicity of Aβ or slightly increased the toxicity (Fig. S4†). The addition of polyvalent trehalose (9-1), however, increased cell viability to 78%, indicating neutralization of the toxicity of Aβ40. We calculated p values of cell viability with Aβ and that with Aβ in the presence of 9-1 using the Student's t-test. The p value (p = 0.010) indicated significant reduction of Aβ cytotoxicity by addition of 9-1.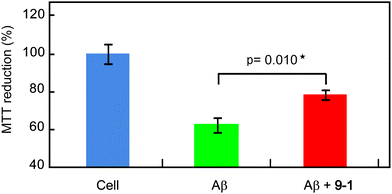 | ||
| Fig. 7 Toxicity of Aβ(1-40) aggregates formed by pre-incubation of Aβ (23 μM) at 37 °C for 24 h in the absence and presence of polyvalent trehalose (10 mM). Values of p were calculated using the Student's t-test. The data represent mean ± standard error of the mean. | ||
Discussion
Addition of disaccharides changed the time-course of ThT fluorescence. Though polyvalent trehalose inhibited the Aβ aggregation trehalose did not inhibit Aβ aggregation. In fact trehalose accelerated aggregation via more rapid nucleation (Fig. 2). The inhibition effect of trehalose was weak even at higher concentration (>100 mM). The results of these experiments differed from previously reported results,11,12 and from our previous results,27 though the experiments were replicated at least three times. A possible source of those differences was the experimental procedure. In the present study, we incubated Aβ40 and trehalose with shaking at 400 rpm without pre-incubation. Incubation with shaking accelerated amyloidosis because of the increased frequency of collisions. Pre-incubation of Aβ40 with trehalose was carried out by the other groups,11,12 which suggests a weak interaction between Aβ and trehalose. Our present procedure might suggest weak interaction of Aβ and trehalose, and another group reported similar efficacy of inhibition to protein aggregation.33In contrast to monomeric trehalose, polyvalent trehalose (9-1) strongly inhibited Aβ aggregation in both nucleation and fibril elongation. The results suggest that the cause of the weak inhibitory effect of trehalose was the lack of affinity of monomeric trehalose with Aβ, and that polyvalent trehalose showed a strong inhibitory effect due to its multivalency. We have reported inhibition of Aβ aggregation using glycopolymers carrying trehalose, maltitol and lactitol with alkyl side chains.27 In that case, the inhibitory effects of glycopolymers were related to micelle formation of glycopolymer and the biological activity of sugars, which was difficult to analyze. In the present study, glycopolymers with shorter linker and different sugar ratios were synthesized in order to estimate the inhibitory effect. The results indicate that the glycopolymer inhibition effect was different for each saccharide and sugar content.
Each disaccharide exhibited an inhibitory effect on Aβ40 aggregation, though the monomeric disaccharides showed weak affinities (Fig. 3). Of the monomeric disaccharides, maltose (10) showed the strongest and trehalose (1) the weakest inhibitory effect. The structures of the disaccharides are similar, but they have hydroxy groups with specific orientations that induce the specific inhibitory effects. However, the inhibitory effects of polyvalent disaccharides were larger than those of the monomeric disaccharides based on the multivalent effect, and were in the order polyvalent trehalose > lactose > maltose. The sugar content of the glycopolymer was also critical; polyvalent trehaloses with moderate sugar content (10 and 20%) showed stronger inhibitory effects than those with higher sugar content (63 and 100%). The polymers with higher sugar content had weaker interaction with Aβ due to the steric hindrance of the bulky side chains. The error bars of the time-course curves (Fig. 2–4) are not shown for the kinetic curve fitting.4,5,27,28,33–36 The repeatability is shown in the ESI† (Fig. S4).
Two kinds of morphologies of fibrils and pseudo-spherical aggregates were observed on addition of trehalose (1) and polyvalent trehalose (9-1), and confirmed by replicated experiments. On the other hand, other saccharides (2 and 3) and their polyvalent counterparts (17-1 and 25-1) induced formation of only fibrils. Since pseudo-spherical aggregates were obtained only with the addition of trehalose and polyvalent trehalose, it is conceivable that the interaction between Aβ and trehalose induced pseudo-spherical aggregates. Both fibril and pseudo-spherical morphologies were found, because the interaction between Aβ and trehalose was weak, and not all of the Aβ was induced to form pseudo-spherical aggregates by trehalose. It is possible that pre-incubation of Aβ with trehalose induces the formation of more pseudo-spherical aggregates.
It is noteworthy that the pseudo-spherical morphologies induced by trehalose and polyvalent trehalose were very different from those produced by the other disaccharide and polyvalent disaccharide additives, although the time-course of ThT fluorescence did not show much difference from that with other disaccharide and polyvalent disaccharide additives.
Polyvalent trehalose has the ability to reduce the cytotoxicity of Aβ, while trehalose had no effect or slightly increased the cytotoxicity. The pseudo-spherical aggregates might be composed of Aβ oligomers which induce cytotoxicity, because it has been reported that amylospheroids exhibit stronger cytotoxicity.37 The size of the rounded objects formed with trehalose and polyvalent trehalose was much larger than that of reported amylospheroids. However, Aβ aggregates with trehalose showed stronger cytotoxicity than the control, because the size of the Aβ aggregates was smaller than in the absence of trehalose and in the presence of polyvalent trehalose. It is suggested that Aβ aggregate cytotoxicity is strongly linked to both aggregate size and morphology. The relationship between cytotoxicity and trehalose additives is currently being investigated.
Specific functionality of trehalose was found by AFM observations and cytotoxicity tests, and the morphology change induced by trehalose was noteworthy. A previous report showed only an inhibitory effect on protein aggregation by trehalose (although spherical morphology was suggested by the reported data).11,33 In our experiments, trehalose not only inhibited aggregation, but also induced a specific morphology. Modification of trehalose at the C6 position did not reduce the inhibitory effect; in fact polyvalent trehalose amplified the inhibitory effect by multivalency. The conformation of trehalose is rigid due to the α(1-1) bond.38 Consequently, the modification of trehalose preserved and amplified its inhibitory effect on protein aggregation. Even though trehalose and maltose are composed of the same saccharide (“glucose”), their effects on protein aggregation were very different. The difference in the structures of trehalose and maltose is the glycoside bond between α(1-1) and α(4-1), hence the position of the glycoside is the most important factor for protein stabilization.
It has been reported, as noted above, that addition of trehalose prevents protein denaturation by hydrogen bonding to proteins15,16 and the hydration shell of proteins.17,39,40 Trehalose does not form strong hydrogen bonds, and so tends to stabilize oligomers of the protein, which is different from the mode of action of the other disaccharides. Previous studies suggested that the addition of trehalose prevents formation of the cross β-sheet structure of amyloid, but induces the parallel β-sheet structure.33 This effect might apply to Aβ aggregation, and the unique characteristics of trehalose induce oligomer formation. Although the conformation of Aβ in the presence of polyvalent trehalose has not been elucidated, the induced conformation should be related to that formed with monomeric trehalose. We propose that the difference in the size of the pseudo-spherical aggregates formed in the presence of polyvalent and monomeric trehalose is due to multivalency and the localized condensed trehalose.
The cytotoxicity reducing ability of polyvalent trehalose was modest, compared with other Aβ inhibitors. But trehalose and polyvalent trehalose have the advantage, compared with other inhibitors, of inhibiting the aggregation of various proteins via hydrogen bonding. The present experiments were limited to inhibition of Aβ aggregation, but the polyvalent trehalose is applicable to various proteins. Polyvalent trehalose can be utilized as a protein stabilizer for applications such as food, biosensing and protein drugs.
Conclusions
In this investigation the biological activity of multivalent trehalose toward Aβ(1-40) was quantified. We synthesized polyvalent disaccharides of trehalose, maltose and lactose, and found that the polyvalent disaccharides showed the stronger inhibitory activity toward aggregation of the protein, because of their multivalency. Of the three polyvalent disaccharides investigated, polyvalent trehalose had the strongest inhibitory activity toward Aβ aggregation, and decreased the cytotoxicity of Aβ(1-40) aggregates. Addition of trehalose and polyvalent trehalose induced the formation of specific pseudo-spherical aggregates, which had very different morphology from that of the aggregates formed by the other disaccharide additives. We believe that the morphology and aggregation are controlled by the addition of polyvalent trehalose.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (20106003).Notes and references
- M. P. Mattson, Nature, 2004, 430, 631 CrossRef CAS.
- H. Li, B. H. Monien, E. A. Fradinger, B. Urbanc and G. Bitan, Biochemistry, 2010, 49, 1259 Search PubMed.
- H. Zhu, J. Yu and M. S. Kindly, Mol. Med., 2001, 7, 517 Search PubMed.
- K. Ono, K. Hasegawa, H. Naiki and M. Yamada, Neurochem. Int., 2006, 48, 275 Search PubMed.
- B. Klajnert, M. Cortijo-Arellano, M. Bryszewska and J. Cladera, Biochem. Biophys. Res. Commun., 2006, 339, 577 CrossRef CAS.
- J. R. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030 CrossRef CAS.
- F. Albertorio, V. A. Chapa, X. Chen, A. J. Diaz and P. S. Cremer, J. Am. Chem. Soc., 2007, 129, 10567 CrossRef CAS.
- B. Zhu, T. Furuki, T. Okuda and M. Sakurai, J. Phys. Chem. B, 2007, 111, 5542 Search PubMed.
- L. M. Crowe, D. S. Reid and J. H. Crowe, Biophys. J., 1996, 71, 2087 CrossRef CAS; A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carroll, Glycobiology, 2003, 13, 17R CrossRef CAS.
- F. Beranger, C. Crozet, A. Goldsborough and S. Lehmann, Biochem. Biophys. Res. Commun., 2008, 374, 44 Search PubMed.
- R. Liu, H. Barkhordarian, S. Emadi, C. B. Park and M. R. Sierks, Neurobiol. Dis., 2005, 20, 74 CrossRef CAS.
- A. Watanbe, Y. Okahata, H. Furusawa, M. Hoshi and M. Sakurai, Cryobiology and Cryotechnology, 2005, 51, 137 Search PubMed.
- A. Arora, C. Ha and C. B. Park, FEBS Lett., 2004, 564, 121 CrossRef CAS.
- M. Tanaka, Y. Machida, S. Niu, T. Ikeda, N. R. Jana, H. Doi, M. Kurosawa, M. Nekooki and N. Nukina, Nat. Med., 2004, 10, 148 CrossRef CAS.
- S. D. Allison, B. Chang, T. W. Randolph and J. F. Carpenter, Arch. Biochem. Biophys., 1999, 365, 289 CrossRef CAS.
- F. Francia, M. Dezi, A. Mallardi, G. Palazzo, L. Cordone and G. Venturoli, J. Am. Chem. Soc., 2008, 130, 10240 CrossRef CAS.
- P. S. Belton and A. M. Gil, Biopolymers, 1994, 34, 957 CrossRef CAS.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321 CrossRef CAS.
- J. E. Gestwicki, C. W. Cario, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922 CrossRef CAS.
- A. Tsuchida, K. Kobayashi, N. Mathubara, T. Suzuki and Y. Suzuki, Glycoconjugate J., 1998, 15, 1047 CrossRef CAS.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823 CrossRef CAS; C. R. Becer, M. I. Gibson, J. Geng, R. llyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130 CrossRef CAS.
- Y. Miura, T. Yamauchi, H. Sato and T. Fukuda, Thin Solid Films, 2008, 516, 2443 CrossRef CAS.
- T. Fukuda, S. Onogi and Y. Miura, Thin Solid Films, 2009, 518, 880 CrossRef CAS.
- C. Unverzagt, S. Kelm and J. C. Paulson, Carbohydr. Res., 1994, 251, 285 CrossRef CAS.
- Y. Miura, K. Yasuda, K. Yamamoto, M. Koike, Y. Nishida and K. Kobayashi, Biomacromolecules, 2007, 8, 2129 CrossRef CAS.
- Y. Miura and H. Mizuno, Bull. Chem. Soc. Jpn., 2010, 83, 1004 Search PubMed.
- Y. Miura, C. You and R. Ohnishi, Sci. Technol. Adv. Mater., 2008, 9, 24407 Search PubMed.
- S. S. S. Wang, Y. T. Chen and S. W. Chou, Biochim. Biophys. Acta, 2005, 1741, 307 Search PubMed.
- N. Tanaka, R. Tanaka, M. Tokuhara, S. Kunugi, Y. F. Lee and D. Hamada, Biochemistry, 2008, 47, 2961 Search PubMed.
- R. Sabate, M. Gallardo and J. Estelrich, Biopolymers, 2003, 71, 190 Search PubMed.
- S. Nishimura, T. Murasugi, T. Kubo, I. Kaneko, M. Meguro, S. Marumoto, H. Kogen, K. Koyama, T. Oda and Y. Nakagami, Basic Clin. Pharmacol. Toxicol., 2003, 93, 29 Search PubMed.
- I. Kaneko, N. Yamada, Y. Sakurada, M. Kamenosono and S. Tutumi, J. Neurochem., 1995, 65, 2585 Search PubMed.
- S. Vilasi, C. Iannuzzi, M. Portaccio, G. Irace and I. Sirangelo, Biochemistry, 2008, 47, 1789 Search PubMed.
- M. R. Nichols, M. A. Moss, D. K. Reed, J. H. Hoh and T. L. Rosenberry, Microsc. Res. Tech., 2005, 67, 164 CrossRef CAS.
- J. Bieschke, S. J. Siegel, Y. W. Fu and J. W. Kelly, Biochemistry, 2008, 47, 50 Search PubMed.
- F. Attanasio, S. Cataldo, S. Fisichella, S. Nicoletti, V. G. Nicoletti, B. Pignataro, A. Savarino and E. Rizzarelli, Biochemistry, 2009, 48, 6522 CrossRef CAS.
- M. Hoshi, M. Sato, S. Matsumoto, A. Noguchi, K. Yasutake, N. Yoshida and K. Sato, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6370 CrossRef CAS.
- Y. Choi, K. W. Cho, K. Jeong and S. Jung, Carbohydr. Res., 2006, 341, 1020 Search PubMed.
- F. F. Liu, X. Y. Dong and Y. Sun, J. Mol. Graphics Modell., 2008, 27, 421 Search PubMed.
- F. F. Liu, L. Ji, X. Y. Dong and Y. Sun, J. Phys. Chem. B, 2009, 113, 11320 Search PubMed.
Footnote |
| † Electronic supplementary information available: The syntheses of samples, and additional data from ThT assays and MTT assay. See DOI: 10.1039/c1py00072a |
| This journal is © The Royal Society of Chemistry 2011 |