Synthesis of water-soluble poly[oligo(ethylene glycol) methacrylate]s by living anionic polymerization of oligo(ethylene glycol) vinyl ether methacrylates
Jun
Yamanaka
a,
Takashi
Kayasuga
a,
Mana
Ito
a,
Hideaki
Yokoyama
b and
Takashi
Ishizone
*a
aDepartment of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1-S1-13 Ohokayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: tishizon@polymer.titech.ac.jp
bDepartment of Advanced Materials Science, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwa-no-ha, Kashiwa, Chiba 277-8561, Japan
First published on 27th May 2011
Abstract
Anionic polymerizations of 2-(2-vinyloxyethoxy)ethyl methacrylate (V2), 2-[2-(2-vinyloxyethoxy)ethoxy]ethyl methacrylate (V3), and 2-[2-(2-(2-vinyloxyethoxy)ethoxy)ethoxy]ethyl methacrylate (V4) were carried out with either 1,1-diphenyl-3-methylpentyllithium/lithium chloride or diphenylmethylpotassium/diethylzinc in THF at −78 °C for 2–20 h. All the anionic polymerizations of V2–V4 chemoselectively proceeded on the methacryloyl groups, while the vinyloxyl groups in the side chains were intact during the polymerization. The resulting polymers possessed the predicted molecular weights based on the molar ratios between monomers and initiators and narrow molecular weight distributions (Mw/Mn < 1.1). The vinyloxyl groups in the side chain terminals of anionically obtained poly(V2–V4) were quantitatively hydrolyzed with aqueous HCl in THF to afford a series of well-defined poly[oligo(ethylene glycol) methacrylate]s, poly(OH2–OH4), possessing OH groups in the side chains. The hydrolyzed poly(OH2–OH4) showed water solubility, while the poly(V2–V4) having vinyloxyl protecting groups in the side chains were insoluble in water. On the other hand, cationic polymerization of V2 exclusively occurred on a vinyloxyl group with 1-isobutoxyethyl acetate in toluene at 25 °C in the presence of ethylaluminium dichloride and THF to give a polymer of controlled Mn and relatively narrow molecular weight distribution (Mw/Mn < 1.2).
Introduction
A variety of water-soluble polymers with precisely controlled chain structures, such as predicted molecular weight and narrow molecular weight distribution (MWD), have been intensely prepared, since they are very attractive for fundamental research as well as industrial applications, such as dispersants, stabilizers, emulsifiers, and flocculants.1 Among the water-soluble polymers, polymethacrylates can be variously designed to provide water solubility by introducing suitable hydrophilic polar functions in the ester moieties. For example, tailored water-soluble polymethacrylates carrying 2,3-dihydroxypropyl2–5 and 2-(N,N-dialkylamino)ethyl groups6–8 and glucose functionality9,10 have been prepared via various living polymerizations.We have successfully synthesized well-defined poly[di(ethylene glycol) methacrylate] poly(OH2) and poly[tri(ethylene glycol) methacrylate] poly(OH3) via living anionic polymerizations of the corresponding tert-butyldimethylsilyl (TBDMS) ether monomers, (Si2) and (Si3), and the following deprotection of the TBDMS protecting groups (Chart 1).11 The resulting poly[oligo(ethylene glycol) methacrylate]s, poly(OEGMA), show excellent solubility in water at any temperature, while the ethylene glycol ester counterpart, poly(2-hydroxyethyl methacrylate), poly(OH1),12–16 is highly hydrophilic but insoluble in water. Protection of the acidic OH functionality prior to anionic polymerization and deprotection of the trialkylsilyl groups after polymerization are required to obtain water-soluble poly(OH2) and poly(OH3). Unfortunately, further investigation of polymers possessing oligo(ethylene glycol) units of more than three has been hindered by the difficulty in purifying the protected monomers because of their very high boiling points. Recently, we succeeded in the living anionic polymerizations of a series of methyl ethers of OEGMA, M1–M4, and their ethyl ether counterparts, E1–E4 (Chart 1).17–20 The resulting poly(M2–M4) and poly(E2–E4) were soluble in water, and their aqueous solutions showed typical reversible cloud points (Tcs) between 4 and 68 °C, depending on the side chain length and the ω-alkyl functionality. Polymers possessing longer side chains show higher Tc values than the shorter ones, and the Tcs of the methyl ethers are ca. 25 °C higher than the ethyl ethers. The polymers of ethylene glycol esters, poly(M1) and poly(E1), are insoluble in water, as predicated from the solubility of poly(OH1). These clearly indicate the significant effect of the side chain length of the oligo(ethylene glycol) unit and the ω-functionality on the water-solubility and the thermosensitivity of polymethacrylates.
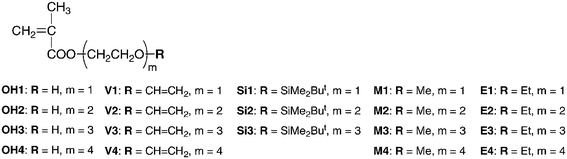 | ||
| Chart 1 Oligo(ethylene glycol) methacrylates. | ||
2-Vinyloxyethyl methacrylate (V1)21–23 having a methacryloyl and a vinyl ether polymerizable moiety is a typical bifunctional monomer similar to glycidyl methacrylate,24,25 1-azilidinylethyl methacrylate,26 and p-styryloxazoline.27 The chemoselective cationic and anionic polymerizations of V1 afforded a well-defined polymer with a methacryloyl side chain and a vinyloxyl side chain, respectively.22 The anionically obtained poly(V1) corresponds to a vinyl ether derivative of poly(OH1). The electron-rich vinyl ether moieties are known to react with alcohols and carboxylic acids to form acetals and hemiacetal esters. Furthermore, the vinyloxyl group can be converted into an OH group via acidic hydrolysis along with the formation of carbonyl compounds. This means that the vinyl ether moiety is a versatile protecting group for the OH functionality, similar to TBDMS ether. In fact, Ruckenstein and Zhang have succeeded in the synthesis of poly(OH1) via the anionic polymerization of V1 and subsequent acidic hydrolysis of the vinyloxyl group.23
In this study, we focus on the chemoselective anionic polymerizations of a series of novel oligo(ethylene glycol) vinyl ether methacrylates, 2-(2-vinyloxyethoxy)ethyl methacrylate (V2), 2-[2-(2-vinyloxyethoxy)ethoxy]ethyl methacrylate (V3), and 2-[2-(2-(2-vinyloxyethoxy)ethoxy)ethoxy]ethyl methacrylate (V4) (Chart 1), to synthesize linear polymethacrylates with tailored chain structures and reactive vinyl ether moieties in their side chains. The following direct acidic hydrolysis of poly(V2–V4) affords well-defined poly(OEGMA)s, poly(OH2–OH4), possessing OH terminals. The present method using the vinyloxyl group provides a novel alternative synthetic route for water-soluble poly(OH2–OH4) as well as the previous pathway using the bulky TBDMS group.11 We also examine the radical and cationic polymerizations of V2 to clarify its polymerizability as a bifunctional monomer. Furthermore, the solubility of anionically produced poly(V2–V4) is of great interest, since the corresponding ethyl ether poly(E2–E4)s show water-solubility and thermosensitivity.18
Results and discussion
Synthesis of monomers
The monomers were synthesized by a reaction of methacryloyl chloride and the corresponding oligo(ethylene glycol) mono vinyl ethers in the presence of triethylamine in diethyl ether (Scheme 1). We synthesized the vinyl ethers of tri(ethylene glycol) and tetra(ethylene glycol) as important precursors for V3 and V4, whereas 2-vinyloxyethanol and 2-(2-vinyloxyethoxy)ethanol were commercially available. At first, we examined the Williamson reaction of di- or tri(ethylene glycol) and 2-chloroethyl vinyl ether with sodium hydride. However, the yields were disappointingly low and the products were contaminated with the starting materials and by-products, such as the divinyl ethers of tetra(ethylene glycol) and penta(ethylene glycol). These troublesome impurities might act as terminators or cross-linking reagents in the polymerizations. We then attempted a two-step synthesis via monochlorination of tetra- or penta(ethylene glycol) with thionyl chloride28 and the following dehydrochlorination of chloroethyl ethers,29 as shown in Scheme 1. Although the first-step reaction suffered from dichlorination, the monochloroethyl ethers could be isolated from the starting diols and the resultant dichlorides by column chromatography. The second-step dehydrochlorination of the monochloroethyl ethers proceeded smoothly with potassium tert-butoxide to give the vinyl ethers of tri(ethylene glycol) and tetra(ethylene glycol) in 32 and 34% yields, respectively. We thus selected the latter synthetic route to prepare alcohols with vinyloxyl terminal groups from the viewpoints of total yield and ease of purification. After the esterification of the alcohols, the resulting methacrylate monomers were thoroughly purified by column chromatography and by fractional vacuum distillation prior to polymerization.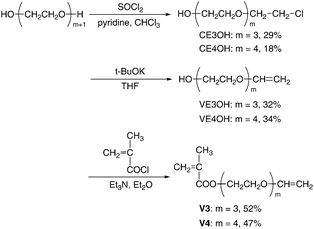 | ||
| Scheme 1 Synthesis of V3 and V4. | ||
Anionic polymerizations of V1–V4
The anionic polymerizations of V2–V4 were carried out in THF at −78 °C for 2–20 h with either diphenylmethylpotassium (Ph2CHK) or 1,1-diphenyl-3-methylpentyllithium (DMPLi), a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct of sec-BuLi and 1,1-diphenylethylene, as the initiator. The polymerization of V1 was also attempted to compare its polymerizability under similar conditions. In most cases, diethylzinc (Et2Zn) or LiCl was added to the initiator, since they were effective to tune the anionic polymerization of (meth)acrylates30–33 and N,N-dialkylacrylamides.34–38 When a THF solution of monomers was added to the initiator solution, the red color of the initiator rapidly disappeared to give a colorless solution, indicating instantaneous initiation. All polymerizations of V1–V4 proceeded homogeneously in THF at −78 °C. After quenching the polymerization with methanol, the conversion of monomers was estimated by 1H NMR spectroscopy. 1H and 13C NMR analysis of the resulting polymers revealed that anionic polymerizations of V1–V4 proceeded exclusively in the methacryloyl moieties, while the vinyloxyl groups in the side chains remained intact (Fig. 1 and 2). The molecular weights of poly(V1–V4) were determined by end-group analysis using the signal intensity ratio between the aromatic proton of the initiator residue and the α-methyl proton of the main chain. The results of polymerization are summarized in Table 1.
:1 adduct of sec-BuLi and 1,1-diphenylethylene, as the initiator. The polymerization of V1 was also attempted to compare its polymerizability under similar conditions. In most cases, diethylzinc (Et2Zn) or LiCl was added to the initiator, since they were effective to tune the anionic polymerization of (meth)acrylates30–33 and N,N-dialkylacrylamides.34–38 When a THF solution of monomers was added to the initiator solution, the red color of the initiator rapidly disappeared to give a colorless solution, indicating instantaneous initiation. All polymerizations of V1–V4 proceeded homogeneously in THF at −78 °C. After quenching the polymerization with methanol, the conversion of monomers was estimated by 1H NMR spectroscopy. 1H and 13C NMR analysis of the resulting polymers revealed that anionic polymerizations of V1–V4 proceeded exclusively in the methacryloyl moieties, while the vinyloxyl groups in the side chains remained intact (Fig. 1 and 2). The molecular weights of poly(V1–V4) were determined by end-group analysis using the signal intensity ratio between the aromatic proton of the initiator residue and the α-methyl proton of the main chain. The results of polymerization are summarized in Table 1.
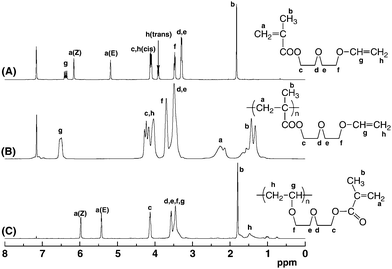 | ||
| Fig. 1 1H NMR spectra of V2 (A) measured in C6D6, anionically obtained poly(V2) (B) measured in C6D6, and cationically obtained poly(V2) (C) measured in CDCl3. | ||
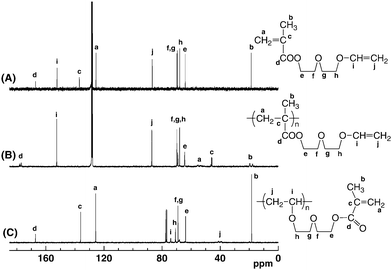 | ||
| Fig. 2 13C NMR spectra of V2 (A) measured in C6D6, anionically obtained poly(V2) (B) measured in C6D6, and cationically obtained poly(V2) (C) measured in CDCl3. | ||
| Run | Monomer type/mmol | Initiator type/mmol | Additive type/mmol | Time/h | Conversiona (%) | 10−3Mn | M w/Mnd | Tacticitye (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calcdb | Obsdc | mm | mr | rr | |||||||
| a Estimated by 1H NMR. b M n(calcd) = (Mw of monomer) × [M]/[I] × conversion/100 + Mw of initiator. c M n(obsd) was determined by end group analysis by 1H NMR. d M w/Mn was determined by SEC calibration using polystyrene standards in THF. e The triad tacticity was determined by the 1H NMR signal intensity of α-methyl proton (mm = 1.25 ppm, mr = 1.05 ppm, rr = 0.90 ppm) of polymers in CDCl3. f Diphenylmethylpotassium. g 1,1-Diphenylethylene. | |||||||||||
| 1 | V1, 3.82 | Ph2CHK,f 0.0739 | 3 | 73 | 5.9 | 7.0 | 1.56 | 13 | 55 | 32 | |
| 2 | V1, 3.41 | Ph2CHK, 0.0532 | Et2Zn, 0.945 | 2 | 90 | 9.0 | 11 | 1.11 | 11 | 55 | 34 |
| 3 | V1, 6.17 | Ph2CHK, 0.108 | Et2Zn, 1.22 | 20 | 100 | 9.0 | 9.2 | 1.17 | 13 | 56 | 31 |
| 4 | V2, 3.25 | Ph2CHK, 0.0634 | 2 | 93 | 9.3 | 11 | 1.53 | 19 | 49 | 33 | |
| 5 | V2, 2.51 | Ph2CHK, 0.0629 | Et2Zn, 0.923 | 3 | 100 | 8.0 | 11 | 1.03 | 16 | 51 | 33 |
| 6 | V2, 4.16 | Ph2CHK, 0.0822 | Et2Zn, 1.25 | 3 | 94 | 9.4 | 10 | 1.06 | 18 | 52 | 30 |
| 7 | V2, 3.95 | Ph2CHK, 0.0387 | Et2Zn, 0.568 | 3 | 100 | 21 | 27 | 1.06 | 10 | 49 | 41 |
| 8 | V3, 1.63 | Ph2CHK, 0.0449 | 3 | 91 | 8.2 | 9.1 | 1.48 | 8 | 37 | 54 | |
| 9 | V3, 2.63 | Ph2CHK, 0.0802 | Et2Zn, 1.04 | 3 | 100 | 8.1 | 7.7 | 1.09 | 6 | 43 | 51 |
| 10 | V3, 3.44 | Ph2CHK, 0.073 | Et2Zn, 1.00 | 6 | 100 | 12 | 14 | 1.05 | 6 | 40 | 54 |
| 11 | V4, 1.90 | Ph2CHK, 0.0831 | Et2Zn, 1.18 | 3 | 100 | 6.8 | 7.2 | 1.07 | 9 | 38 | 53 |
| 12 | V4, 2.04 | Ph2CHK, 0.0437 | Et2Zn, 0.455 | 3 | 100 | 14 | 17 | 1.01 | 9 | 34 | 57 |
| 13 | V1, 3.29 | sec-BuLi, 0.0629/DPE,g 0.113 | LiCl, 0.257 | 20 | 100 | 8.4 | 10 | 1.16 | 5 | 28 | 67 |
| 14 | V2, 3.13 | sec-BuLi, 0.0627/DPE, 0.113 | 2 | 95 | 9.5 | 12 | 1.24 | 6 | 32 | 62 | |
| 15 | V2, 2.74 | sec-BuLi, 0.0512/DPE, 0.112 | LiCl, 0.302 | 3 | 86 | 9.5 | 13 | 1.03 | 1 | 26 | 73 |
| 16 | V2, 3.38 | sec-BuLi, 0.0609/DPE, 0.0957 | LiCl, 0.441 | 20 | 100 | 11 | 14 | 1.03 | 1 | 29 | 70 |
| 17 | V2, 4.61 | sec-BuLi, 0.0427/DPE, 0.111 | LiCl, 0.213 | 20 | 100 | 22 | 42 | 1.06 | |||
| 18 | V3, 2.62 | sec-BuLi, 0.0622/DPE, 0.0844 | LiCl, 0.201 | 20 | 100 | 12 | 13 | 1.05 | 4 | 27 | 69 |
| 19 | V3, 2.93 | sec-BuLi, 0.0675/DPE, 0.163 | LiCl, 0.225 | 20 | 88 | 9.7 | 13 | 1.06 | 7 | 28 | 65 |
| 20 | V4, 2.46 | sec-BuLi, 0.0762/DPE, 0.114 | LiCl, 0.372 | 20 | 100 | 9.6 | 17 | 1.07 | 5 | 25 | 70 |
We first attempted to polymerize V1–V3 with Ph2CHK in THF at −78 °C. The conversions of V1–V3 were 73–93% within 3 h. The resulting poly(V1–V3) had the predicted molecular weights based on the molar ratio between the monomers and Ph2CHK. Size exclusion chromatography (SEC) traces of the polymers showed that the MWDs were broad (Mw/Mn = 1.48–1.56). On the other hand, the addition of Lewis acidic Et2Zn to the polymerization system induced the quantitative conversions and effectively narrowed the MWDs of poly(V1–V3) in addition to the attainment of molecular weight control.30,31 The Mw/Mn values were reduced to less than 1.1 in most cases. This binary initiator system of Ph2CHK/Et2Zn is also effective to afford poly(V4)s with tailored molecular weights and narrow MWDs. The polymerization behavior of V1–V4 is in good accordance with our previous results of various (meth)acrylate monomers including trialkylsilyl-protected OEGMAs, Si2 and Si3,11 and the methyl and ethyl ethers of OEGMAs, M1–M4 and E1–E4.17,20 The drastic narrowing of MWDs could be attained, since the added Lewis acidic Et2Zn associated with the propagating enolate anions reduces the polymerization rate and the nucleophilicity.
The polymerizations of V2–V4 were next attempted with an organolithium initiator, DMPLi, in THF at −78 °C. Controlled polymerization of V1 has been already achieved with 1,1-diphenylhexyllithium/LiCl in THF at −60 °C.22 Similar to the results of V1, V2–V4 also underwent anionic polymerizations quantitatively in the methacryloyl moieties (runs 15–20). The polymerizations were completed in THF at −78 °C within 20 h, when a 3–8-fold amount of LiCl was added to DMPLi. SEC traces of the polymers showed unimodal and narrow MWDs, and the Mw/Mn values were 1.03–1.07. The observed Mns of the polymers agreed well with the predicted ones from the molar ratios between the monomers and DMPLi.39 On the other hand, poly(V2), having a rather broad MWD, Mw/Mn = 1.24, was produced in 95% yield within 2 h, when we carried out the polymerization of V2 in the absence of LiCl (run 14). Similar additive effect of LiCl on the narrowing of MWDs has been previously reported in the anionic polymerizations of various (meth)acrylate monomers, including V1,22Si2 and Si3,11M1–M4, and E1–E4.17,20 The plausible explanation for the additive effect of LiCl involves the reduction of the polymerization rate, the lowered nucleophilicity of the propagating chain end, and the equilibrium shift from the associated state toward the non-associated species in the presence of a common ion salt.40,41
We then estimated the stereoregularity of the polymers by the relative signal intensity of an α-methyl proton appearing at 0.90–1.25 ppm in the 1H NMR spectra measured in CDCl3. The tacticities in the triad are also shown in Table 1. The poly(V1–V4) produced with an organolithium initiator in THF always possessed rr-rich configurations regardless of the side chain lengths of the oligo(ethylene glycol) units. The presence of LiCl as the common ion did not affect the stereoregularity of poly(V2). These findings are consistent with previous reports for a variety of polymethacrylates produced under similar polymerization conditions (obtained with organolithium in THF at −78 °C).11,17,20,22,33
On the other hand, poly(V1) and poly(V2) obtained with either Ph2CHK or Ph2CHK/Et2Zn had mr-rich configurations. By contrast, the rr-triads were dominant in the cases of poly(V3) and poly(V4) carrying longer oligo(ethylene glycol) units. Similar tendency in changing the stereoregularity in the side chain length has been observed in the polymerizations of M1–M4 and E1–E4 initiated with organopotassium initiator systems.17,20 If the number of ethylene glycol units was below three, the tacticity of polymer was always mr-rich, similar to the various poly(methyl methacrylate)s produced with organopotassium initiators.30 On the other hand, the polymers having longer side chain lengths had rr-rich configurations regardless of the ω-functionality of the oligo(ethylene glycol) unit. A possible explanation for this polymerization behavior is that an association of the multidentate oligo(ethylene glycol) ether moiety occurs with the potassium ion at the propagating chain end (Scheme 2), as is considered in cases of glymes. Since this association might be strongly affected by the side chain length, a clear effect on the stereoregularity of poly(OEGMA) is observed.
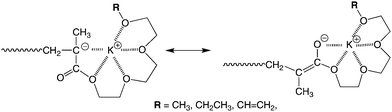 | ||
| Scheme 2 Plausible coordination structure at propagating chain end. | ||
Radical polymerization of V2
Free radical polymerization of V2 was next attempted in benzene at 70 °C for 3–24 h with azobis(isobutyronitrile) (AIBN) as the initiator (Table 2). The reaction mixture was homogeneous within 3 h, and 75% of V2 was consumed. However, an insoluble gel formed in the systems after 6 and 24 h. The 1H NMR spectrum of the THF-soluble part (run 22) was in accordance with that of poly(V2) obtained by anionic polymerization. For instance, the signals at 5.2 and 6.2 ppm derived from the methacryloyl group of V2 disappeared, and the signals of the vinyloxyl group (OCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) at 6.4–6.7 ppm were still present. The SEC traces of the soluble part of the resulting polymer showed multimodal peaks, and the Mw/Mn values were 6.8–12, indicating very broad MWDs. These facts suggest that the methacryloyl groups in V2 preferentially react to afford polymethacrylate at an early stage in the radical polymerization. However, the vinyloxyl groups in the side chain of poly(V2) were not completely intact toward the radical propagating species, and started to react after the consumption of methacryloyl group. The reaction between the vinyloxyl group and the propagating radical eventually gave an insoluble gel through cross-linking, although the reactivity of the vinyl ether moiety was significantly lower than that of the methacryloyl group. In fact, several research groups have succeeded in the chemoselective radical polymerization of the methacryloyl group in V1 monomer,21,22 similar to anionic polymerization.
CH2) at 6.4–6.7 ppm were still present. The SEC traces of the soluble part of the resulting polymer showed multimodal peaks, and the Mw/Mn values were 6.8–12, indicating very broad MWDs. These facts suggest that the methacryloyl groups in V2 preferentially react to afford polymethacrylate at an early stage in the radical polymerization. However, the vinyloxyl groups in the side chain of poly(V2) were not completely intact toward the radical propagating species, and started to react after the consumption of methacryloyl group. The reaction between the vinyloxyl group and the propagating radical eventually gave an insoluble gel through cross-linking, although the reactivity of the vinyl ether moiety was significantly lower than that of the methacryloyl group. In fact, several research groups have succeeded in the chemoselective radical polymerization of the methacryloyl group in V1 monomer,21,22 similar to anionic polymerization.
| Run | V2/mmol | AIBNa/mmol | Time/h | Conversionb (%) | 10−3Mn(obsd)c | M w/Mnc |
|---|---|---|---|---|---|---|
| a 2,2′-Azobisisobutyronitrile. b Estimated by 1H NMR. c M n(obsd) and Mw/Mn were determined by SEC calibration using polystyrene standards in THF. d Polymer yield was 75%. e The insoluble gel formed in the polymerization system. f M n(obsd) and Mw/Mn were determined by SEC calibration using polystyrene standards in DMF containing 0.01 M LiBr. | ||||||
| 21 | 7.30 | 0.616 | 3 | 76d | 11 | 12.2 |
| 22 | 7.00 | 0.175 | 6 | ∼100e | 40f | 6.77f |
| 23 | 2.10 | 0.0463 | 20 | ∼100e | — | — |
Cationic polymerization of V2
Cationic polymerization of V2 was finally examined in toluene at 25 °C for 24 h with 1-isobutoxyethyl acetate (IEA) in the presence of ethylaluminium dichloride (EtAlCl2) as the activator. A small amount of Lewis basic THF was added in order to tune the polymerization of the vinyl ether.22,42,43 After quenching the polymerization with methanol containing a small amount of aqueous ammonia, the conversion of the monomer was estimated by 1H NMR spectroscopy. The molecular weights of the resulting polymers were estimated by SEC measurement in THF. The results of the cationic polymerization of V2 are summarized in Table 3.| Run | Monomer type/mmol | Initiator type/mmol | THF/mmol | M/I | Time/h | Conversiona (%) | 10−3Mn | M w/Mnd | |
|---|---|---|---|---|---|---|---|---|---|
| Calcdb | Obsdc | ||||||||
| a Estimated by 1H NMR. b M n(calcd) = (Mw of monomer) × [M]/[I] × conversion/100 + Mw of initiator. c M n(obsd) was determined by end group analysis by 1H NMR. d M n(obsd) and Mw/Mn were determined by SEC calibration using polystyrene standards in THF. e 1-Isobutoxyethyl acetate. f At 0 °C. | |||||||||
| 24 | V2, 3.50 | IEA,e 0.184/EtAlCl2, 0.166 | 2.59 | 19 | 24 | 84 | 3.3 | 4.2 | 1.20 |
| 25 | V2, 4.02 | IEA, 0.163/EtAlCl2, 0.205 | 2.96 | 25 | 24 | 100 | 5.1 | 5.6 | 1.18 |
| 26 | V2, 4.31 | IEA, 0.0907/EtAlCl2, 0.126 | 1.90 | 48 | 24 | 85 | 8.2 | 8.0 | 1.18 |
| 27 | V2, 3.01 | IEA, 0.126/EtAlCl2, 0.662 | 11.1 | 24 | 24 | 100 | 4.9 | 3.6 | 1.52 |
| 28f | V2, 3.46 | IEA, 0.403/ZnCl2, 0.547 | — | 8 | 15 | 100 | 1.9 | 2.0 | 1.38 |
| 29f | V2, 3.06 | IEA, 0.168/ZnCl2, 3.41 | — | 18 | 15 | 100 | 3.8 | 1.7 | 1.61 |
The cationic polymerization of V2 in the vinyloxyl group proceeded exclusively, similar to the previous report for V1.22 As can be seen in Fig. 1C and 2C, the methacryloyl group of V2 is intact during the course of the cationic polymerization, in sharp contrast to the anionic and radical polymerization. A series of poly(vinyl ether)s possessing controlled Mns and relatively narrow MWDs were obtained in 84–100% yields from the reaction mixture (runs 24–26). However, the MWD of polymer became broad when the content of activator (EtAlCl2) increased (run 27). This is probably due to the rapid propagation of V2 under the aforementioned conditions. A similar cationic polymerization of V2 successfully proceeded with IEA/ZnCl2 in toluene at 0 °C for 15 h, although the molecular weight controls were not realized. These results clearly indicate that V2 certainly undergoes cationic polymerization in a chemoselective fashion in the vinyloxyl groups, as expected. The methacryloyl groups of V2 and the resulting poly(V2) were not attacked by the propagating cationic species during the course of polymerization.
Acetoxylation of vinyloxyl groups in poly(V2–V4)
The vinyloxyl groups, vinyl ether moieties, are known to readily react with various carboxylic acids to form corresponding hemiacetal ester functionalities through an addition reaction. Furthermore, the resulting hemiacetal esters are widely recognized as typical dormant species and versatile initiators for the cationic polymerization of vinyl ethers by activating them with Lewis acids.The reaction of the vinyloxyl groups in poly(V2–V4) with acetic acid proceeded in toluene at 70 °C for 5 h, as shown in Scheme 3. After the acetoxylation, the polymers were obtained in good yields and were characterized by 1H and 13C NMR measurements. The vinyloxyl groups in poly(V2) and poly(V3) were completely converted into hemiacetal esters after the addition of acetic acid, similar to the previous report for poly(V1).22Fig. 3A and B show the 1H NMR spectra of poly(V2) and the acetoxylated poly(V2). The proton signals due to the vinyloxyl group at 6.4 and 4.1 ppm disappear completely, and new proton signals of the OCH(OCOCH3)CH3 moiety alternatively appear at 6.1 (OCH), 1.8 (OCOCH3), and 1.2 ppm (CH3). The results of acetoxylation indicate the possibility of chemical modification of the polymers by treating them with various carboxylic acids. On the other hand, the acetoxylation reaction of poly(V4) was not complete, even if the reaction was attempted for a longer time in the presence of a large excess of acetic acid. The highest conversion of the vinyloxyl group in poly(V4) was 62%, while 38% of the vinyloxyl terminal groups were present. Although the reason for the incomplete acetoxylation is not clear now, the vinyloxyl terminal groups in the longer side chains might have lower reactivity compared to those in the shorter side chains.
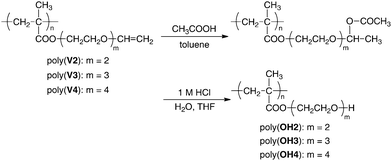 | ||
| Scheme 3 Reaction of polymers with acetic acid and subsequent acid hydrolysis. | ||
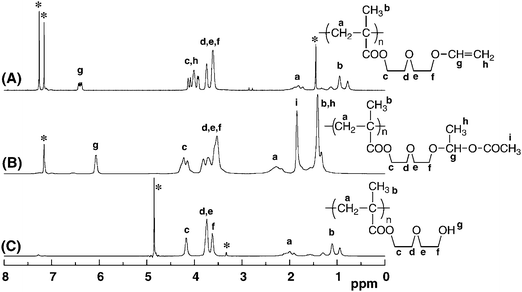 | ||
| Fig. 3 1H NMR spectra of anionically obtained poly(V2) (A) measured in C6D6, poly(V2) after reaction with acetic acid (B) measured in C6D6, and poly(OH2) (C) measured in CD3OD. The signals attributable to solvents are marked with asterisks (*). | ||
We then carried out the following acidic hydrolysis of acetoxylated polymers having a hemiacetal ester moiety to generate the hydroxyl functionalities, as shown in Scheme 3. The hydrolysis was performed with 2 M aqueous hydrochloric acid in THF at 0 °C for 1 h. Fig. 3B and C show the 1H NMR spectra before and after the acidic hydrolysis of acetoxylated poly(V2). After the acidic hydrolysis, the 1H NMR signals derived from the 1-acetoxyethoxyl group of the acetoxylated polymers at 1.2, 1.8, and 6.1 ppm completely disappear. In addition, broad IR absorption characteristic of the OH groups newly appeared at 3100–3700 cm−1 after hydrolysis of the polymers. It should be emphasized that the 1H and 13C NMR and IR spectra after the two-step hydrolysis of poly(V2) and poly(V3) were in good accordance with those obtained by deprotection of the TBDMS ether moiety for poly(Si2) and poly(Si3), respectively.11 Similar to the cases of poly(V2) and poly(V3), the complete acidic hydrolysis of the partially acetoxylated poly(V4) proceeded, although the yield of the first-step acetoxylation was 62%. This indicates that direct acidic hydrolysis of the vinyloxyl moiety is also possible to form the OH group, as well as the hydrolysis of the 1-acetoxyethoxyl group, as shown later.
We performed SEC measurements of the polymers in DMF containing 0.01 M LiBr. Fig. 4 shows typical SEC traces of poly(V2), the acetoxylated polymer, and poly(OH2) after the hydrolysis. The SEC trace of poly(V2) shifts toward a higher molecular weight region after acetoxylation by keeping a unimodal and narrow shape. The SEC trace of the hydrolyzed poly(OH2) again shows a unimodal and narrow MWD, as can be seen in Fig. 4C. The chromatogram of poly(OH2) slightly shifts from that of the acetoxylated poly(V3) to a higher molecular weight side in DMF, although the theoretical molecular weight certainly decreases via the transformation of the 1-acetoxyethoxyl group into an OH group. This curious elution behavior observed in SEC measurement is probably due to the difference in the hydrodynamic volume of the polymers before and after deprotection in a highly polar DMF solvent, as previously reported.11 A similar phenomenon was also observed during the course of the hydrolysis of acetoxylated poly(V3) and poly(V4) into poly(OH3) and poly(OH4), respectively. The intriguing point is that the SEC traces of poly(OH2–OH4) possess unimodal and narrow shapes similar to the chromatograms of the starting poly(V2–V4), indicating that no side reaction such as main chain degradation and/or cross-linking takes place during the course of acetoxylation and acid hydrolysis. These observations substantiate that a series of well-defined poly(OEGMA)s, poly(OH2–OH4), are obtained by the two-step polymer reaction. To the best of our knowledge, poly(OH4), poly[tetra(ethylene glycol) methacrylate], is newly obtained in the present paper.
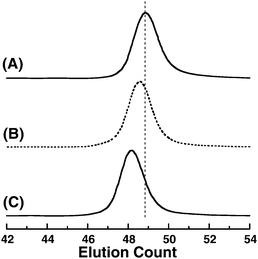 | ||
| Fig. 4 SEC traces of anionically obtained poly(V2) (peak A, Mn(SEC) = 23000, Mw/Mn = 1.09), poly(V2) after reaction with acetic acid (peak B, Mn(SEC) = 25000, Mw/Mn = 1.09), and, poly(OH2) (peak C, Mn(SEC) = 27000, Mw/Mn = 1.08) measured in DMF containing 0.01 M LiBr. | ||
Acidic hydrolysis of vinyloxyl group in poly(V2–V4)
In this section, the direct hydrolysis of the vinyloxyl group of poly(V2–V4) was examined to generate the OH functionalities by treatment with 1 M aqueous hydrochloric acid in THF at 0 °C for 1 h, as shown in Scheme 4. The solubility of poly(V2–V4) drastically changed after the acidic treatment, as shown in the next section. It was revealed by the 1H and 13C NMR and IR measurements that the polymers were completely hydrolyzed. For example, Fig. 5 clearly shows that the 1H NMR signals derived from the vinyloxyl group of the original poly(V4) at 4.1 and 6.4 ppm disappear after acidic hydrolysis. The proton signals of the tetra(ethylene glycol) units were observed at 3.4–4.1 ppm after deprotection, while the shape of signals slightly changed from that of poly(V4). The observed intensity relative to other signals suggested that the cleavage occurred exclusively in the vinyloxyl group but not on the ester linkage. In fact, the 1H NMR spectrum after acid hydrolysis was in good accordance with that after the two-step reaction of poly(V4), as discussed above. These points strongly support the formation of novel poly(OH4) via deprotection of the vinyloxyl groups in poly(V4).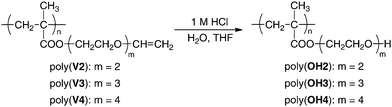 | ||
| Scheme 4 Acid hydrolysis of polymers. | ||
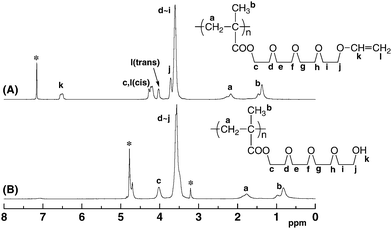 | ||
| Fig. 5 1H NMR spectra of anionically obtained poly(V4) (A) measured in C6D6 and poly(OH4) (B) measured in CD3OD. The signals attributable to solvents are marked with asterisks (*). | ||
Fig. 6 shows the typical SEC traces of poly(V4) and poly(OH4) after acidic hydrolysis. The SEC trace of poly(OH4) shifts from that of poly(V4) toward a higher molecular weight region after acid hydrolysis by keeping a unimodal and narrow shape, although the molecular weight of the hydrolyzed poly(OH4) theoretically decreases after the removal of the vinyl group. This strange elution behavior shown by SEC analysis is again due to the difference in the hydrodynamic volume of the polymers before and after deprotection in DMF, as discussed above in regard to the acidic hydrolysis of the acetoxylated polymers. In fact, the degree of polymerization of poly(OH4) estimated by 1H NMR (DP = 25.3) was in accordance with that of the starting poly(V4) (DP = 25.2). A similar elution behavior was also observed during the course of the transformation of poly(V2) and poly(V3) into poly(OH2) and poly(OH3), respectively. We thus confirmed that direct acidic hydrolysis of poly(V2–V4) proceeded smoothly without side reactions such as main chain degradation and/or cross-linking, since the deprotected poly(OH2–OH4) maintained unimodal and narrow MWDs. These spectroscopic observations substantiate that complete removal of the vinyl protecting groups in poly(V2–V4)s is attained to afford well-defined poly(OH2–OH4) under acidic conditions.
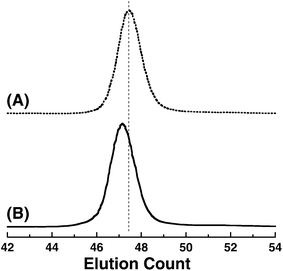 | ||
| Fig. 6 SEC traces of anionically obtained poly(V4) (peak A, Mn(SEC) = 34000, Mw/Mn = 1.07) and poly(OH4) obtained by the direct acidic hydrolysis (peak B, Mn(SEC) = 36000, Mw/Mn = 1.10) measured in DMF containing 0.01 M LiBr. | ||
Solubility and glass transition temperature of polymers
The solubilities of polymers obtained in this study are shown in Table 4 with those of poly(M1–M4), poly(E1–E4), and poly(OH1) as the references. Poly(V2–V4)s were soluble in most aprotic organic solvents but insoluble in hexane and Et2O. Interestingly, poly(V2–V4)s possessing vinyl terminal groups in their side chains were insoluble in water at any temperature. It should be emphasized that all corresponding poly(M2–M4) and poly(E2–E4) possessing methyl and ethyl terminals were soluble in water, and the aqueous solutions showed typical Tcs between 4 and 67 °C.17,20 These contrastive solubilities in water indicate that the vinyl terminal group is more hydrophobic than the methyl or ethyl groups. Although the number of carbon atoms in the vinyl group is the same as the ethyl group, the flexibility of the ethyl group might play an important role in the water solubility of poly(E2–E4). Another plausible factor is that the π-electron interaction between the planar vinyloxyl groups with restricted conformation prevents the solubilization of poly(V2–V4) in water, while the interaction of the flexible ethyl substituent is rather negligible. It is evident that among poly(V2–V4), the polarity of the polymer increases with the number of ethylene glycol units, since the polymers carrying longer side chains, poly(V3) and poly(V4), become soluble in alcohols. A similar tendency in polarity was observed in the increasing effect of the Tcs of poly(M2–M4) and poly(E2–E4).17,20 On the other hand, deprotected poly(OH2), poly(OH3), and poly(OH4) showed water solubility at any temperature, whereas poly(OH1), having the shortest side chain, was insoluble in water. The solubility of newly synthesized poly(OH4) in other solvents is the same as poly(OH3).| Polymer | Solvent | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hexane | Benzene | CHCl3 | Acetone | Et2O | THF | DMF | DMSO | EtOH | MeOH | H2O | |
| a S: soluble, I: insoluble. b Obtained by IEA/EtAlCl2. c T c = 26 °C. d T c = 52 °C. e T c = 67 °C. f T c = 4 °C. g T c = 27 °C. h T c = 42 °C. | |||||||||||
| Poly(V1) | I | S | S | S | I | S | S | S | I | I | I |
| Poly(V2) | I | S | S | S | I | S | S | S | I | I | I |
| Poly(V2)-cationicb | I | S | S | S | I | S | S | S | I | I | I |
| Poly(V3) | I | S | S | S | I | S | S | S | I | S | I |
| Poly(V4) | I | S | S | S | I | S | S | S | S | S | I |
| Poly(M1) | I | S | S | S | I | S | S | S | I | I | I |
| Poly(M2) | I | S | S | S | I | S | S | S | S | S | Sc |
| Poly(M3) | I | S | S | S | I | S | S | S | S | S | Sd |
| Poly(M4) | I | S | S | S | I | S | S | S | S | S | Se |
| Poly(E1) | I | S | S | S | S | S | S | S | S | S | I |
| Poly(E2) | I | S | S | S | S | S | S | S | S | S | Sf |
| Poly(E3) | I | S | S | S | S | S | S | S | S | S | Sg |
| Poly(E4) | I | S | S | S | S | S | S | S | S | S | Sh |
| Poly(OH1) | I | I | I | I | I | I | S | S | S | S | I |
| Poly(OH2) | I | I | S | I | I | S | S | S | S | S | S |
| Poly(OH3) | I | I | S | S | I | S | S | S | S | S | S |
| Poly(OH4) | I | I | S | S | I | S | S | S | S | S | S |
The newly obtained poly(V2–V4)s were colorless and sticky solids, while poly(V1) was white powder. Their glass transition temperatures (Tgs) were measured by differential scanning calorimetry (DSC). Poly(V1), poly(V2), poly(V3), and poly(V4) presented Tg values at 15, −33, −47, and −54 °C, respectively. It is apparent that the Tg of the polymers decreases by increasing the side chain length, the number of oligo(ethylene glycol) units. On the other hand, deprotected poly(OH2), poly(OH3), and poly(OH4) presented Tg values at −25, −59, and −64 °C, respectively.
In conclusion, we have succeeded in the chemoselective anionic polymerization of novel oligo(ethylene glycol) vinyl ether methacrylates, V2–V4, to give linear polymethacrylates with the predicted molecular weights and narrow MWDs. On the other hand, poly(vinyl ether) with pendant methacryloyl moieties was obtained by the chemoselective cationic polymerization of V2. The anionically obtained poly(V2–V4)s with vinyloxyl terminals in the side chains are insoluble in water, while the methyl- or ethyl ether counterparts, poly(M2–M4) and poly(E2–E4), show water solubility, indicating the highly hydrophobic property of planar vinyloxyl terminals. The water-soluble polymethacrylates, poly(OH2–OH4), are readily obtained by the simple acidic hydrolysis of the vinyloxyl groups of poly(V2–V4). It was thus demonstrated that the vinyloxyl groups of V2–V4 are capable of masking the OH moieties in OEGMAs during anionic polymerization.
Experimental
Materials
V1 was synthesized as previously reported.22V2 (Nippon Shokubai) was purified by fractional distillation from CaH2in vacuo. Commercially available methacryloyl chloride (Tokyo Kasei), acetic acid (Kanto Chemical), potassium tert-butoxide (Tokyo Kasei), and penta(ethylene glycol) (Aldrich) were used as received. Tetra(ethylene glycol) (Tokyo Kasei) was dried and distilled over CaH2 under the reduced pressure. Triethylamine (Sigma-Aldrich) and pyridine (Kanto Chemical) were dried and distilled over CaH2. LiCl (Kanto Chemical) was dried in vacuo for 2 days under heating and used as a THF solution. Diethylzinc (Tosoh-Akzo) was distilled under the reduced pressure and diluted with dry THF. Trioctylaluminium (Sumitomo Chemical Industry) was diluted with dry n-heptane. 1,1-Diphenylethylene (Aldrich) was distilled from CaH2 and then distilled in the presence of 1,1-diphenylhexyllithium on a vacuum line. THF as the polymerization solvent was refluxed over sodium wire, distilled from LiAlH4 under nitrogen, and finally distilled from sodium naphthalenide solution on a vacuum line. n-Heptane was washed with concentrated H2SO4 for 1 day and dried over anhydrous MgSO4, and it was further dried over P2O5 for 1 day under reflux. It was then distilled in the presence of 1,1-diphenylhexyllithium under nitrogen. ZnCl2 (Aldrich, 1.0 M in ether) was diluted with dry ether. EtAlCl2 (Aldrich, 1.8 M in toluene) was diluted with dry toluene.Initiators
Commercially available sec-BuLi (1.3 M in cyclohexane, Nakarai Tesque Inc.) was used without purification and diluted with dry n-heptane. Ph2CHK was prepared by the reaction of potassium naphthalenide with 1.1-fold diphenylmethane in THF at room temperature for 48 h. The concentrations of anionic initiators were determined by colorimetric titration using standardized 1-octanol in THF in an all-glass reactor in vacuo as previously reported.44 1-Isobutoxyethyl acetate (IEA) was prepared by the reaction of acetic acid with isobutyl vinyl ether at 60 °C for 3 h.2-[2-(2-(2-Chloroethoxy)ethoxy)ethoxy]ethanol (CE3OH)
Thionyl chloride (56.6 g, 476 mmol) was added dropwise to a mixture of tetra(ethylene glycol) (74.7 g, 385 mmol), pyridine (40.3 g, 509 mmol), and chloroform (95 mL) with stirring at 0 °C under nitrogen.28 The reaction mixture was stirred for 3 h at 90 °C and concentrated under reduced pressure to remove thionyl chloride and chloroform. Then, white solid of pyridinium chloride was precipitated in the reaction mixture. After filtration to remove pyridinium chloride, the filtrate was further concentrated under reduced pressure. The residue was purified by column chromatography (silica gel pretreated with triethylamine, hexane/ethyl acetate = 1/1–0/1) to remove tetra(ethylene glycol) and bis[2-(2-chloroethoxy)ethyl] ether. Subsequent vacuum distillation over calcium hydride gave a colorless liquid of CE3OH (23.3 g, 110 mmol, 29%, bp 104–108 °C/0.3 mmHg).1H NMR (CDCl3): δ 3.66 (m, 16H, ClCH2CH2OCH2CH2OCH2CH2OCH2CH2OH).
13C NMR (CDCl3): δ 42.8 (CH2Cl), 61.8 (CH2OH), 70.4, 70.6, 70.7, 70.8, 71.5, 72.6 (ClCH2CH2OCH2CH2OCH2CH2OCH2CH2OH).
Anal. Calcd for C8H17ClO4: C, 45.18, H, 8.06, Cl, 16.67. Found: C, 44.54, H, 8.35, Cl, 16.41%.
2-[2-(2-Vinyloxyethoxy)ethoxy]ethanol (VE3OH)
A solution of CE3OH (23.0 g, 108 mmol) in THF (40 mL) was added dropwise to a solution of potassium tert-butoxide (17.6 g, 157 mmol) in THF (180 mL) with stirring at 0 °C under nitrogen.29 The reaction mixture was stirred for 2 h at 90 °C and filtered to remove a precipitated potassium chloride. The filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (silica gel pretreated with triethylamine, hexane/ethyl acetate = 3/1–1/1). Vacuum distillation in the presence of methylene blue gave a colorless liquid of VE3OH (6.24 g, 35 mmol, 32%, bp 71–78 °C/0.5 mmHg).
1H NMR (C6D6): δ 2.51 (m, 1H, OH), 3.33 (m, 8H, HOCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.51 (m, 4H, CH2OH, CH2OCHCH2), 3.93 (d, J = 7 Hz, 1H, trans CH2CH), 4.14 (d, J = 14 Hz, 1H, cis CH2CH), 6.41 (dd, 1H, OCHCH2).
13C NMR (C6D6): δ 61.9 (CH2OH), 67.6 (CH2OCHCH2), 69.7, 70.5, 70.9, 72.9 (HOCH2CH2OCH2CH2OCH2CH2OCHCH2), 86.6 (CH2CH), 152.2 (OCHCH2).
Anal. Calcd for C8H16O4: C, 54.53, H, 9.15. Found: C, 52.82, H, 9.28%.
2-[2-(2-Vinyloxyethoxy)ethoxy]ethyl methacrylate (V3)
A solution of methacryloyl chloride (3.40 g, 32.5 mmol) in ether (5 mL) was added dropwise to a mixture of VE3OH (5.87 g, 33.3 mmol), triethylamine (5.20 g, 51.4 mmol), and ether (15 mL) with stirring at 0 °C under nitrogen. The reaction mixture was stirred overnight at room temperature and filtered to remove a precipitated triethylamine hydrochloride. After filtration, the filtrate was concentrated under reduced pressure, and the residue was purified by column chromatography (silica gel pretreated with triethylamine, hexane/ethyl acetate = 8/2). Vacuum distillation in the presence of methylene blue gave a colorless liquid of V3 (4.12 g, 16.9 mmol, 52%, bp 93–101 °C/0.3 mmHg).
1H NMR (C6D6): δ 1.83 (s, 3H, α-CH3), 3.37 (m, 8H, COOCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.53 (t, J = 5 Hz, 2H, CH2OCHCH2), 3.93 (d, J = 7 Hz, 1H, trans CH2CH), 4.14 (m, 3H, COOCH2 and cis CH2CH), 5.19 (s, 1H, (E)CH2C), 6.17 (s, 1H, (Z)CH2C), 6.41 (dd, 1H, OCHCH2).
13C NMR (C6D6): δ 18.4 (α-CH3), 64.0 (COOCH2), 67.7 (CH2OCHCH2), 69.3, 69.8, 70.9, 71.0 (COOCH2CH2OCH2CH2OCH2CH2O– CHCH2), 86.5 (CH2CH), 125.3 (CH2C), 136.8 (CH2CCH3), 152.3 (OCHCH2), 167.0 (CO).
IR (neat, cm−1): 598, 756, 816, 945, 1172, 1298, 1321, 1454, 1619, 1719 (CO), 2876.
Anal. Calcd for C12H20O5: C, 59.00, H, 8.25. Found: C, 58.64, H, 8.31%.
2-[2-(2-(2-(2-Chloroethoxy)ethoxy)ethoxy)ethoxy]ethanol (CE4OH)
A procedure similar to that described above for CE3OH was followed using penta(ethylene glycol) (76.3 g, 321 mmol), thionyl chloride (42.6 g, 358 mmol), and pyridine (30.0 g, 378 mmol). Column chromatography (silica gel, hexane/ethyl acetate = 3/7–0/1) and the subsequent vacuum distillation gave CE4OH (14.9 g, 58.0 mmol, 18%, bp 120–124 °C/0.4 mmHg) as a colorless liquid.1H NMR (CDCl3): δ 3.75 (m, 20H, HOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCH2CH2Cl).
13C NMR (CDCl3): δ 42.8 (CH2Cl), 61.8 (CH2OH), 70.4, 70.6, 70.7, 71.4, 72.5 (HOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCH2CH2Cl).
Anal. Calcd for C10H21ClO5: C, 46.78, H, 8.19, Cl, 13.84. Found: C, 45.49, H, 8.39, Cl, 13.33%.
2-[2-(2-(2-Vinyloxyethoxy)ethoxy)ethoxy]ethanol (VE4OH)
The same procedure was followed as described above for VE3OH using potassium tert-butoxide (9.61 g, 85.6 mmol) and CE4OH (14.8 g, 57.4 mmol). Column chromatography (silica gel pretreated with triethylamine, hexane/ethyl acetate = 1/1–0/1) and the subsequent vacuum distillation gave VE4OH (4.33 g, 19.7 mmol, 34%, bp 103–108 °C/0.4 mmHg) as a colorless liquid.
1H NMR (C6D6): δ 2.51 (m, 1H, OH), 3.29 (m, 12H, HOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.56 (m, 4H, CH2OH, CH2OCHCH2), 3.93 (d, J = 7 Hz, 1H, trans CH2CH), 4.14 (d, J = 14 Hz, 1H, cis CH2CH), 6.41 (dd, 1H, OCHCH2).
13C NMR (C6D6): δ 61.9 (CH2OH), 67.7 (CH2OCHCH2), 69.8, 70.6, 70.9, 71.0, 71.3, 72.9 (HOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 86.6 (CH2CH), 152.2 (OCHCH2).
Anal. Calcd for C10H20O5: C, 54.55, H, 9.09. Found: C, 53.82, H, 9.32%.
2-[2-(2-(2-Vinyloxyethoxy)ethoxy)ethoxy]ethyl methacrylate (V4)
A solution of methacryloyl chloride (1.93 g, 18.5 mmol) in ether (10 mL) was added dropwise to a mixture of VE4OH (4.21 g, 19.1 mmol), triethylamine (2.86 g, 28.3 mmol), and ether (10 mL) with stirring at 0 °C under nitrogen. The reaction mixture was stirred overnight at room temperature and filtered to remove a precipitated triethylamine hydrochloride. The filtrate was washed with water, aqueous sodium bicarbonate, and brine, and was dried over anhydrous MgSO4. After filtration, the filtrate was concentrated under the reduced pressure, and the residue was purified by column chromatography (silica gel pretreated with triethylamine, hexane/ethyl acetate = 2/1). Vacuum distillation in the presence of methylene blue gave a colorless liquid of V4 (2.51 g, 8.71 mmol, 47%, bp 100–110 °C/0.4 mmHg).
1H NMR (C6D6): δ 1.82 (s, 3H, α-CH3), 3.39 (m, 12H, COOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.55 (t, J = 5 Hz, 2H CH2OCHCH2), 3.91 (d, J = 6 Hz, 1H, trans CH2CH), 4.13 (m, 3H, COOCH2 and cis CH2CH), 5.21 (s, 1H, (E)CH2C), 6.14 (s, 1H, (Z)CH2C), 6.41 (dd, 1H, OCHCH2).
13C NMR (C6D6): δ 18.4 (α-CH3), 64.0 (COOCH2), 67.7 (CH2OCHCH2), 69.3, 69.8, 70.9, 70.9, 71.0, 71.0 (COOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 86.5 (CH2CH), 125.2 (CH2C), 136.8 (CH2CCH3), 152.3 (OCHCH2), 166.9 (CO).
IR (neat, cm−1): 664, 729, 816, 943, 1041, 1136, 1296, 1319, 1452, 1637, 1720 (CO), 2873.
Anal. Calcd for C14H24O6: C, 58.33, H, 8.33. Found: C, 57.70, H, 8.94%.
Purification of monomers for anionic polymerization
After careful fractional distillation, monomers V1–V4 were degassed and sealed off in an all-glass apparatus equipped with a break-seal in the presence of CaH2 and diluted with dry n-heptane. The monomer solution in n-heptane was distilled from CaH2 on a vacuum line into ampules fitted with break-seals. In the cases of V1–V3, the distilled monomers were treated with 1–2 mol% of trioctylaluminium in n-heptane for 3 min and again distilled under high vacuum conditions. The purified monomers were diluted with dry THF. The resulting monomer solutions (0.2–0.4 M) in THF were stored at −30 °C until ready to use for the anionic polymerization.Anionic polymerization
All polymerizations were carried out at −78 °C in an all-glass apparatus equipped with break-seals under high-vacuum conditions as previously reported.44 A typical polymerization procedure was as follows: A THF solution (2.52 mL) of 1,1-diphenylethylene (0.0335 M, 0.0844 mmol) was added to a n-heptane solution (3.36 mL) of sec-BuLi (0.0185 M, 0.0612 mmol) through the break-seal at −78 °C. After 15 min, LiCl (0.0853 M, 0.201 mmol) in THF (2.36 mL) was added to the mixture at −78 °C for 15 min. Then, V3 (0.640 g, 2.62 mmol) in THF (11.7 mL) was added rapidly to the initiator system at −78 °C through the break-seal with vigorous shaking of the apparatus. After standing at −78 °C for 20 h, the polymerization was terminated with methanol. After concentration of reaction mixture in vacuo, the residue was poured into a large excess of hexane to precipitate a poly(V3) (0.65 g, 100%, Mn = 13000, Mw/Mn = 1.05). The molecular weight and MWD of precipitated samples were identical to the values before precipitation. The resulting polymers were further purified by reprecipitations in a THF/hexane system and by freeze-drying from benzene solution. Polymers thus obtained were characterized by 1H and 13C NMR and IR spectroscopies. The following is the full list.
Anionically obtained poly(V2)
1H NMR (C6D6): δ 1.1–1.9 (m, 3H, α-CH3), 2.0–2.7 (m, 2H, main chain CH2), 3.50 (bs, 4H, COOCH2CH2OCH2CH2OCHCH2), 3.71 (bs, 2H, CH2OCHCH2), 3.9–4.5 (m, 4H, COOCH2 and CH2CH), 6.4–6.6 (broad, 1H, OCHCH2).
13C NMR (C6D6): δ 16–22 (α-CH3), 45–46 (main chain quaternary), 55 (main chain CH2), 64 (COOCH2), 68, 69, 70 (COOCH2CH2OCH2CH2OCHCH2), 87 (CH2CH), 152 (OCHCH2), 177 (CO).
IR (neat, cm−1): 816, 946, 983, 1044, 1135, 1172, 1249, 1321, 1454, 1619, 1719(CO), 2929.
Anionically obtained poly(V3)
1H NMR (C6D6): δ 1.1–1.8 (m, 3H, α-CH3), 2.0–2.7 (m, 2H, main chain CH2), 3.3–3.6 (bs, 8H, COOCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.72 (bs, 2H, CH2OCHCH2), 3.9–4.4 (m, 4H, COOCH2 and CH2CH), 6.4–6.6 (bs, 1H, OCHCH2).
13C NMR (C6D6): δ 16–22 (α-CH3), 45–46 (main chain quaternary), 55 (main chain CH2), 64 (COOCH2), 68, 69, 69, 70, 71 (COOCH2CH2OCH2CH2OCH2CH2OCHCH2), 87 (CH2CH), 152 (OCHCH2), 177 (CO).
IR (neat, cm−1): 684, 746, 815, 966, 1037, 1120, 1199, 1243, 1321, 1449, 1480, 1617, 1725 (CO), 2930.
Anionically obtained poly(V4)
1H NMR (C6D6): δ 1.1–1.8 (m, 3H, α-CH3), 2.0–2.8 (m, 2H, main chain CH2), 3.3–3.8 (m, 14H, COOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 3.9–4.3 (broad, 4H, CH2CH and COOCH2), 6.4–6.6 (broad, 1H, OCHCH2).
13C NMR (C6D6): δ 16–22 (α-CH3), 45–46 (main chain quaternary), 55 (main chain CH2), 64 (COOCH2), 68, 69, 69, 70, 71, 71, 71 (COOCH2CH2OCH2CH2OCH2CH2OCH2CH2OCHCH2), 86.7 (CH2CH), 152 (OCHCH2), 178 (CO).
IR (neat, cm−1): 684, 748, 816, 964, 1038, 1106, 1200, 1245, 1321, 1453, 1619, 1726 (CO), 2872.
Radical polymerization
All polymerizations were carried out in a round-bottom flask equipped with a three-way stopcock under nitrogen. A typical polymerization procedure was as follows: V2 (1.46 g, 7.30 mmol) and AIBN (0.090 g, 0.616 mmol) were dissolved in benzene (17.5 mL), and the mixture was degassed through three freeze–thaw cycles. The mixture was heated to 70 °C for 3 h, and the polymerization was terminated by cooling to room temperature. After concentration of the reaction mixture in vacuo, the residue was poured into a large excess of hexane to precipitate poly(V2) (1.10 g, 75%, Mn = 11000, Mw/Mn = 12.2).
Cationic polymerization
Cationic polymerization was carried out in an all-glass apparatus equipped with break-seals under high vacuum conditions. A typical polymerization procedure was as follows: A toluene solution (11.7 mL) of V2 (0.804 g, 0.343 M, 4.01 mmol) was added to a mixture of 1-isobutoxyethyl acetate (IEA) (0.163 mmol) and THF (2.96 mmol) in toluene (6.20 mL) at room temperature. After cooling to −78 °C, a toluene solution (3.71 mL) of EtAlCl2 (0.0552 M, 0.205 mmol) was immediately added to the mixture through the break-seal. The polymerization system was then warmed to 25 °C and stood for 24 h. After 24 h, the polymerization was terminated with methanol containing a small amount of aqueous ammonia. The conversion of V2 was analyzed by 1H NMR. The reaction mixture was washed with 1 M HCl and water and dried over MgSO4. After concentration of reaction mixture in vacuo, the residue was poured into hexane to precipitate a poly(V2) (0.87 g, 109%, Mn = 5200, Mw/Mn = 1.18).Cationically obtained poly(V2)
1H NMR (CDCl3): δ 1.4–1.7 (bs, 2H, main chain CH2), 1.90 (s, 3H, α-CH3), 3.4–3.6 (m, 5H, main chain CH and COOCH2CH2OCH2CH2), 3.67 (t, J = 5 Hz, 2H, COOCH2CH2OCH2CH2), 4.23 (t, J = 5 Hz, 2H, COOCH2), 5.66 (s, 1H, (E)CH2C), 6.07 (s, 1H, (Z) CH2C).
13C NMR (CDCl3): δ 18.3 (α-CH3), 39–41 (main chain CH2), 63.8 (COOCH2), 68–74 (main chain CH), 69.0 (COOCH2CH2OCH2CH2), 70.7 (COOCH2CH2OCH2CH2), 125.7 (CH2C), 136.1 (CH2C), 167.1 (CO).
IR (neat, cm−1): 669, 756, 930, 1215, 1425, 1520, 1715(CO), 3019.
Acetoxylation of polymer
Acetic acid (0.90 g, 15.0 mmol) was added by a syringe through rubber septum to a solution of poly(V2) (0.30 g, 1.50 mmol based on the monomer unit, Mn = 10000, Mw/Mn = 1.06) in toluene (10 mL) with stirring at 0 °C under nitrogen. The reaction mixture was stirred for 5 h at 70 °C and concentrated under the reduced pressure. The residue was poured into a large excess of hexane to precipitate poly[2-(2-(1-acetoxyethoxy)ethoxy)ethyl methacrylate] (0.33 g, 85%, Mn = 25000, Mw/Mn = 1.09 (SEC in DMF)).
Acidic hydrolysis of acetoxylated polymer
Aqueous HCl (1 M, 0.96 mL, 0.96 mmol) was added to a solution of poly[2-(2-(1-acetoxyethoxy)ethoxy)ethyl methacrylate] (0.25 g, 0.96 mmol based on the monomer unit, Mn = 25000, Mw/Mn = 1.09) in THF (10 mL) with stirring at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and concentrated under the reduced pressure. The residue was poured into a large excess of hexane to precipitate a poly[di(ethylene glycol) methacrylate] (Poly(OH2)) (0.27 g, Mn = 27000, Mw/Mn = 1.08 (SEC in DMF)). The resulting polymers were further purified by reprecipitations in an ethanol/hexane system and by freeze-drying from 1,4-dioxane solution containing small amount of methanol. Polymers thus obtained were characterized by 1H and 13C NMR and IR spectroscopies. The following is the full list.
13C NMR (CD3OD): δ 16–22 (α-CH3), 46–47 (main chain quaternary), 56 (main chain CH2), 62 (CH2CH2OH), 65 (COOCH2), 70 (COOCH2CH2), 74 (CH2OH), 179 (CO).
IR (neat, cm−1): 887, 1063, 1123, 1248, 1354, 1453, 1721 (CO), 2873, 2942, 3100–3700 (OH).
13C NMR (CD3OD): δ 16–22 (α-CH3), 46 (main chain quaternary), 56 (main chain CH2), 62 (CH2CH2OH), 65 (COOCH3), 70 (COOCH2CH2), 72 (OCH2CH2O), 74 (CH2OH), 179 (CO).
IR (neat, cm−1): 793, 886, 1062, 1240, 1348, 1579, 1719 (CO), 2865, 2996, 3100–3700 (OH).
13C NMR (CD3OD): δ 16–22 (α-CH3), 46 (main chain quaternary), 56 (main chain CH2), 62 (CH2CH2OH), 65 (COOCH3), 70 (COOCH2CH2), 72 (OCH2CH2OCH2CH2O), 74 (CH2OH), 179 (CO).
IR (neat, cm−1): 798, 884, 934, 1063, 1223, 1346, 1657, 1719 (CO), 2861, 2995, 3100–3700 (OH).
Direct acidic hydrolysis of polymers
Aqueous HCl (1 M, 0.34 mL, 0.34 mmol) was added to a solution of poly(V4) (0.11 g, 0.36 mmol based on the monomer unit, Mn = 34000, Mw/Mn = 1.07) in THF (10 mL) with stirring at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and concentrated under the reduced pressure. The residue was poured into a large excess of hexane to precipitate a poly(OH4) (0.07 g, 84%, Mn = 36000, Mw/Mn = 1.10 (SEC in DMF)). The polymer was further purified by freeze-drying from 1,4-dioxane solution containing small amount of methanol.
Measurements
1H and 13C NMR spectra were recorded on a Bruker DPX300 (300 MHz for 1H and 75 MHz for 13C) in CDCl3, C6D6, or CD3OD. Tacticity of polymers was determined by the 1H NMR integral ratio of three split α-methyl proton signals appearing at 0.90–1.25 ppm according to the assignment of poly(methyl methacrylate). Three signals were assigned as mm (1.25 ppm), mr (1.05 ppm), rr (0.90 ppm) triads. Infrared spectra were recorded on a JASCO FT/IR-4100 instrument. SEC chromatograms for determination of MWD were obtained in THF at 40 °C at a flow rate of 1.0 mL min−1 with a TOSOH HLC-8020 instrument with three polystyrene gel columns (TOSOH G4000HXL, G3000HXL, and G2000HXL) with either ultraviolet (254 nm) absorption or refractive index detection. SEC measurements were also performed in DMF containing 0.01 M LiBr at 40 °C at a flow rate of 1.0 mL min−1 with a TOSOH HLC-8120 instrument with three polystyrene gel columns (TOSOH GMHXL × 2 and G2000HXL) with refractive index detection. The Tgs of the polymers were measured by DSC using a Seiko instrument DSC6220 apparatus under nitrogen. The polymer sample was first heated to 100 °C, cooled to −150 °C with liquid nitrogen, and then scanned at a rate of 10 °C min−1.Acknowledgements
This work was supported by a Grant-in Aid (No. 20550108) from the Ministry of Education, Science, Sports, and Culture, Japan. The authors appreciate Professor R. Faust at University of Massachusetts Lowell for his insightful suggestion on the cationic polymerizability of V2.References and notes
- B. A. Bolto, Prog. Polym. Sci., 1995, 20, 987 CrossRef CAS.
- H. Mori, A. Hirao and S. Nakahama, Macromolecules, 1994, 27, 35 CrossRef.
- H. Mori, A. Hirao, S. Nakahama and K. Senshu, Macromolecules, 1994, 27, 4093 Search PubMed.
- N. G. Hoogeveen, M. A. Cohen Stuart, G. J. Fleer, W. Frank and M. Arnold, Macromol. Chem. Phys., 1996, 197, 2553 CrossRef CAS.
- H. Zhang and E. Ruckenstein, Macromolecules, 2000, 33, 4738 Search PubMed.
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 930 CrossRef CAS.
- S. Creutz, Ph. Teyssié and R. Jérôme, Macromolecules, 1997, 30, 6 CrossRef CAS.
- M. Vamvakaki, N. C. Billingham and S. P. Armes, Macromolecules, 1999, 32, 2088 CrossRef CAS.
- K. Ohno, Y. Tsuji and T. Fukuda, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2473 CrossRef CAS.
- M. Ejaz, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 2000, 33, 2870 CrossRef CAS.
- T. Ishizone, S. Han, S. Okuyama and S. Nakahama, Macromolecules, 2003, 36, 42 Search PubMed.
- A. Hirao, H. Kato, K. Yamaguchi and S. Nakahama, Macromolecules, 1986, 19, 1294 Search PubMed.
- H. Mori, O. Wakisaka, A. Hirao and S. Nakahama, Macromol. Chem. Phys., 1994, 195, 3213 Search PubMed.
- C. Nojiri, T. Okano, H. Koyanagi, S. Nakahama, K. D. Park and S. W. Kim, J. Biomater. Sci., Polym. Ed., 1992, 4, 75 Search PubMed.
- K. Senshu, S. Yamashita, M. Ito, A. Hirao and S. Nakahama, Langmuir, 1995, 11, 2293 Search PubMed.
- K. Senshu, M. Kobayashi, N. Ikawa, S. Yamashita, A. Hirao and S. Nakahama, Langmuir, 1999, 15, 1763 Search PubMed.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312 CrossRef CAS.
- H. Yokoyama, T. Miyamae, S. Han, T. Ishizone, K. Tanaka, A. Takahara and N. Torikai, Macromolecules, 2005, 38, 5180 Search PubMed.
- R. Zhang, A. Seki, T. Ishizone and H. Yokoyama, Langmuir, 2008, 24, 5527 CrossRef CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963 CrossRef CAS.
- S. Aoshima, O. Hasegawa and T. Higashimura, Polym. Bull., 1985, 13, 229 Search PubMed.
- H. Zhang and E. Ruckenstein, Macromolecules, 1998, 31, 746 Search PubMed.
- E. Ruckenstein and H. Zhang, Polym. Bull., 2001, 47, 113 Search PubMed.
- L. Leemans, R. Fayt, P. Teyssié, H. Uytterhoeven and W. De Winter, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2187 Search PubMed.
- G. Hild, J. P. Lamps and P. Rempp, Polymer, 1993, 34, 2875 Search PubMed.
- T. Ishizone, T. Takata and M. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1335 Search PubMed.
- M. Miyamoto, K. Hayashizaki, M. Tokimizu and T. Saegusa, Macromolecules, 1991, 24, 3988 Search PubMed.
- D. B. Amabilino, P. R. Ashton, C. L. Brown, E. Córdova, L. A. Godinez, T. T. Goodnow, A. E. Kaifer, S. P. Newton, M. Pietraszkiewicz, D. Philip, F. M. Raymo, A. S. Reder, J. F. Stoddert and D. J. Williams, J. Am. Chem. Soc., 1995, 117, 1271 CrossRef CAS.
- B.-A. Feit and B. Halak, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2171 Search PubMed.
- H. Ozaki, A. Hirao and S. Nakahama, Macromol. Chem. Phys., 1987, 20, 1442 Search PubMed.
- T. Ishizone, K. Yoshimura, A. Hirao and S. Nakahama, Macromolecules, 1998, 31, 8706 CrossRef CAS.
- R. Fayt, R. Forte, C. Jacobs, R. Jérôme, T. Ouhadi and P. Teyssié, Macromolecules, 1987, 20, 1442 Search PubMed.
- S. K. Varshney, J. P. Hautekeer, R. Fayt, R. Jérôme and P. Teyssié, Macromolecules, 1990, 23, 2618 Search PubMed.
- M. Kobayashi, S. Okuyama, T. Ishizone and S. Nakahama, Macromolecules, 1999, 32, 6466 Search PubMed.
- T. Ishizone and M. Ito, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4328 CrossRef CAS.
- M. Ito and T. Ishizone, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4832 Search PubMed.
- T. Suzuki, J. Kusakabe and T. Ishizone, Macromolecules, 2008, 41, 1929 Search PubMed.
- T. Suzuki, J. Kusakabe, K. Kitazawa, T. Nakagawa, S. Kawauchi and T. Ishizone, Macromolecules, 2010, 43, 107 Search PubMed.
- The observed Mn values increased with increasing the monomer-to-initiator ratios, while the apparent deviation in Mn value was observed (Table 1, runs 17 and 20). This is probably due to the trace amount of impurity in the monomer, resulting in the partial loss of initiator at the initial stage of polymerization.
- D. Kunkel, A. H. E. Müller, M. Janata and L. Lochmann, Makromol. Chem., Macromol. Symp., 1992, 60, 315 Search PubMed.
- A. V. Yakimansky, A. H. E. Müller and M. Van Beylen, Macromolecules, 2000, 33, 5686 CrossRef CAS.
- S. Aoshima and T. Higashimura, Macromolecules, 1989, 22, 1009 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245 CrossRef CAS.
- A. Hirao, K. Takenaka, S. Packrisamy, K. Yamaguchi and S. Nakahama, Makromol. Chem., 1985, 186, 1157 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |