Self-association behavior in water of an amphiphilic diblock copolymer comprised of anionic and dendritic blocks
Shin-ichi
Yusa
*a,
Yoshihiko
Shimada
a,
Toyoko
Imae
b and
Yotaro
Morishima
c
aDepartment of Materials Science and Chemistry, Graduate School of Engineering, University of Hyogo, 2167 Shosha, Himeji, Hyogo 671-2201, Japan. E-mail: yusa@eng.u-hyogo.ac.jp; Fax: +81 79 266 8868; Tel: +81 79 267 4954
bGraduate Institute of Engineering, Jing-Cheng Honors College, National Taiwan University of Science and Technology, 43, section 4, Keelung Road, Taipei, 10607, Taiwan. E-mail: imae@mail.ntust.edu.tw
cFaculty of Engineering, Fukui University of Technology, 6-3-1 Gakuen, Fukui, 910-8505, Japan. E-mail: morisima@fukui-ut.ac.jp
First published on 20th May 2011
Abstract
Well-defined amphiphilic diblock copolymers (PAMPS-b-PG2) composed of a hydrophilic linear polyelectrolyte and hydrophobic blocks bearing pendent dendritic moieties was synthesized via reversible addition–fragmentation chain transfer (RAFT) controlled radical polymerization of 3,5-bis[3′,5′-bis(benzyloxy)benzyloxy]benzyl 11-acrylamidoundecanoate (G2) using a poly(sodium 2-(acrylamido)-2-methylpropanesulfonate) (PAMPS) based macro-chain transfer agent. Degrees of polymerization of PAMPS and PG2 blocks were 71 and 3, respectively. The polydispersity index was 1.25 as estimated from GPC. Hydrophobic association of the PG2 blocks in aqueous solutions was suggested from restricted motions of the dendritic moieties as observed by 1H NMR in D2O. A fluorescence spectrum of 8-anilino-1-naphthalenesulfonic acid, ammonium salt hydrate indicated that the fluorescence probe was solubilized in the hydrophobic domain formed from the polymer aggregate in water. The formation of polymer aggregates was indicated by light scattering data. The hydrodynamic radius (Rh) of the polymer aggregate was independent of the polymer concentration.
Introduction
It is well established that amphiphilic block copolymers form micelles where hydrophobic sequences constitute cores and hydrophilic sequences form shells.1 Considerable attention, both from scientific and technological points of view, has been focused on amphiphilic block copolymer micelles because they are expected to be a potentially useful material for practical applications such as drug delivery,2–4 separation,5 and catalysis.6Dendrimers, including hyperbranched macromolecules, with a tailor made structure have been investigated as attractive compounds for various fields such as nano-scale reactors, diagnostics, gene delivery vectors, magnetic resonance imaging agents, and homogeneous catalysts.7,8 Furthermore, dendrimers are ideal building blocks for polymer architecture, because their structure can be controlled precisely. Xi et al.9 reported synthesis of methacrylates with a dendritic moiety as a pendent group and their radical polymerization. Stupp and Zubarev10 reported synthesis of a series of triblock structures containing dendritic, rodlike, and coillike blocks by a combination of anionic polymerization and step-by-step condensation reactions. Fréchet et al.11–13 reported the synthesis and association behavior of linear poly(ethylene oxide)-dendritic benzyl ether amphiphilic block copolymers. Imae et al.14 also reported the preparation and physicochemical properties of amphiphilic polymers including dendritic moiety. The large degree of vacancies in the interior of dendrons offers the ability to encapsulate many guest molecules. Their numerous end groups can be modified with various chemical functional groups.
In the past decade, the development of the controlled radical polymerization methods allows the synthesis of a wide variety of block copolymers under mild conditions. Nitroxide-mediated or stable free radical polymerization (SFRP),15 atom transfer radical polymerization (ATRP),16,17 and reversible addition–fragmentation chain transfer (RAFT) radical polymerization18 are the most studied methods. These methods are based on a reversible activation/deactivation cycle of propagating polymer radicals, resulting in a small fraction of propagating polymer radicals and a large fraction of reversible deactivated dormant polymer chains. The low concentration of active propagating polymer radicals suppresses possible termination and side reactions. Recently, well defined linear-dendritic block copolymers are prepared by SFRP19,20 and ATRP.21
Among the available controlled radical polymerization methods, RAFT polymerization is applicable to a wide range of monomers, functional groups, and reaction conditions including aqueous media. The RAFT process is achieved simply by the addition of a suitable chain transfer agent (CTA) to an ordinary radical polymerization system. The polymers obtained by the RAFT process can be used as precursors to block copolymers by the addition of further monomer. From these advantages, many research groups have focused on the employment of the RAFT process in the controlled polymerization.21–24
In this paper, we report the controlled synthesis of an AB diblock copolymer (PAMPS-b-PG2, Fig. 1) of sodium 2-(acrylamido)-2-methylpropanesulfonate (AMPS) and a methacrylamide-based dendritic monomer by the RAFT process using an PAMPS macro-chain transfer agent (macro-CTA). The association behavior of the amphiphilic diblock copolymer in water was characterized with 1H NMR, fluorescence, dynamic light scattering (DLS), static light scattering (SLS), and transmission electron microscopy (TEM) techniques.
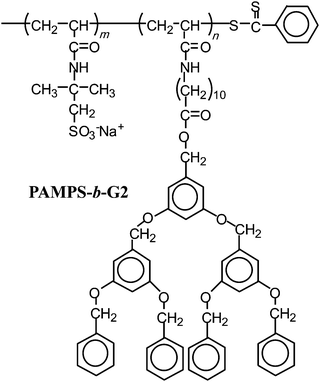 | ||
| Fig. 1 Chemical structure of PAMPS-b-PG2. | ||
Experimental
Materials
4-Cyanopentanoic acid dithiobenzoate was synthesized according to the method reported by McCormick and co-workers.25 Poly(sodium 2-(acrylamido)-2-methylpropanesulfonate) macro-chain transfer agent (PAMPS macro-CTA) was prepared by RAFT radical polymerization using 4-cyanopentanoic acid dithiobenzoate as a CTA.25,26Sodium 11-acrylamidoundecanoate was prepared according to the method of Gan and co-workers.273,5-Bis[3′,5′-bis(benzyloxy)benzyloxy]benzyl bromide (>98%) from Tokyo Kasei Kogyo Co., 8-anilino-1-naphthalenesulfonic acid, ammonium salt hydrate (97%) (ANS) from Aldrich, and 4,4′-azobis-4-cyanovaleric acid (98%) from Wako Pure Chemicals Co. were used as received without further purification. Dimethylsulfoxide (DMSO) was dried over 4 Å molecular sieves and distilled under reduced pressure. Water was purified with a Millipore Milli-Q system. Other reagents were used as received.Synthesis of 3,5-bis[3′,5′-bis(benzyloxy)benzyloxy]benzyl 11-acrylamidoundecanoate (G2)
Sodium 11-acrylamidoundecanoate (3.10 g, 11.2 mmol), 3,5-bis[3′,5′-bis(benzyloxy)benzyloxy]benzyl bromide (3.06 g, 3.79 mmol), tetra-n-butylammonium bromide (234 mg, 0.744 mmol), and 2,6-di-tert-butylcresol (18.5 mg, 0.0840 mmol) in a mixture of water (40 cm3) and chloroform (20 cm3) were heated to 110–115 °C in an oil bath for 24 h. The reaction mixture was diluted with chloroform (300 cm3), washed gently twice with water, and the organic layer was dried over anhydrous sodium sulfate. The solvent was removed by evaporation, and the crude product was purified by silica-gel column chromatography with chloroform as an eluent. The first fraction was collected, and condensation gave a white solid (G2); yield 2.69 g (72.4%); mp 50–51 °C. 1H NMR (500 MHz, CDCl3): δ = 1.12–1.36 (m, 12H, CH2), 1.41–1.58 (m, 2H, CH2), 1.59–1.72 (m, 2H, CH2), 2.32–2.41 (t, 2H, CH2), 3.24–3.35 (q, 2H, CH2), 4.97 (s, 4H, CH2), 5.04 (s, 8H, CH2), 5.05 (s, 2H, CH2), 5.52–5.60 (bs, 1H, CONH), 5.60–5.66 (m, 1H, vinyl CH), 6.01–6.11 (m, 1H, vinyl CH), 6.22–6.30 (m, 1H, vinyl CH), 6.55 (s, 1H, Ar CH), 6.59 (s, 4H, Ar CH), 6.69 (s, 4H, Ar CH), 7.29–7.48 (m, 20H, Ar CH). 13C NMR (DEPT) (126 MHz, CDCl3): δ = 25.04 (CH2), 27.03 (CH2), 29.22 (CH2), 29.33 (CH2), 29.37 (CH2), 29.47 (CH2), 29.55 (CH2), 29.65 (CH2), 34.39 (CH2), 39.71 (CH2), 65.97 (CH2), 70.07 (CH2), 70.19 (CH2), 101.67 (Ar CH), 101.75 (Ar CH), 106.50 (Ar CH), 107.14 (Ar CH), 126.10 (vinyl CH2), 127.66 (Ar CH), 128.13 (Ar CH), 128.70 (Ar CH), 131.17 (vinyl CH), 136.86 (Ar C), 138.58 (Ar C), 139.29 (Ar C), 160.09 (Ar C), 160.28 (Ar C), 165.61 (CONH), 173.71 (COO).Preparation of block copolymers (PAMPS-b-PG2)
DMSO was used as solvent for the polymerization to proceed in a homogeneous solution. A typical procedure for the block copolymerization of G2 by the RAFT process is as follows: PAMPS macro-CTA (1.57 g, 0.0709 mmol, Mw (SLS) = 2.21 × 104, Mw/Mn (GPC) = 1.28), G2 (2.00 g, 2.04 mmol), and 4,4′-azobis-4-cyanovaleric acid (3.96 mg, 0.0141 mmol) were dissolved in DMSO (25 cm3). The solution was degassed by Ar bubbling for 30 min. Polymerization was carried out at 70 °C for 24 h. A clear solution of the reaction mixture was poured into a large excess of chloroform to precipitate resulting polymer. The polymer was purified by reprecipitating from a methanol solution into a large excess of chloroform and collected by filtration; yield 0.88 g.Characterization
To obtain the relaxation time distribution by QELS, τA(τ), the inverse Laplace transform (ILT) analysis was performed using the algorithm REPES.28–30
| g(1)(t) = ∫τA(τ)exp (−t/τ)dln τ | (1) |
| D = D0(1 + kdCp) | (2) |
The weight-average molecular weight (Mw), z-average radius of gyration (Rg), and second virial coefficient (A2) values were estimated from the relation34
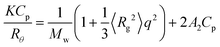 | (3) |
Results and discussion
The conversion of G2 in the presence of PAMPS macro-CTA (Mw (SLS) = 2.21 × 104, Mw/Mn (GPC) = 1.28) was estimated to be 6.3% from 1H NMR of the obtained block copolymer. The low conversion of G2 may be attributed to the steric hindrance of the bulky pendent dendritic groups of the monomer. Number-average degrees of polymerization (DPn) for the PAMPS and PG2 block sequences in the obtained diblock copolymer were determined to be 71 and 3, respectively, on the basis of SLS and 1H NMR data. The Mw/Mn value for the diblock copolymer determined from GPC analysis was 1.25, suggesting that the polymerization proceeded in accordance with a “living” fashion. The amphiphilic diblock copolymer obtained is soluble in water. We also prepared a diblock copolymer with the different block lengths using relatively smaller molecular weight PAMPS macro-CTA (Mw (SLS) = 4.29 × 103, Mw/Mn (GPC) = 1.18).Values of DPn for the PAMPS and PG2 blocks were 19 and 3, respectively, with an Mw/Mn value of 1.15 for the diblock copolymer. This block copolymer was insoluble in water. Therefore, we investigated the association behavior of PAMPS-b-PG2 where DPn values for the PAMPS and PG2 blocks were 71 and 3, respectively. The molecular characteristics of the diblock copolymer are presented in Table 1.
| PAMPS | PAMPS-b-PG2 | |
|---|---|---|
| a Determined at 40 °C in methanol containing 0.2 M LiClO4 by GPC relative to standard poly(ethylene oxide) (PEO). b Estimated from 1H NMR in DMSO-d6 at 100 °C along with Mw (SLS). c Determined at 25 °C in 0.1 M NaCl by static light scattering (SLS). d Determined at 488 nm. e Determined at 25 °C in 0.1 M NaCl by dynamic light scattering (DLS). f Calculated from a ratio of Mw (SLS) and Mn (NMR). | ||
| M n (GPC)a | 4.88 × 103 | 5.04 × 103 |
| M w/Mn (GPC)a | 1.28 | 1.25 |
| M n (NMR)b | — | 2.49 × 104 |
| M w (SLS) c | 2.21 × 104 | 2.60 × 106 |
| dn/dCpd/cm3 g−1 | 0.153 | 0.160 |
| A 2 c/cm3 g−2 mol | 3.08 × 10−3 | 1.04 × 10−5 |
| R h e/nm | 3.52 | 32.5 |
| R g c/nm | — | 26.2 |
| R g/Rh | — | 0.804 |
| N agg f | — | 104 |
Fig. 2 compares 1H NMR spectrum of PAMPS-b-PG2 in D2O at 27 °C with that in DMSO-d6 at 100 °C. In D2O, 1H NMR spectrum of PAMPS-b-PG2 is almost the same as that of the PAMPS macro-CTA (PAMPS homopolymer). The resonance bands observed at 1.2–1.8 ppm are attributed to the pendent methyl protons and the methylene protons of the main chain, bands at 1.9–2.4 ppm to the methine proton of the main chain, bands at 3.1–3.7 ppm to the pendent methylene protons, and bands at 7.1–7.8 ppm to the amide proton. The resonance bands due to the PG2 block were not discernible, suggesting the hydrophobic association of PG2 in water. On the other hand, in DMSO-d6 at 100 °C, the resonance bands related to PG2 were clearly observed at 4.8–5.2 and 6.5–6.8 ppm. The DPn value for the PG2 block is 3, which was calculated from the integral area intensity ratio of the 1H NMR resonance band for the pendent methylene protons in the AMPS unit and that for the benzyl protons in the G2 unit in DMSO-d6 at 100 °C.
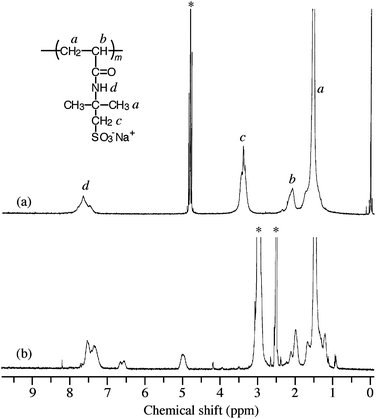 | ||
| Fig. 2 Comparison of 500 MHz 1H NMR spectra of PAMPS-b-PG2 in D2O containing 0.1 M NaCl at 25 °C (a) and in DMSO-d6 at 100 °C (b). The polymer concentration is 5.0 mg cm−3. Asterisks represent solvent bands. | ||
The fluorescence spectra of the ANS probe depend on microscopic polarity around the probe.35–38 ANS emits fluorescence around 510 nm weakly in hydrophilic media, e.g., in water but strongly in hydrophobic media accompanying a significant blue shift. Therefore, increased fluorescence intensity along with a blue shift of the emission maximum indicates that the probe is located in hydrophobic media.
The relative fluorescence intensity (I/I0) of an emission band of ANS (2.0 × 10−5 M) solubilized in aqueous 0.1 M NaCl solutions is plotted against polymer concentration (Cp) in Fig. 3a, where I and I0 are the fluorescence intensities in the presence and absence of the polymer, respectively. The I/I0 ratios of PAMPS are constant around 1.0 in the whole range of Cp studied. In the case of PAMPS-b-PG2, the I/I0 ratios are around 1.0 at Cp ≤ 0.07 mg cm−3. As Cp is increased, the I/I0 ratios begin to increase at Cp > 0.07 mg cm−3. These observations suggest that the polymer aggregates are able to incorporate ANS molecules into the hydrophobic microdomain formed from the association of the PG2 blocks.
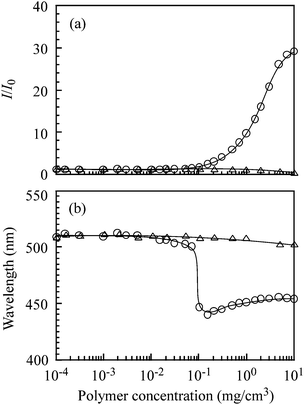 | ||
| Fig. 3 Relative fluorescence intensity (I/I0) (a) and emission maxima (b) in fluorescence spectra of 8-anilino-1-naphthalenesulfonic acid, ammonium salt hydrate (ANS) (2.0 × 10−5 M) as a function of concentrations of PAMPS (△) and PAMPS-b-PG2 (○) in aqueous 0.1 M NaCl solutions. | ||
Fig. 3b shows the Cp dependence of the ANS emission maximum wavelengths in the presence of PAMPS and PAMPS-b-PG2. The maximum wavelength for PAMPS is nearly constant at ca. 508 nm independent of Cp over the whole range of Cp depicted in Fig. 2b, which indicates the ANS probes existing in the bulk water phase. On the other hand, the maximum wavelengths in the presence of PAMPS-b-PG2 are constant at 508 nm in a low Cp region (Cp < 0.07 mg cm−3), however the emission maxima begin to decrease in the Cp region of 0.07–1.0 mg cm−3 with increasing Cp, reaching a blue-shifted wavelength around 450 nm at higher Cp. This blue shift suggests the formation of hydrophobic microdomains, in which ANS probes are solubilized when Cp is higher than 1.0 mg cm−3. A polymer concentration at the onset of the increase in I/I0 (Fig. 3a) agrees with that at the onset of the blue shift of the emission maximum (Fig. 3b).
Fig. 4a compares DLS relaxation time distributions for PAMPS and PAMPS-b-PG2 at Cp = 5.0 mg cm−3 in aqueous 0.1 M NaCl solutions at a fixed θ of 90°. These polymers exhibit unimodal distributions with different relaxation times. The relaxation time (τ) at the peak top of the distribution for PAMPS-b-PG2 is longer than that of PAMPS.
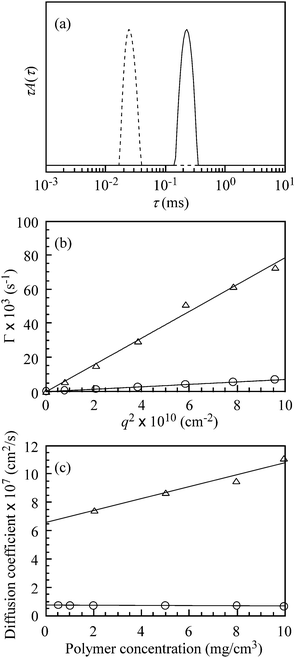 | ||
Fig. 4 (a) Typical examples of dynamic light scattering (DLS) relaxation time distributions for PAMPS (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and PAMPS-b-PG2 ( ) and PAMPS-b-PG2 (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) in 0.1 M NaCl aqueous solutions at 25 °C, where the polymer concentration (Cp) is 5.0 mg cm−3. (b) Relationship between the relaxation rate (Γ) and the square of the scattering vector (q2) for PAMPS (△) and PAMPS-b-PG2 (○) in aqueous 0.1 M NaCl solutions at Cp = 5.0 mg cm−3. (c) Plots of the diffusion coefficient (D) for PAMPS (△) and PAMPS-b-PG2 (○) in aqueous 0.1 M NaCl solutions at 25 °C as a function of Cp at a scattering angle (θ) of 90°. ) in 0.1 M NaCl aqueous solutions at 25 °C, where the polymer concentration (Cp) is 5.0 mg cm−3. (b) Relationship between the relaxation rate (Γ) and the square of the scattering vector (q2) for PAMPS (△) and PAMPS-b-PG2 (○) in aqueous 0.1 M NaCl solutions at Cp = 5.0 mg cm−3. (c) Plots of the diffusion coefficient (D) for PAMPS (△) and PAMPS-b-PG2 (○) in aqueous 0.1 M NaCl solutions at 25 °C as a function of Cp at a scattering angle (θ) of 90°. | ||
In Fig. 4b, the relaxation rates (Γ = τ−1) for PAMPS and PAMPS-b-PG2 at Cp = 5.0 mg cm−3 estimated from DLS data at different measuring angles are plotted as a function of the square of the scattering vector (q2). The linear relationship passing through the origin indicates that all the relaxation modes are due to translational diffusion. The diffusion coefficients (D) estimated from the slope of the Γ–q2 plot for PAMPS andPAMPS-b-PG2 at Cp = 5.0 mg cm−3 are 8.71 × 10−7 and 7.34 × 10−8 cm2s−1, respectively.
The D values for PAMPS increase with an increase in Cp (Fig. 4c), being a normal tendency for simple polyelectrolytes.39–41 In contrast, the D values for PAMPS-b-PG2 are practically constant independent of Cp in the whole range of Cp studied (Fig. 4c). Therefore, the hydrodynamic virial coefficient (kd) in eqn (2) can be regarded as virtually zero. No further aggregation between the polymer aggregates was recognized upon further increase in the polymer concentration beyond the Cp range shown in Fig. 3c. Consequently, the hydrodynamic size of the polymer aggregates, and hence the aggregation number (Nagg), is constant over a wide range of the polymer concentrations. Values of the hydrodynamic radii (Rh) can be calculated from the Stokes–Einstein relation along with the translational diffusion coefficient at limiting dilution (D0) determined by the extrapolation of Cp to zero in D–Cp plots. Values of Rh for PAMPS and PAMPS-b-PG2 thus estimated are listed in Table 1.
Fig. 5 compares Zimm plots for PAMPS and PAMPS-b-PG2 in aqueous 0.1 M NaCl solutions. The measurements of SLS were performed in a 2.0–10 mg cm−3 range of Cp. Values of dn/dCp for PAMPS and PAMPS-b-PG2 at 488 nm are listed in Table 1. The Mw values are estimated by the extrapolation of θ and Cp to zero, and values of Rg and A2 are estimated from the slope of the angular and concentration dependence in the Zimm plots, respectively. The SLS data for PAMPS and PAMPS-b-PG2 are presented in Table 1. The Mw value for PAMPS-b-PG2 is much lager than that for PAMPS, indicative of the multipolymer aggregates of the PG2 blocks. The Nagg value of the PAMPS-b-PG2 aggregate was calculated from the ratio of the apparent Mw values of the polymer aggregate and a single polymer chain. The former was estimated from Mw and Mw/Mn determined by SLS and GPC, respectively. The molecular weight of the single polymer chain of PAMPS-b-PG2 was calculated from 1H NMR spectra measured in DMSO-d6 at 100 °C. The Nagg value thus estimated for PAMPS-b-PG2 is 104 (Table 1). Unfortunately, the Rg value for PAMPS was unable to be determined with SLS measurements, because angular dependence in the Zimm plot was too small to determine the value of Rg (<10 nm). The A2 values for PAMPS and PAMPS-b-PG2 were estimated to be 3.08 × 10−3 and 1.04 × 10−5 cm3 g−2 mol, respectively. Amphiphilic polyelectrolyte aggregates often exhibit a small A2 value owing to an electrostatic shielding effect by counter ions condensed on the aggregates.42,43
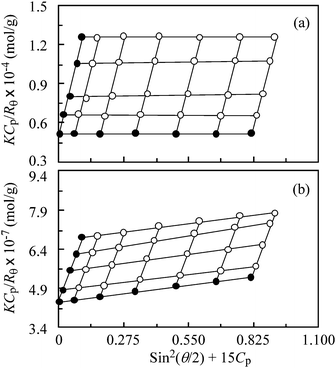 | ||
| Fig. 5 Zimm plots at 25 °C for PAMPS (a) and PAMPS-b-PG2 (b) in aqueous 0.1 M NaCl solutions at scattering angles from 30° to 130° with a 20° interval. The polymer concentrations were varied from 2.0 to 10 mg cm−3. | ||
The Rg/Rh ratio (ρ) is an important parameter that reflects a particle size distribution and conformation. The theoretical value of the ρ parameter for a homogeneous hard sphere is 0.788, and it increases substantially for less dense structures and polydisperse systems, e.g., ρ = 1.5–1.7 for flexible linear chains in a good solvent while ρ ≥ 2 for a rigid rod.44–47 The ρ value for PAMPS-b-PG2 was found to be 0.804 (Table 1).
Fig. 6 shows a TEM image of the PAMPS-b-PG2 aggregates. Spheres with radii ranging 27–31 nm are observed. The size observed by TEM is almost the same as that estimated from light scattering measurements.
 | ||
| Fig. 6 TEM photograph of the PAMPS-b-PG2 aggregates. | ||
Assuming that the diblock copolymer forms a core–shell type polymer micelle with a core formed from the hydrophobic PG2 blocks and a shell formed from permanently charged PAMPS blocks, the Rh value is the sum of the radius of the core (Rc) and the corona thickness (L). The Rc can be calculated from eqn (4):48,49
| Rc = [3Mw,micwG2/(4πNAρG2xG2)]1/3 | (4) |
Conclusions
The use of PAMPS macro-CTA allowed the synthesis of the novel amphiphilic diblock copolymer with a linear PAMPS block and a hydrophobic block bearing pendent dendritic moieties. It was suggested that production of PAMPS-b-PG2 with relatively low molecular weight distribution (Mw/Mn = 1.25) comes from the living nature of the polymerization process. In water, the amphiphilic nature of the amphiphilic diblock copolymer enabled to form aggregates. The motion of the bulky dendritic moieties was restricted because of hydrophobic association in water. Fluorescence data indicated that the hydrophobic microdomain could incorporate hydrophobic molecules such as ANS. The hydrodynamic radius of the polymer aggregate was 32.5 nm with a narrow unimodal distribution. The value of ρ parameter suggested that the polymer aggregates were nearly uniform. Further research of application of the PAMPS-b-PG2 aggregates to encapsulate hydrophobic molecules such as drugs is ongoing. The large degree of vacancies in the interior of dendrons can provide the ability to encapsulate many guest molecules. Furthermore, the terminal groups of dendrons can be modified with various functional groups.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Soft-Interface Science” from the Ministry of Education, Culture, Sports, Science and Technology of Japan.References
- Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, Wiley, New York, 1985 Search PubMed.
- G. Kown, S. Suwa, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 1994, 29, 17–23 CrossRef CAS.
- Y. Nagasaki, T. Okada, C. Scholz, M. Iijima, M. Kato and K. Kataoka, Macromolecules, 1998, 31, 1473–1479 CrossRef CAS.
- A. Rolland, J. O'Mullane, L. Brookman and K. Petrak, J. Appl. Polym. Sci., 1992, 44, 1195–1203 CrossRef CAS.
- P. N. Hurter and T. A. Hatton, Langmuir, 1992, 8, 1291–1299 Search PubMed.
- A. Kitahara, Adv. Colloid Interface Sci., 1980, 12, 109–140 CrossRef CAS.
- J. Issberner, R. Moors and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1994, 33, 2413–2420 CrossRef.
- D. A. Tomalia and P. R. Dvornic, Nature, 1994, 372, 617–618 CrossRef CAS.
- Y. M. Chen, C. F. Chen, W. H. Liu, Y. F. Li and F. Xi, Macromol. Rapid Commun., 1996, 17, 401–407 Search PubMed.
- E. R. Zubarev and S. I. Stupp, J. Am. Chem. Soc., 2002, 124, 5762–5773 CrossRef CAS.
- J. M. J. Fréchet, Science, 1994, 263, 1710–1715 CrossRef CAS.
- I. Gitsov, K. L. Wooley and J. M. J. Fréchet, Angew. Chem., Int. Ed. Engl., 1992, 31, 1200–1202 CrossRef.
- I. Gitsov and J. M. J. Fréchet, Macromolecules, 1993, 26, 6536–6546 CrossRef CAS.
- T. Imae, in Structure–Performance Relationship in Surfactants, ed. K. Esumi and M. Ueno, CRC Press, 2nd edn, 2003 Search PubMed.
- M. K. Georges, P. M. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572–7573 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, T. P. Le, R. T. A. Mayadunne, G. F. Meijs, G. Moad, C. L. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- K. Matyjaszewski, T. Shigemoto, J. M. J. Fréchet and M. R. Leduc, Macromolecules, 1996, 29, 4167–4171 CrossRef CAS.
- M. R. Leduc, C. J. Hawker, J. Dao and J. M. J. Fréchet, J. Am. Chem. Soc., 1996, 118, 11111–11118 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, D. B. Thomas and C. L. McCormick, Macromolecules, 2003, 36, 1436–1439 Search PubMed.
- B. Ray, Y. Isobe, K. Morioka, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2003, 36, 543–545 CrossRef CAS.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef CAS.
- J. F. Lutz, D. Neugebauer and K. Matyjaszewski, J. Am. Chem. Soc., 2003, 125, 6986–6993 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2003, 36, 4208–4215 CrossRef CAS.
- K. W. Yeoh, C. H. Chew, L. M. Gan, L. L. Koh and H. H. Teo, J. Macromol. Sci., Part A: Pure Appl. Chem., 1989, 26, 663–680 Search PubMed.
- J. Jakes, Czech. J. Phys., 1988, B38, 1305–1316 Search PubMed.
- K. Schillén, W. Brown and R. M. Johnsen, Macromolecules, 1994, 27, 4825–4832 CrossRef CAS.
- W. Brown, K. Schillén, M. Almgren, S. Hvidt and P. Bahadur, J. Phys. Chem., 1991, 95, 1850–1858 CrossRef CAS.
- W. H. Stockmayer and M. Schmidt, Pure Appl. Chem., 1982, 54, 407–414 CrossRef CAS.
- Dynamic Light Scattering, ed. R. Pecora and J. Berne, Plenum Press, New York, 1976 Search PubMed.
- Laser Light Scattering, ed. B. Chu, Academic Press, New York, 2nd edn, 1991 Search PubMed.
- B. H. Zimm, J. Chem. Phys., 1948, 16, 1099–1116 CrossRef CAS.
- J. Slavik, Biochim. Biophys. Acta, 1982, 694, 1–25 CAS.
- A. H. Hunt and B. Jirgensons, Biochemistry, 1973, 12, 4435–4441 Search PubMed.
- G. Weber and L. B. Young, J. Biol. Chem., 1964, 239, 1415–1423 CAS.
- L. Stryer, J. Mol. Biol., 1965, 13, 482–495 CAS.
- W. Peiqiang, M. Siddiq, C. Huiying, Q. Di and C. Wu, Macromolecules, 1996, 29, 277–281 Search PubMed.
- S. Yusa, M. Kamachi and Y. Morishima, Macromolecules, 2000, 33, 1224–1231 Search PubMed.
- J. Fundin, W. Brown and M. S. Vethamuthu, Macromolecules, 1996, 29, 1195–1203 Search PubMed.
- J. R. Quintana, M. D. Jánez, M. Villacampa and I. Katime, Macromolecules, 1995, 28, 4139–4143 Search PubMed.
- M. Villacampa, E. D. Apodaca, J. R. Quintana and I. Katime, Macromolecules, 1995, 28, 4144–4149 Search PubMed.
- E. Nordmeier and M. D. Lechner, Polym. J., 1989, 21, 623–632 Search PubMed.
- T. Konishi, T. Yoshizaki and H. Yamakawa, Macromolecules, 1991, 24, 5614–5622 CrossRef CAS.
- A. Qin, M. Tian, C. Ramireddy, S. E. Webber, P. Munk and Z. Tuzar, Macromolecules, 1994, 27, 120–126 CrossRef CAS.
- Modern Theory of Polymer Solutions, ed. H. Yamakawa, Harper & Row, New York, 1971 Search PubMed.
- S. Pispas, R. Hadjichristidis, I. Potemkin and A. Khokhlov, Macromolecules, 2000, 33, 1741–1746 CrossRef CAS.
- S. Keki, G. Deak, A. Kuki and M. Zsuga, Polymer, 1998, 39, 6053–6055 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |