Synthesis of reversible and irreversible cross-linked (M)PEG-(meth)acrylate based functional copolymers
W.
Steinhauer
,
H.
Keul
* and
M.
Möller
*
DWI an der RWTH Aachen e.V. and Institute of Technical and Macromolecular Chemistry, RWTH Aachen, Pauwelsstrasse 8, 52056, Aachen, Germany. E-mail: keul@dwi.rwth-aachen.de
First published on 13th May 2011
Abstract
Copolymers of poly(ethylene glycol) methyl ether methacrylates (MW ≈ 300 and 1100) and poly(ethylene glycol) acrylate (MW ≈ 375) with N-acryloxysuccinimide and pentafluorophenyl methacrylate were synthesized by free radical copolymerization in dioxane (80 °C) or THF (60 °C) using azobisisobutyronitrile as initiator. Converting the N-acryloxysuccinimide or pentafluorophenyl methacrylate repeating units with cystamine followed by TCEP·HCl treatment at pH < 7, water soluble, functional copolymers with thiol groups in the side chains were obtained. Reversibly and irreversibly cross-linked gels were prepared by oxidation of the thiol groups to disulfide bonds using H2O2 or by reacting the precursor copolymers with ethylene bisacrylate or poly(ethylene glycol) bisacrylate, via Michael-type addition reaction. The obtained gels were compared with regards to their chemical composition and their ability to absorb water by determination of the equilibrium water content of each gel viathermogravimetric analysis.
Introduction
Poly(ethylene oxide) (PEO), also known as poly(ethylene glycol) (PEG), has attracted considerable interest for its unique properties: unlike homologous polyethers, PEG is soluble in water at moderate temperatures for a wide range of molecular weights: from oligomers up to a molecular weight of a few million.1–3 Furthermore, PEG is biocompatible and inhibits protein adsorption,4,5 making polymeric micelles with a PEG corona, PEG gels and other aggregates of PEG good candidates for drug delivery5–7 or other biomedical purposes,8,9 among others in biomimetics10 or for protein protection by PEGylation.11–14 However, because each linear PEG molecule possesses only two attachment sites (the end groups), its efficient use as carrier for active components is limited. A number of attempts to increase the loading capacity of PEG through chemical modifications have been made.15–27 The most recent modifications of PEGs refer to increase of the end group functionality16–18 as for example by etherification of the end groups with 5-hydroxyisophthalic acid,19 or by the attachment of dendritic units with multiple alcohol functionalities.20–27 Another approach to increase the end group functionality is linked to the modification of the polymer architecture from linear to star shaped molecules.28,29 The most relevant strategies for the preparation of PEG derivatives with functional groups along the backbone are linear copolymers of EO and glycidol as well as polyglycidol [a polyether with hydroxymethyl side groups] itself.30–32The synthesis of polymer brushes with PEG in the side chains combines two main advantages of poly(ethylene glycol): first, the number of the terminal functional groups can be increased ad libitum by variation of the number of PEG side chains and second, the carrier backbone, which due to its chemical origin is not or only slightly soluble in water, can completely be made water soluble by attaching an adequate number of PEG molecules in the side chains.
In general, there are three main synthetic routes for the preparation of polymer brushes with covalently linked side chains to a linear backbone: “grafting onto”,33–36 “grafting from”37–40 and “grafting through”.41–44
In the case of “grafting onto” both backbone and side chains are prepared separately, followed by the grafting of side chains onto a backbone via a coupling reaction between the pendant functional groups of the backbone and the end—functional groups of the grafts. The grafting density of the polymer brushes in this procedure is limited for both kinetic and thermodynamic reasons. First, the diffusion of the unreacted grafts to the reactive sites on the backbone slows down with increasing the grafting density because of increasing steric hindrance. Second, the attachments of grafts to polymer brushes with a high grafting density become entropically unfavourable because the graft must change from a random coil conformation to a more stretched conformation once it is attached to the backbone. Thus, it is difficult to achieve complete substitution of reactive sites on the backbone.
The “grafting from” technique is based on the growth of side chains from the initiating groups attached to the polymer backbone, making this method the “method of choice” for the preparation of cylindrical polymer brushes with defined grafting density and well-defined backbones and side chains.37–40,45–54 A high density of functional groups along the side chains allows the usage of these molecular brushes for many applications.55–65
The “grafting through” method comprises homo- and copolymerizations of macromonomers leading also to cylindrical polymer brushes.41–44 However, due to the inherently low concentration of polymerizable groups and the steric hindrance of the side chains the synthesis of brushes with a high degree of polymerization is difficult. Thus, much higher DP for the main chain than that of the side chains can mainly be achieved by the radical polymerization of highly concentrated macromonomer solutions or bulk polymerization.66,67 Furthermore, the poor size control of the resulting polymer brushes and the incomplete conversion of the macromonomers, causing difficulties in the purification, are the major limitations for the radical polymerization of macromonomers. Attempts to synthesize cylindrical polymer brushes through living/controlled polymerizations, such as anionic,68–70 cationic,71group transfer,72 ring-opening73–76 and atom transfer radical77 polymerizations of macromonomers, have failed, not achieving a high DP of the main chain, resulting in structures resembling stars rather than cylindrical brushes. Polymerization of macromonomers is connected with diffusion and chemical controlled kinetic events, making it different from those of low molecular weight monomer polymerizations, resulting in linear polymer chains. Dilution of the macromonomer with low molecular weight comonomers decreases the effect of high segment density; in order to get similar final molecular weights the concentration of the low molecular weight comonomer must be increased the higher the molecular weight of the macromonomer is.78 Furthermore, adding low molecular weight active comonomers to the reaction mixture containing the macromonomer reactive copolymers are obtained which upon polymer analogues reaction lead to multifunctional copolymers.
Multifunctional polymers with different reactive repeating units have been prepared, such as polymers based on maleic anhydrid,79vinyl isocyanate80 and (meth)acrylate active ester monomers.81–88 Among the active esters (aE), the succinimide-based monomers, namely N-methacryloxysuccinimide (N-HS-MA) and acryloxysuccinimide (N-HS-A), have been used exclusively until recently. They proved to be suitable for the preparation of multifunctional poly((meth)acrylamides),81–83 hydrogels,84 materials for controlled drug release85 and chromatography supports.86 Alternatively pentafluorophenyl methacrylates (PFP-MA) and acrylates (PFP-A) were used as active ester building blocks. Poly(pentafluorophenyl(meth)acrylate)s are known to provide a better solubility and increased reactivity compared to N-hydroxysuccinimide-based polymers.87 Using active esters as comonomers, (meth)acrylamides, functionalized for example with thiols,88 can be obtained by polymer analogous reactions with thiol functional primary or secondary amines.87Thiols are known to be highly reactive and to have a strong tendency to form disulfide bonds; this makes them difficult in handling. However, by choosing the reaction conditions carefully, controlled oxidation of the thiol-groups to disulfide-bonds can be performed, yielding 3D networks of different cross-linking density, depending on the concentration of thiol-groups. This cross-linking reaction is fully reversible: by treating the product with a suitable reducing agent i.e.dithiothreitol89,90 or tris(2-carboxyethyl)phosphine,91,92thiol-groups are recovered.90 The intermediate formation of 3D networks can be used for purification of the synthesized functional polymers.
Furthermore, conjugate addition reactions between thiols and acrylates (also termed Michael-type addition reactions) are currently investigated,93–95 yielding 3D networks which are irreversibly cross-linked, when bis- or multifunctional acrylates are used.88 The high reactivity of thiols towards metals such as gold or copper has brought interest to the use of these macromolecules for functionalization.96–103
Experimental section
Materials
Poly(ethylene glycol) methyl ether methacrylates (MPEG300-MA and MPEG1100-MA, Aldrich) and poly(ethylene glycol) acrylate (PEG375-A, Aldrich) were passed through an aluminium oxide (Fluka, for chromatography) column to remove the inhibitor prior to use. N-Acrylsuccinimide (N-HS-A, 99%, Acros Organics), pentafluorophenyl methacrylate (PFP-MA, Polysciences, Inc.), cysteamine hydrochloride (min. 98%, AppliChem), cystamine dihydrochloride (98+%, Alfa Aesar), tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl, ≥ 98%, Fluka-BioChemika), H2O2 (30% solution in water, Merck), ethylene diacrylate (EG-bis-A, ABCR GmbH & Co. KG), poly(ethylene glycol) diacrylate (PEG575-bis-A, Aldrich), triethylamine (TEA, ≥99.5%, Sigma-Aldrich), dioxane (absolute, over molecular sieve (H2O < 0.01%), Fluka), and pyridine (Py, absolute, over molecular sieve H2O < 50 ppm, Acros Organics) were used as received. Tetrahydrofuran (THF) was dried over sodium, distilled under nitrogen and stored over molecular sieves under argon. 2,2′-Azobisisobutylronitrile (AIBN, Aldrich) was purified by double recrystallization in methanol.Instruments
1H and 19F NMR spectra were recorded on a Bruker DPX-300 FTNMR spectrometer at 300 MHz and 282 MHz, respectively. Deuterated chloroform (CDCl3) or deuterated dimethyl sulfoxide (DMSO-d6) were used as solvents, and tetramethylsilane (TMS) served as an internal standard.
Size exclusion chromatography (SEC) measurements were performed in THF with 250 mg L−12,6-di-tert-butyl-4-methylphenol using a high-performance liquid chromatography pump (ERC HPLC 64200) equipped with a RI Jasco detector and the PSS WinGPC Unity software program. Adequate molecular weight separation was achieved using five MZ gel columns in series at a flow rate of 1.0 mL min−1 and a temperature of 20 °C. The diameter of each column was 8 mm, the nominal pore width were 50, 50, 100, 1000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å, respectively. A calibration curve was obtained with PMMA standards.
000 Å, respectively. A calibration curve was obtained with PMMA standards.
Dynamic rheological measurements were performed with Rheometric Scientific DSR operating in the oscillation mode. Gelation measurements were carried out between the parallel plates (20 mm) with a gap size of 1000 μm. The rheometer is equipped with a water bath system for temperature control over an extended time. Deformation amplitude and frequency were set 0.01 and 1 Hz respectively, to ensure that the oscillatory deformation is within the linear viscoelastic regime, where the dynamic elastic modulus (G′) and viscous modulus (G′′) are independent of strain amplitude. To measure chemical gelation all polymer samples were dissolved at a concentration of 50 wt% in degassed water or methylene chloride and triethylamine (1.1 eq., with respect to SH-groups) was added. The solutions were mixed with 2.0 eq. H2O2 (30 wt% water solution) or stoichiometrically with EG-bis-A and PEG575-bis-Avia syringes. After mixing, 1 mL of the polymer sample was placed on the rheometer and time sweeps were performed at RT for 24 h.
For swelling tests gel samples prepared as mentioned above were stored in distilled water at RT for 24 h. After removing from water the samples were beated on a paper tissue to remove nonbonded water and the equilibrium water content of each gel sample was determined by thermogravimetric analysis (TGA) using a Netzsch TG 209 system (with a TA System-Controller TASC 4414) and the NETZSCH Proteus Thermal Analysis software program.
Synthesis
M
n,SEC = 29400; Mw/Mn = 2.0 monomodal.
1H NMR (DMSO-d6): δ 0.7–1.2 (b, -CH2-C(-CH3, -COOR)-), 1.6–2.2 (b, -CH2-CH(-COOR)-, -CH2-C(-CH3, -COOR)-), 2.8 (b, -CO-CH2-CH2-CO-), 3.4 (b, -OCH3), 3.5–3.8 (m, -O-CH2-CH2-O-), 4.0–4.4 (b, -COO-CH2-CH2-O-).
Polymers based on MPEG1100-MA, PEG375-A and different content of N-HS-A or PFP-MA were synthesized according to the above-described procedure (Table 1). The results are summarized in Table 2.
| Polymer | PEG-substrate | Active ester | AIBN [g]/[mmol] | Solvent/[mL] | T/t [°C]/[h] | ||
|---|---|---|---|---|---|---|---|
| Monomer | [g]/[mmol] | Monomer | [g]/[mmol] | ||||
| 1 | MPEG300-MA | 7.20/24.00 | N-HS-A | 1.02/6.05 | 0.05/0.30 | THF/45 | 60/4 |
| 2 | MPEG300-MA | 2.40/8.00 | N-HS-A | 0.34/2.00 | 0.02/0.10 | Dioxane/15 | 80/4 |
| 3 | MPEG300-MA | 7.20/24.0 | N-HS-A | 1.02/6.00 | 0.05/0.30 | Dioxane/45 | 80/4 |
| 4 | MPEG300-MA | 7.19/23.97 | N-HS-A | 1.02/6.02 | 0.05/0.30 | Dioxane/45 | 80/4 |
| 5 | MPEG300-MA | 2.70/9.00 | N-HS-A | 0.17/1.00 | 0.02/0.11 | Dioxane/15 | 80/4 |
| 6 | MPEG1100-MA | 5.50/5.00 | N-HS-A | 0.85/5.00 | 0.02/0.10 | THF/15 | 60/24 |
| 7 | MPEG1100-MA | 17.67/16.06 | N-HS-A | 0.68/4.01 | 0.03/0.20 | THF/40 | 60/4 |
| 8 | PEG375-A | 9.01/24.00 | N-HS-A | 1.02/6.00 | 0.05/0.30 | THF/45 | 60/4 |
| 9 | MPEG300-MA | 2.40/8.00 | PFP-MA | 0.50/2.00 | 0.02/0.10 | Dioxane/15 | 80/4 |
| 10 | MPEG300-MA | 2.40/8.00 | PFP-MA | 0.50/2.00 | 0.02/0.10 | Dioxane/15 | 80/4 |
| 11 | MPEG300-MA | 9.61/32.02 | PFP-MA | 2.02/8.00 | 0.07/0.40 | Dioxane/60 | 80/6 |
| Polymer | Active ester | M n c/[g mol−1] | PDIc | ||
|---|---|---|---|---|---|
| No. | Name | F a | f b | ||
| a F = mol fraction of the active ester in the feed. b f = mol fraction of the active ester in the copolymer determined by 1H NMR. c Number-average molecular weight (Mn) and polydispersity index (PDI) determined by SEC (THF) using PMMA standards. | |||||
| 1 | MPEG300-MA-co-N-HS-A | 0.20 | 0.24 | n.d. | n.d. |
| 2 | MPEG300-MA-co-N-HS-A | 0.20 | 0.21 | 40800 |
2.1 |
| 3 | MPEG300-MA-co-N-HS-A | 0.20 | 0.19 | 29400 |
2.0 |
| 4 | MPEG300-MA-co-N-HS-A | 0.20 | 0.14 | 11400 |
2.2 |
| 5 | MPEG300-MA-co-N-HS-A | 0.10 | 0.10 | 19300 |
2.7 |
| 6 | MPEG1100-MA-co-N-HS-A | 0.50 | 0.59 | 13700 |
1.6 |
| 7 | MPEG1100-MA-co-N-HS-A | 0.20 | 0.26 | 27600 |
1.3 |
| 8 | PEG375-A-co-N-HS-A | 0.20 | 0.24 | 7100 | 3.1 |
| 9 | MPEG300-MA-co-PFP-MA | 0.20 | 0.41 | 44900 |
1.7 |
| 10 | MPEG300-MA-co-PFP-MA | 0.20 | 0.37 | 44800 |
1.7 |
| 11 | MPEG300-MA-co-PFP-MA | 0.20 | 0.23 | n.d. | n.d. |
1H NMR (CDCl3): δ 0.7–1.2 (b, -CH2-C(-CH3, -COOR)-), 1.6–2.2 (b, -CH2-CH(-COOR)-, -CH2-C(-CH3, -COOR)-), 2.6 (b, -COO-CH2-CH2-SH), 3.2–3.5 (b, -COO-CH2-CH2-SH), 3.4 (b, -OCH3), 3.5–3.8 (m, -O-CH2-CH2-O-), 4.0–4.4 (b, -COO-CH2-CH2-O-).
Polymers with MPEG1100-MA and different content of thiol-groups were synthesized according to the above-described procedure with different amounts of cysteamine or cysteamine hydrochloride, triethylamine and TCEP·HCl in the feed (Table 3). The results are summarized in Table 4.
| Polymer no. | Precursor polymer, no., aE/[mmol] | Et3N [g]/[mmol] | Cys·HCl [g]/[mmol] | DCM/[mL] | t/[h] | TCEP·HCl [g]/[mmol] | H2O a/[mL] | t/[h] |
|---|---|---|---|---|---|---|---|---|
| a Adjusted to pH = 8 with TEA. b The free base of cysteamine (Cys) was used. | ||||||||
| 12 | MPEG1100-MA-co-N-HS-A, 6, 1.18 | — | 0.75/9.68b | 30 | 24 | 1.40/4.84 | 50 | 3 |
| 13 | MPEG1100-MA-co-N-HS-A, 6, 1.33 | 1.10/10.86 | 1.23/10.86 | 30 | 41 | 1.56/5.43 | 50 | 3 |
| 14 | MPEG300-MA-co-N-HS-A, 1, 2.22 | 4.49/44.34 | 0.54/4.88 | 25 | 68 | 0.76/2.66 | 40 | 16 |
| Polymer no. | Precursor polymer name (no.), fa | SH-functional PEG-copolymer name, fb | Conversionc (%) |
|---|---|---|---|
| a f = mol fraction of the active ester in the copolymer determined by 1H NMR. b f = mol fraction of the active ester functionalized with SH-groups in the product copolymer determined by 1H NMR. c Conversion of the active esters into the corresponding SH-functional amides determined by 1H NMR. d Literature known side reactions took place. | |||
| 12 | MPEG1100-MA-co-N-HS-A (6), 0.59 | MPEG1100-MA-co-N-ME-AA, 0.45 | 76.3d |
| 13 | MPEG1100-MA-co-N-HS-A (6), 0.59 | MPEG1100-MA-co-N-ME-AA, 0.22 | 37.3 |
| 14 | MPEG300-MA-co-N-HS-A (1), 0.24 | MPEG1100-MA-co-N-ME-AA, 0.15 | 62.5 |
1H NMR (CDCl3): δ 0.7–1.2 (b, -CH2-C(-CH3, -COOR)-), 1.6–2.2 (b, -CH2-CH(-COOR)-, -CH2-C(-CH3, -COOR)-), 2.6 (b, -COO-CH2-CH2-SH), 3.2–3.5 (b, -COO-CH2-CH2-SH), 3.4 (b, -OCH3), 3.5–3.8 (m, -O-CH2-CH2-O-), 4.0–4.4 (b, -COO-CH2-CH2-O-).
Polymers with MPEG1100-MA, PEG375-A but also PFP-MA and different content of thiol-groups were synthesized according to the above-described procedure using cystamine dihydrochloride and triethylamine in the feed (Table 5). The results are summarized in Table 6.
| Polymer no. | Precursor polymer, no., aE/[mmol] | Et3N [g]/[mmol] | Cys·2HCl [g]/[mmol] | Dioxane/[mL] | T/t [°C]/[h] | TCEP·HCl [g]/[mmol] | H2O a/[mL] | t/[h] |
|---|---|---|---|---|---|---|---|---|
| a Adjusted to pH ≈ 8 with TEA. b According to the preparation procedure of the precursor polymer. | ||||||||
| 15 | MPEG300-MA-co-N-HS-A, 2, 2.0b | 10.12/100.0 | 0.50/2.20 | 50 | 80/72 | 0.69/2.42 | 50 | 22 |
| 16 | MPEG300-MA-co-N-HS-A, 3, 5.12 | 27.24/269.17 | 1.27/5.64 | 120 | 80/70 | 1.77/6.20 | 150 | 18 |
| 17 | MPEG300-MA-co-N-HS-A, 4, 6.00b | 30.36/300.00 | 1.49/6.63 | 150 | 80/63 | 2.09/7.28 | 60 | 15 |
| 18 | MPEG300-MA-co-N-HS-A, 5, 1.00b | 5.06/50.00 | 0.25/1.10 | 25 | 80/68 | 0.35/1.21 | 22 | 16 |
| 19 | MPEG1100-MA-co-N-HS-A, 7, 4.00b | 21.89/216.30 | 0.99/4.40 | 200 | 80/70 | 1.38/4.84 | 75 | 4 |
| 20 | PEG375-A-co-N-HS-A, 8, 6.00b | 30.10/297.50 | 1.49/6.60 | 240 | 80/68 | 2.10/7.26 | 100 | 39 |
| 21 | MPEG300-MA-co-PFP-MA, 10, 3.04 | 8.31/82.10 | 0.40/1.75 | 50 | 80/93 | 0.56/1.97 | 50 | 24 |
| 22 | MPEG300-MA-co-PFP-MA, 11, 3.90 | 12.53/123.80 | 0.63/2.80 | 80 | 75/70 | 1.10/3.83 | 100 | 6 |
| Polymer no. | Precursor polymer name (no.), fa | SH-functional PEG-copolymer name, fb | Conversion [%] |
|---|---|---|---|
| a f = mol fraction of the active ester in the copolymer determined by 1H NMR. b f = mol fraction of the active ester functionalized with SH-groups in the product copolymer determined by 1H NMR. | |||
| 15 | MPEG300-MA-co-N-HS-A (2), 0.21 | MPEG300-MA-co-N-ME-AA, 0.21 | >99 |
| 16 | MPEG300-MA-co-N-HS-A (3), 0.19 | MPEG300-MA-co-N-ME-AA, 0.17 | 89.5 |
| 17 | MPEG300-MA-co-N-HS-A (4), 0.14 | MPEG300-MA-co-N-ME-AA, 0.15 | >99 |
| 18 | MPEG300-MA-co-N-HS-A (5), 0.10 | MPEG300-MA-co-N-ME-AA, 0.11 | >99 |
| 19 | MPEG1100-MA-co-N-HS-A (7), 0.26 | MPEG1100-MA-co-N-ME-AA, 0.26 | >99 |
| 20 | PEG375-A-co-N-HS-A (8), 0.24 | PEG375-A-co-N-ME-AA, 0.26 | >99 |
| 21 | MPEG300-MA-co-PFP-MA (10), 0.37 | MPEG300-MA-co-N-ME-MAA, 0.11 | 29.7 |
| 22 | MPEG300-MA-co-PFP-MA (11), 0.23 | MPEG300-MA-co-N-ME-MAA, 0.19 | 82.6 |
Reversibly cross-linked gels (G1, G2, G5, G6 and G7) containing MPEG1100-MA and PEG375-A were synthesized according to the above described procedure with different amounts of N-(2-mercaptoethyl)acrylamide (N-ME-AA) or N-(2-mercaptoethyl)methacrylamide (N-ME-MAA). The results are summarized in Table 7.
| Gel | Precursor polymer, no., mmol of N-ME-(M)AA | Solvent/[mL] | Cross-linker [g]/[mmol] | EWC a | |
|---|---|---|---|---|---|
| Totalb [%] | Swellingc [%] | ||||
| a Equilibrium water content. b Total water content: TWC = (1 − mdry/mtotal) × 100. c Calculated according to: ((mtotal − mdry)/mdry) × 100. d 0.01 M phosphate buffered solution (pH 7.4). e A 30 wt% solution of H2O2 in water was used. f According to the preparation procedure of the precursor polymer. | |||||
| G1 | MPEG300-MA-co-N-ME-AA, 16, 3.37 | PBS d/35.0 | H2O2,e 7.86/69.34 | 84.8 | 557.9 |
| G2 | MPEG300-MA-co-N-ME-AA, 17, 0.75 | H2O/0.8 | H2O2,e 0.05/1.51 | 28.4 | 40.0 |
| G3 | MPEG300-MA-co-N-ME-AA, 17, 1.47 | DCM/3.0 | EG-bis-A, 0.12/0.73 | 35.1 | 54.1 |
| G4 | MPEG300-MA-co-N-ME-AA, 17, 1.30 | DCM/2.5 | PEG575-bis-A, 0.37/0.65 | 61.8 | 161.8 |
| G5 | MPEG1100-MA-co-N-ME-AA, 19, 4.50 | H2O/4.5 | H2O2,e 0.93/8.99 | 80.7 | 418.1 |
| G6 | PEG375-A-co-N-ME-AA, 20, 6.00f | H2O/15.0 | H2O2,e 0.49/14.52 | 65.2 | 187.4 |
| G7 | MPEG300-MA-co-ME-MAA, 21, 1.35 | H2O/5.0 | H2O2,e 0.31/2.71 | 41.9 | 72.1 |
Irreversibly cross-linked gel (G4) obtained by cross-linking of poly(MPEG300-MA-co-N-ME-AA) (17) using PEG575-bis-A was synthesized according to the above-described procedure. The results are summarized in Table 7.
Results and discussion
The present work describes the free radical copolymerization of (M)PEG-(meth)acrylates of different molecular weights (MPEG300-MA, MPEG1100-MA and PEG375-A) with N-acrylsuccinimide (N-HS-A) or pentafluorophenyl methacrylate (PFP-MA) active esters, resulting in reactive copolymers with different ratios of PEG side chains and active esters in the backbone (Scheme 1). By conversion of the active ester in the precursor copolymers with aminothiols, such as cysteamine or cystamine, water soluble copolymers functionalized with thiol groups were obtained (after reducing the disulfide bonds). Treatment of the thiol functional copolymers with hydrogen peroxide or reaction with bis-acrylates (ethylene glycol-bisacrylate or oligo (ethylene glycol-bisacrylate)) results in reversible or irreversible cross-linked hydrogels. A comparison of the synthesized gels with respect to their chemical composition and their ability to absorb water was performed.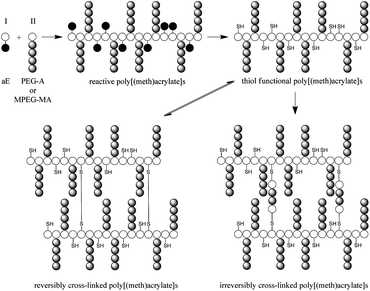 | ||
| Scheme 1 Concept for the preparation of reversibly and irreversibly cross-linked, (M)PEG-(meth)acrylate based hydrogels. | ||
N-Acrylsuccinimide and pentafluorophenyl methacrylate were previously homo- and copolymerized in THF and dioxane at different concentrations and temperatures of 60 respectively 80 °C.87,88,104 For copolymerization of these active esters with the macromonomers mentioned before we choose the same conditions, a molar ratio of active ester to macromonomer of 20/80 and a monomer to initiator ratio of 100. The monomer ratio of 20/80 assures water solubility of the copolymers and a suitable cross-linking density. In the following the results obtained for all possible combinations of reactive- and macro-monomers are described.
Free radical copolymerization of N-HS-A or PFP-MA with (M)PEG-(meth)acrylate macromonomers
Preliminary polymerization experiments of N-HS-A with MPEG300-MA (molar concentration of monomers [ΣM] = 0.67 M) in dioxane at 80 °C yielded a copolymer of Mn = 40800 g mol−1 and Mw/Mn = 2.1 (Scheme 2). However, when MPEG1100-MA was copolymerized under the same conditions ([ΣM] = 0.67 M) an insoluble polymer was obtained. Even when the overall monomer concentration was reduced to 0.25 M cross-linking could not be avoided. However, when THF was used as solvent ([ΣM] = 0.5 M) at 60 °C a copolymer was obtained with Mn = 27600 g mol−1 and Mw/Mn = 1.3.
![Preparation of poly[(meth)acrylate] copolymers with PEG side chains and active ester repeating units.](/image/article/2011/PY/c1py00087j/c1py00087j-s2.gif) | ||
| Scheme 2 Preparation of poly[(meth)acrylate] copolymers with PEG side chains and active ester repeating units. | ||
Copolymerization of PEG375-A and N-HS-A could not be performed in dioxane at 80 °C and [ΣM] = 0.67 M, however at [ΣM] = 0.33 M and in THF at 60 °C and a monomer concentration of 0.67 M polymerization was possible (Mn = 7100 g mol−1 and Mw/Mn = 3.1).
Copolymerization of PFP-MA with (M)PEGx-(M)As was more problematic: with MPEG1100-MA and PEG375-A under all conditions applied non-soluble products were obtained. The only successful polymerization was that of MPEG300-MA with PFP-MA in dioxane at 80 °C and a monomer concentration of 0.67 M (Mn = 44900 g mol−1; Mw/Mn = 1.7).
The composition of the copolymers was proven by 1H NMR analysis (Fig. 1a and b). In addition the presence of PFP-groups was proven by 19F NMR spectroscopy (Fig. 1c). The mole ratio of MPEG300-MA, MPEG1100-MA or PEG375-A and N-HS-A was calculated from the ratio of the integrals of the methylene protons (signal h, h′) of the succinimide and the methyl protons (signal e) of the MPEG300-MA and MPEG1100-MA or the methylene protons attached to the ester group (signal c) of PEG375-A, respectively. Furthermore, the mole ratio of MPEG300-MA and PFP-MA repeating units was calculated from the ratio of integrals of the methyl protons (signal B, G) and the methyl protons (signal E) of the MPEG300-MA, respectively.
![1H NMR spectra in CDCl3 of: (a) MPEG300-MA-co-PFP-MA, (b) MPEG300-MA-co-N-HS-A and (c) 19F NMR spectrum in CDCl3 of MPEG300-MA-co-PFP-MA copolymers synthesized by free radical copolymerization in dioxane at 80 °C and [ΣM] = 0.67 mol L−1.](/image/article/2011/PY/c1py00087j/c1py00087j-f1.gif) | ||
| Fig. 1 1H NMR spectra in CDCl3 of: (a) MPEG300-MA-co-PFP-MA, (b) MPEG300-MA-co-N-HS-A and (c) 19F NMR spectrum in CDCl3 of MPEG300-MA-co-PFP-MA copolymers synthesized by free radical copolymerization in dioxane at 80 °C and [ΣM] = 0.67 mol L−1. | ||
In order to get information on the reactivity of the monomers used, kinetic studies were performed in dioxane at 80 °C and THF at 60 °C. Copolymerization of MPEG300-MA and N-HS-A in dioxane ([ΣM] = 0.67 mol L−1, Fig. 2a) clearly shows a slightly lower conversion of the macromonomer. By increasing the molecular weight of the macromonomer by a factor of 3.6 polymerization in dioxane at 80 °C was not possible anymore (cross-linking was observed). Reducing the polarity of solvent from 4.8 (dioxane) to 4.0 (THF) and decreasing the temperature from 80 to 60 °C polymerization of MPEG1100-MA and N-HS-A became possible (Fig. 2b). The difference in reactivity of the macromonomer and N-HS-A becomes more pronounced: after 6 h the conversion of N-HS-A was 94.4% (compared to 99.9) and that of MPEG1100-MA 72.6% (compared to 94.5). Copolymerization of PEG375-A and N-HS-A (Fig. 2c) could not be performed under the conditions used for MPEG300-MA/N-HS-A due to cross-linking. Obviously, under the reaction conditions the OH-groups at the end of the PEG-side chains react with repeating units of the activated ester forming a polymer network. However, by reducing the concentration of monomer from [ΣM] = 0.67 to [ΣM] = 0.33 mol L−1 linear copolymers were obtained, the reactivity of N-HS-A being higher than that of macromonomer PEG375-A. After 3 h and 6 h the conversion of PEG375-A was 82.8 and 91.2%, that of N-HS-A 93.8 and 99.9%.
![(Top) Kinetic studies of the copolymerization reactions of (a) MPEG300-MA/N-HS-A (80/20, dioxane, 80 °C, [ΣM] = 0.67 mol L−1), (b) MPEG1100-MA/N-HS-A (80/20, THF, 60 °C, [ΣM] = 0.5 mol L−1), (c) PEG375-A/N-HS-A (80/20, dioxane, 80 °C, [ΣM] = 0.33 mol L−1) and (d) MPEG300-MA/PFP-MA (80/20, dioxane, 80 °C, [ΣM] = 0.67 mol L−1). (Bottom) Total monomer conversion vs. time plot for different copolymerizations in THF (60 °C) or dioxane (80 °C) with a (M)PEGx-(M)A/aE-ratio of 80/20 and the initial monomer/initiator ratio of 100 adjusted in the feed.](/image/article/2011/PY/c1py00087j/c1py00087j-f2.gif) | ||
| Fig. 2 (Top) Kinetic studies of the copolymerization reactions of (a) MPEG300-MA/N-HS-A (80/20, dioxane, 80 °C, [ΣM] = 0.67 mol L−1), (b) MPEG1100-MA/N-HS-A (80/20, THF, 60 °C, [ΣM] = 0.5 mol L−1), (c) PEG375-A/N-HS-A (80/20, dioxane, 80 °C, [ΣM] = 0.33 mol L−1) and (d) MPEG300-MA/PFP-MA (80/20, dioxane, 80 °C, [ΣM] = 0.67 mol L−1). (Bottom) Total monomer conversion vs. time plot for different copolymerizations in THF (60 °C) or dioxane (80 °C) with a (M)PEGx-(M)A/aE-ratio of 80/20 and the initial monomer/initiator ratio of 100 adjusted in the feed. | ||
Finally, the copolymerization of MPEG300-MA and PFP-MA could be performed under the same conditions as MPEG300-MA and N-HS-A proving again the lower reactivity of the macromonomer. After 3 h and 6 h the conversion of MPEG300-MA was 74.8 and 86.8%, that of PFP-MA 88.8 and 97.3%.
In Fig. 2e the overall monomer conversion–time plots reveal characteristic differences after 3 h reaction time, however, after 6 h the overall monomer conversions are between 77.0 and 95.6%. Thus a comparison of the thiol functional polyacrylates should give information on the influence of the length of the PEG-chain, the influence of the PEG-chain end-group and on the reactivity of the two active esters.
Four series of (M)PEG-(meth)acrylates with N-HS-A and PFP-MA copolymers were prepared for further modification by polymer analogues reactions. A summary of the characteristics of the synthesized copolymers is given in Table 2 (Experimental part). Changing the active ester/macromonomer ratio in the feed the amount of the active ester repeating units in the backbone of each copolymer could be varied in a broad range. With some exceptions, the active ester/macromonomer ratio adjusted in the feed could be observed in the copolymers.
Synthesis of hydrophilic thiol functional copolymers by polymer analogues reactions with cysteamine or cystamine
Thiol functional copolymers were synthesized via nucleophilic substitution of N-hydroxysuccinimide or pentafluorophenol with aminethiols, such as cysteamine or cystamine, followed by the TCEP·HCl treatment of the resulting product polymers at pH > 7 in degassed water to get the water soluble SH-functional copolymers (Scheme 3). In first experiments cysteamine was reacted with MPEG300-MA-co-N-HS-A and MPEG1100-MA-co-N-HS-A in methylene chloride at room temperature and subsequently treated with TCEP·HCl. The yield was relatively high (76.3%), however, side reactions occurred. As reported in the literature nucleophilic attack occurred not only at the ester carbonyl but also at the imide carbonyl (Scheme 4).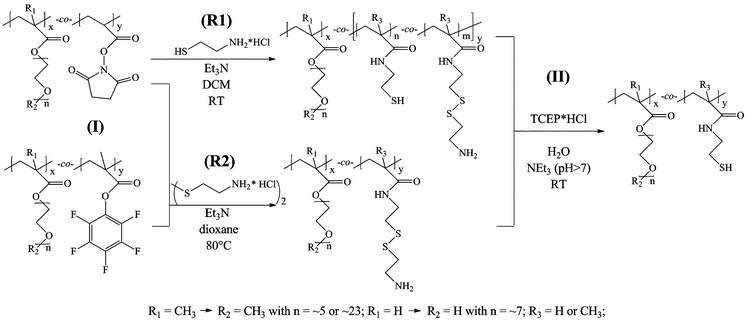 | ||
| Scheme 3 Synthesis route of thiol-functional copolymers using (R1) cysteamine (hydrochloride) or (R2) cystamine dihydrochloride (step I) followed by TCEP·HCl treatment (step II). | ||
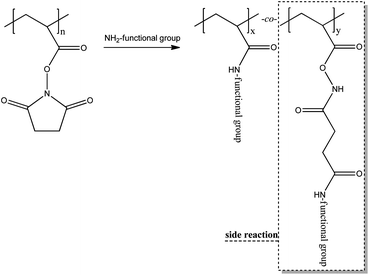 | ||
| Scheme 4 A side reaction known by the literature104 was found to take place converting the N-HS-A repeating units with the free base cysteamine (Table 4, polymer 12). | ||
This side reaction could be avoided if cysteamine hydrochloride and a mixture of methylene chloride and triethylamine were used. This way the free amine group is produced in situ in equilibrium (R–NH2·HCl + Et3N ⇌ R–NH2 + Et3N·HCl). The lower concentration of free amine might be the reason for the higher selectivity. Based on these results MPEG300-MA-co-N-HS-A, MPEG1100-MA-co-N-HS-A, PEG375-A-co-N-HS-A and MPEG300-MA-co-PFP-MA were reacted with cystamine dihydrochloride followed by reduction with TCEP·HCl. After purification of the products by dialysis the corresponding copolymers (M)PEGx-(M)A-co-N-ME-(M)AA were obtained in good yields (Table 6, Experimental part). In opposite to the literature the TCEP·HCl was found not to be able to reduce the disulfide bonds within minutes at pH < 7.105
The mole ratio of MPEGx-MA and N-ME-AA was determined from 1H NMR spectra by comparing the ratio between the integrals of the methylene protons of the N-ME-AA (Fig. 3b, –CH2–SH, signal I) and the methyl protons of the MPEGx-MA (Fig. 3b, –O–CH3, signal E) appearing at δ = 2.6 and 3.4 ppm. In analogy to this, the mole ratio of MPEGx-MA and N-ME-MAA was determined. Furthermore, the mole ratio of PEG375-A and N-ME-AA was determined from 1H NMR spectra by comparing the ratio between the integrals of the methylene protons of the N-ME-AA (Fig. 3a, –CH2–SH, signal i) and the methylene protons of PEG375-A (Fig. 3a, –COO–CH2–, signal c) appearing at δ = 2.6 or 4.2 ppm, respectively.
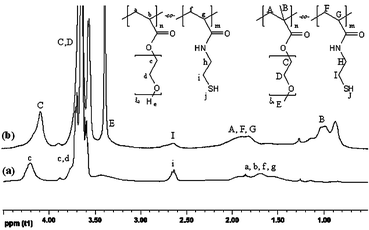 | ||
| Fig. 3 1H NMR spectra of (a) poly(PEG375-A-co-N-ME-AA) and (b) poly(MPEG300-MA-co-N-ME-AA). | ||
Chemical gelation of SH-functional copolymers by disulfide bonds formation or by Michael-type addition reaction
Chemical gelation by oxidation of the SH-groups to disulfide bonds by H2O2 or by reaction of the thiol functional copolymer with EG-bis-A or PEG575-bis-A, via a Michael-type addition reaction, is shown in Scheme 5. Each cross-linking reaction was carried out in the presence of triethylamine to adjust the pH ≈ 8 in order to activate the thiol groups of the corresponding precursor copolymers for the oxidation to disulfide bonds or the Michael-type addition reaction.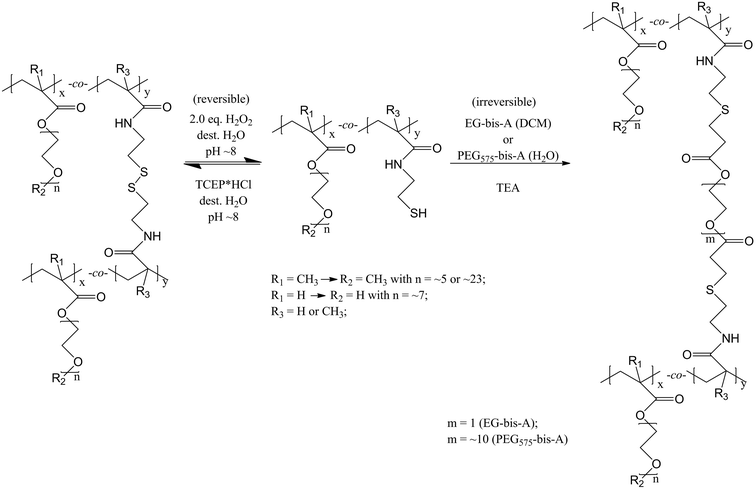 | ||
| Scheme 5 Gelation by oxidation of the SH-groups to disulfide bonds by H2O2 (reversible) or by reaction of the functional copolymer with EG-bis-A or PEG575-bis-A (irreversible), via a Michael-type addition reaction. | ||
The synthesized gels are summarized in Table 7 (see Experimental part). In order to explore the cross-linking behaviour of the thiol functional copolymers by oxidationviaH2O2 to disulfide bonds different ratios of SH-groups/H2O2 were used. Using ≤2.0 eq. of H2O2 (with respect to the thiol groups) the cross-linking was found to be fully reversible. Cleaving the gels cross-linked by disulfide bonds using 1.1 eq. of TCEP·HCl (with respect to the amount of possible disulfide bonds) the corresponding water soluble copolymers were obtained again, observing the same content of thiol groups compared to the corresponding precursor copolymer detected using 1H NMR spectroscopy. Cross-linking with 20.6 eq. of H2O2 (Table 7, G1) the gelation process was found to be not reversible anymore. Analyzing the obtained gel using Raman spectroscopy different oxidation states of the sulfur atoms in the S–S bonds, such as sulfinothioates and sulfonothioates, were observed. Thus, a recovery of water soluble, thiol functional copolymers using TCEP·HCl was not possible anymore. Therefore, in order to get reversibly cross-linkable gels a strict control of the amount of H2O2 used is necessary.
In order to determine the cross-linking kinetics using H2O2, EG-bis-A and PEG575-bis-A rheological measurements of the cross-linking reactions of poly(MPEG300-MA-co-N-ME-AA) as a representative example were performed. Using H2O2 (30 wt% H2O2 in H2O) or PEG575-bis-A as cross-linking agents hard gels were obtained within few minutes, making the kinetic studies under the chosen reaction conditions viarheometry impossible (Table 7, G1, G2, G5, G6 and G7).
Only the rate of the cross-linking reaction using EG-bis-A as the cross-linking agent (Table 7, G3) was found to be low enough to determine the dependency of the elastic modulus G′, the viscous modulus G′′ and the loss factor tan δ = G′′/G′ on time at 25 °C. A semilogarithmic plot (Fig. 4) was chosen for a clear representation of the changes in the dynamic moduli of the reaction, especially at the beginning of the cross-linking reaction. Directly after mixing of the reactants, the reaction mixture exhibited an initial (after 2 min of the preparation time) elastic modulus G′ of about 250 Pa and a viscous modulus G′′ of 100 Pa. The cross-linking of the poly(MPEG300-MA-co-N-ME-AA) with EG-bis-A via Michael-type addition reaction started immediately, as represented by the rapid increase of both moduli (G′ and G′′), achieving the final elastic modulus G′ of ca. 48000 Pa after about 16 h.
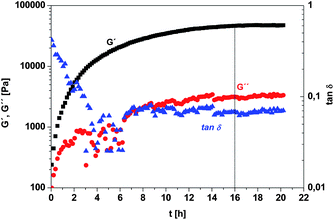 | ||
| Fig. 4 Elastic modulus G′, viscous modulus G′′ and loss factor tan δ during cross-linking of poly(MPEG300-MA-co-N-ME-AA) (Table 7, G3) with 0.5 eq. of EG-bis-A and 1.1 eq. of TEA (with respect to thiol groups content), via a Michael-type addition reaction at room temperature. After 16 h the final hardness/cross-linking density was achieved (dashed line). | ||
The use of different graft copolymers with (M)PEG- and thiol side groups and of different cross-linking agents (procedures) resulted in gels of different chemical compositions (Table 8). To characterize these gels and to determine the influence of changes in the composition on the equilibrium water content (EWC) thermogravimetric analyses of the gels were performed.
| Gel | Precursor polymer, (no.) composition [mol%:mol%] |
Cross-linker (SH-eq.) | EWCa,b [%] | Swellingc [%] | (M)PEG d [wt%] |
|---|---|---|---|---|---|
| a Equilibrium water content (EWC) determined by TGA (10 K min−1). b Total water content in the resulting gel determined by TGA (10 K min−1). c Calculated swelling at the equilibrium water content. d Calculated amount of (M)PEG exclusively contributed by the side-chains with respect to total weight of the gel. e Calculated amount of (M)PEG exclusively contributed by the cross-linking agent with respect to total weight of the gel. | |||||
| G1 | MPEG300-MA-co-N-ME-AA (16), 83:17 |
H2O2 (20.5) | 84.8 | 557.9 | 65.7 |
| G2 | MPEG300-MA-co-N-ME-AA (17), 85:15 |
H2O2 (2.0) | 28.4 | 40.0 | 66.5 |
| G3 | MPEG300-MA-co-N-ME-AA (17), 85:15 |
EG-bis-A (0.5) | 35.1 | 54.1 | 63.6(+1.2)e |
| G4 | MPEG300-MA-co-N-ME-AA (17), 85:15 |
PEG575-bis-A (0.5) | 61.8 | 161.8 | 57.5(+10.6)e |
| G5 | MPEG1100-MA-co-N-ME-AA (19), 74:26 |
H2O2 (2.0) | 80.7 | 418.1 | 88.6 |
| G6 | PEG375-A-co-N-ME-AA (20), 74:26 |
H2O2 (2.4) | 65.2 | 187.4 | 68.9 |
| G7 | MPEG300-MA-co-ME-MAA (21), 89:11 |
H2O2 (2.0) | 41.9 | 72.1 | 67.6 |
The TGA traces of the gels, obtained by disulfide bond formation using about 2 eq. (G2, G5, G6 and G7) or 20.6 eq. (G1) of H2O2 (Fig. 5, left) clearly demonstrate that the ability of the gels to absorb water is dependent on several parameters: (i) the length of PEG side chains (G2vs.G5), (ii) the end-group of the PEG side chain (G2vs.G6), (iii) the sequence of the comonomer units in the backbone (methacrylate-co-acrylate or methacrylate-co-methacrylate; G2vs.G7) and (iv) the amount of H2O2 used for the oxidation of the thiol groups (G2vs.G1).
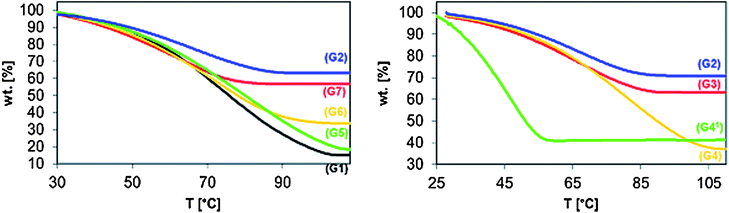 | ||
| Fig. 5 TGA curves of the synthesized gels (heating rate 10 K min−1): determination of the equilibrium water content (EWC). Left: poly(MPEG300-MA-co-N-ME-AA) G2, poly(MPEG300-MA-co-N-ME-MAA) G7, poly(PEG375-A-co-N-ME-AA) G6, poly(MPEG1100-MA-co-N-ME-AA) G5 using 2.0 eq. H2O2 and poly(MPEG300-MA-co-N-ME-AA) G1 using 20.6 eq. H2O2. Right: poly(MPEG300-MA-co-N-ME-AA) based gels cross-linked by H2O2G2, EG-bis-A G3, PEG575-bis-A G4, and in addition G411 with a heating rate of 1 K min−1. | ||
The loss of water begins in all cases already at low temperatures and becomes faster when the temperature is increasing, achieving the dry state latest at 105 °C represented by the final plateau (this strongly depends on the heating rate).
By increasing the length of the MPEG side chains by a factor of 4.6 from ∼5 to ∼23 ethylene glycol repeating units (G2vs.G5) the swelling ability of the gels increases from 40.0 to 418.1% (∼10.5 times). Keeping the length of the (M)PEG side chain (more or less) constant, while the terminal methoxy group was replaced by a hydroxyl group (G2, n ≈ 5 vs.G6, n ≈ 7) the swelling ability of the gels increased from 40.0 (G2) to 187.4% (G6), 4.7 times. Furthermore, changing the sequence of the monomer units in the backbone from methacrylate-co-acrylate (G2) to methacrylate-co-methacrylate (G7) the ability of swelling slightly increased (G2 40.0% vs.G7 72.1%), which was unexpected, considering the acrylates being more polar (hydrophilic) than the corresponding methacrylates. Finally, by increasing the amount of H2O2 used for the oxidation of the thiol groups beside disulfide bonds sulfinothioates and sulfonothioates were obtained (G2 2.0 eq. vs.G1 20.6 eq.). This not only leads to the change to an irreversible cross-linking but also to an increase in the swelling ability of the gels obtained by the factor of 13.9 (G2 40.0 vs.G1 557.9%).
The TGA traces of the gels, obtained by cross-linking of poly(MPEG300-MA-co-N-ME-AA) (17) using different cross-linking agents, such as H2O2 (2.0 eq., G2), EG-bis-A (0.5 eq., G3) and PEG575-bis-A (0.5 eq., G4) (Fig. 5, right) clearly demonstrate that by increasing the spacer length (length of the cross-linking bridges) between the main chains of the polymer network (G2 –S–S– < G3 EG-bis-A < G4PEG575-bis-A) the amount of absorbed water increases.
Using EG-bis-A as the cross-linking agent instead of H2O2, not only the length of the bridges—the distance between the main chains of the polymer network—increases slightly, but also additional polar groups are introduced into the network, leading to an increase of the swelling ability (G2 40.0 vs.G3 54.1%). Furthermore, increasing the number of ethylene glycol repeating units in the cross-linking agent by a factor of 10 (EG-bis-A: n = 1 vs.PEG575-bis-A: n ≈ 10) the observed swelling ability was found to increase by a factor of 3.0 (G3 54.1 vs.G4 161.8%) and a factor of 4.0 in comparison to gel obtained using H2O2 as cross-linking agent (G2 40.0 vs.G4 161.8%) of the corresponding copolymer.
In order to distinguish between (but also to quantify) different types of absorbed water, such as free, slightly bounded and strongly bounded water, the TGA measurement of the swollen gel obtained from the Michael-type addition reaction of poly(MPEG300-MA-co-N-ME-AA) to PEG575-bis-A (G4) was repeated (Fig. 5, right) using a lower heating rate (1 K min−1, curve 3). The swelling ability was found to be of the same order of magnitude as for the measurement with a heating rate of 10 K min−1 however, the plateau was reached at a much lower temperature (∼55 °C at 1 K min−1vs. ∼100 °C at 10 K min−1). From the shape of the curve—the uniform decrease of the weight—no difference in the strength of the absorbed water could be detected.
Summary and conclusion
Copolymers based on (M)PEGx-(meth)acrylates were prepared by free radical copolymerization of MPEG300-MA, MPEG1100-MA or PEG375-A with N-acrylsuccinimide or pentafluorophenyl methacrylate. Kinetic studies of the copolymerizations with the initial macromonomer/active ester ratio of 80/20 mol% in the feed were performed, showing a higher reactivity of the active esters in comparison to the macromonomers. The average number of active groups per polymer chain can be adjusted by the comonomer ratio in the feed. Furthermore, a synthetic procedure was developed for the preparation of water soluble poly(meth)acrylates with reactive thiol groups in the side chain. Finally, chemically cross-linked gels were prepared by oxidation of the thiol groups in the precursor copolymers with H2O2 and formation of disulfide bonds, making the cross-linking of the corresponding gels reversible using up to 2.0 eq. of the H2O2 and irreversible in the case of the use of 20.6 eq. of H2O2 (eq. amounts are given with respect to the number of thiol groups in the precursor copolymer). By reacting the SH-functional copolymers with EG-bis-A or PEG575-bis-Avia a Michael-type addition reaction irreversibly cross-linked gels were obtained. A kinetic study of the cross-linking reaction of poly(MPEG300-MA-co-N-ME-AA) with EG-bis-A was performed using rheometry. The characteristics of the gels with respect to the equilibrium water content (EWC) was determined using thermogravimetric analysis, showing a brought range of the degree of swelling depending on the chemical composition of the precursor copolymers and the cross-linking agent.References
- F. E. Bailey, Jr and J. V. Koleske, Poly(ethylene oxide), Academic Press, New York, 1976 Search PubMed.
- N. G. Gaylard, Polyethers, part 1, Interscience, New York, 1963 Search PubMed.
- J. Furukawa and T. Saegusa, Polymerization of Aldehydes and Oxides, Interscience, New York, 1963 Search PubMed.
- J. H. Lee, H. B. Lee and J. D. Andrade, Prog. Polym. Sci., 1995, 20, 1043 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3 CrossRef CAS; G. S. Kwon and T. Okano, Adv. Drug Delivery Rev., 1996, 21, 107 CrossRef CAS.
- S. B. La, T. Okano and K. Kataoka, J. Pharm. Sci., 1996, 85, 85 CrossRef CAS.
- T. Riley, T. Govender, S. Stolnik, C. D. Xiong, M. C. Garnett, L. Illum and S. S. Davis, Colloids Surf., B, 1999, 16, 147 CrossRef CAS.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143 CrossRef.
- S. Zalipsky, Adv. Drug Delivery Rev., 1995, 16, 157 CrossRef CAS.
- P. Bailon, A. Palleroni, C. A. Schaffer, C. L. Spence, W.-J. Fung, J. E. Porter, G. K. Ehrlich, W. Pan, Z.-X. Xy, M. W. Modi, A. Farid and W. Berthold, Bioconjugate Chem., 2001, 12, 195 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS.
- In Poly(ethylene glycol) Chemistry and Biological Applications, ed. J. M. Harris and S. Zalipsky, ACS, Washington, DC, USA, 1997, p. 1 Search PubMed.
- R. Knischka, P. J. Lutz, A. Sunder, R. Mulhaupt and H. Frey, Macromolecules, 2000, 33, 315 CrossRef CAS; T. K. Bera, D. Taton and Y. Gnanou, Polym. Mater. Sci. Eng., 1997, 77, 126 Search PubMed.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933 CrossRef CAS.
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150 CrossRef CAS.
- S. Herman, G. Hooftman and E. Schlacht, J. Bioact. Compat. Polym., 1995, 10, 145 Search PubMed.
- M. Benaglia, R. Annunziata, M. Cinquini, F. Cozzi and S. Ressel, J. Org. Chem., 1998, 63, 8628 CrossRef CAS.
- R. P. Lanza, R. Langer and W. L. Chick, Principles of Tissue Engineering, R.G. Landes/Academic Press, San Diego, CA, 1997 Search PubMed.
- B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Biomaterials Science: An Introduction to Material in Medicine, Academic Press, San Diego, CA, 2000 Search PubMed.
- N. A. Peppas and R. Langer, Science, 1994, 263, 1715 CrossRef CAS.
- L. Christenson, A. G. Mikos, D. F. Gibbons and G. L. Picciolo, Tissue Eng., 1997, 3, 71 Search PubMed.
- J. L. Hernandez-Lopez, R. E. Bauer, W.-S. Chang, G. Glasser, D. Grebel-Koehler, M. Klapper, M. Kreiter, J. Leclaire, J.-P. Majoral, S. Mittler, K. Mullen, K. Vasilev, T. Weil, J. Wu, T. Zhu and W. Knoll, Mater. Sci. Eng., C, 2003, 23(1–2), 267 Search PubMed.
- M. J. Cloninger, Curr. Opin. Chem. Biol., 2002, 6(6), 742 CrossRef CAS.
- R. Van Heerbeek, P. C. J. Kamer, P. W. N. M. Van Leeuwen and J. N. H. Reek, Chem. Rev., 2002, 102, 3717 CrossRef.
- M. A. Carnahan, C. Middleton, J. Kim, T. Kim and M. W. Grinstaff, J. Am. Chem. Soc., 2002, 124, 5291 CrossRef CAS.
- D. Taton, M. Saule, J. Logan, R. Duran, S. Hou, E. L. Chaikof and Y. Gnanou, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1669 CrossRef CAS.
- N. N. Reed and K. D. Janda, Org. Lett., 2000, 2, 1311 CrossRef CAS.
- M. Hans, H. Keul and M. Moeller, Polymer, 2009, 50, 1103 CrossRef CAS.
- A. Fitton, J. Hill, D. Jane and R. Miller, Synthesis, 1987, 1140 CrossRef CAS.
- D. Taton, A. Le Borgne, M. Sepulchre and N. Spassky, Macromol. Chem. Phys., 1994, 195, 139 CrossRef CAS.
- M. Schappacher, C. Billaud, C. Paulo and A. Deffieux, Macromol. Chem. Phys., 1999, 200, 2377 CrossRef CAS.
- M. Schappacher, J. Bernard and A. Deffieux, Macromol. Chem. Phys., 2003, 204, 762 CrossRef CAS.
- A. Deffieux and M. Schappacher, Macromolecules, 1999, 32, 1797 CrossRef.
- S. W. Ryu and A. Hirao, Macromolecules, 2000, 33, 4765 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Möller, Macromolecules, 1998, 31, 9413 CrossRef CAS.
- G. Cheng, A. Böker, M. Zhang, G. Krausch and A. H. E. Müller, Macromolecules, 2001, 34, 6883 CrossRef CAS.
- H. G. Börner, K. Beers, K. Matyjaszewski, S. S. Sheiko and M. Möller, Macromolecules, 2001, 34, 4375 CrossRef.
- M. Zhang, T. Breiner, H. Mori and A. H. E. Müller, Polymer, 2003, 44, 1449 CrossRef CAS.
- M. Wintermantel, M. Gerle, K. Fischer, M. Schmidt, I. Wataoka, H. Urakawa, K. Kajiwara and Y. Tsukahara, Macromolecules, 1996, 29, 978 CrossRef CAS.
- Y. Tsukahara, K. Mizuno, A. Segawa and Y. Yamashita, Macromolecules, 1989, 22, 1546 CrossRef CAS.
- Y. Tsukahara, K. Tsutsumi, Y. Yamashita and S. Shimada, Macromolecules, 1990, 23, 5201 CrossRef CAS.
- M. Wintermantel, M. Schmidt, Y. Tsukahara, K. Kajiwara and S. Kohjiya, Macromol. Rapid Commun., 1994, 15, 279 CrossRef CAS.
- S. Qin, H. G. Börner, K. Matyjaszewski and S. S. Sheiko, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 237 Search PubMed.
- R. Venkatesh, L. Yajjou, C. E. Koning and B. Klumperman, Macromol. Chem. Phys., 2004, 205, 2161 CrossRef CAS.
- K. Ishizu and H. Kakinuma, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 63 Search PubMed.
- A. Zhang, J. Barner, I. Goessl, J. P. Rabe and A. D. Schlüter, Angew. Chem., Int. Ed., 2004, 43, 5185 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- K. A. Davis and K. Matyjaszewski, Adv. Polym. Sci., 2002, 159, 2 CAS.
- H. G. Börner, D. Duran, K. Matyjaszewski, M. da Silva and S. S. Sheiko, Macromolecules, 2002, 35, 3387 CrossRef.
- D. Neugebauer, K. Matyjaszewski, M. da Silva and S. S. Sheiko, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2002, 43, 239 Search PubMed.
- C. Li, N. Gunari, K. Fischer, A. Janshoff and M. Schmidt, Angew. Chem., Int. Ed., 2004, 43, 1101 CrossRef CAS.
- S. Muthukrishnan, M. Zhang, M. Burkhardt, M. Drechsler, H. Mori and A. H. E. Müller, Macromolecules, 2005, 38, 7926 CrossRef CAS.
- J. Tao, S. Stewart, G. Liu and M. Yang, Macromolecules, 1997, 30, 2738 CrossRef CAS.
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960 CrossRef CAS.
- G. Liu, Z. Li and X. Yan, Polymer, 2003, 44, 7721 CrossRef CAS.
- G. Liu, L. Qiao and A. Guo, Macromolecules, 1996, 29, 5508 CrossRef CAS.
- G. Liu, Adv. Mater., 1997, 9, 437 CrossRef CAS.
- K. Ishizu, T. Ikemoto and A. Ichimura, Polymer, 1999, 40, 3147 Search PubMed.
- M. Zhang, M. Drechsler and A. H. E. Müller, Chem. Mater., 2004, 16, 537 CrossRef CAS.
- M. Zhang, P. Teissier, M. Krekhov, V. Cabuil and A. H. E. Müller, Prog. Colloid Polym. Sci., 2004, 126, 35 Search PubMed.
- M. Zhang, C. Estournes, W. Bietsch and A. H. E. Müller, Adv. Funct. Mater., 2004, 14, 871 CrossRef CAS.
- C. Price, Pure Appl. Chem., 1983, 55, 1563 CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS.
- P. Dziezok, S. S. Sheiko, K. Fischer, M. Schmidt and M. Möller, Angew. Chem., Int. Ed. Engl., 1997, 36, 2812 CrossRef CAS.
- M. Gerle, K. Fischer, S. Roos, A. H. E. Müller, M. Schmidt, S. S. Sheiko, S. Prokhorova and M. Möller, Macromolecules, 1999, 32, 2629 CrossRef CAS.
- D. Pantazis, I. Chalari and N. Hadjichristidis, Macromolecules, 2003, 36, 3783 CrossRef CAS.
- Y. Tsukahara, J. Inoue, Y. Ohta, S. Kohjiya and Y. Okamoto, Polym. J., 1994, 26, 1013 CrossRef CAS.
- P. Rempp, P. Lutz, P. Masson, P. Chaumont and E. Franta, Makromol. Chem., Suppl., 1985, 13, 47 Search PubMed.
- S. Kanaoka, M. Sueoka, M. Sawamoto and T. Higashimura, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2513 Search PubMed.
- R. Asami, M. Takaki and Y. Moriyama, Polym. Bull., 1986, 16, 125 Search PubMed.
- K. Nomura, S. Takahashi and Y. Imanishi, Macromolecules, 2001, 34, 4712 CrossRef CAS.
- W. J. Feast, V. C. Gibson, A. F. Johnson, E. Khosravi and M. A. Mohsin, Polymer, 1994, 35, 3542 Search PubMed.
- V. Heroguez, S. Breunig, Y. Gnanou and M. Fontanille, Macromolecules, 1996, 29, 4459 CrossRef CAS.
- V. Heroguez, Y. Gnanou and M. Fontanille, Macromolecules, 1997, 30, 4791 CrossRef CAS.
- K. Yamada, M. Miyazaki, K. Ohno, T. Fukuda and M. Minoda, Macromolecules, 1999, 32, 290 Search PubMed.
- M. Riza, S. Mustapha and R. Dewi, Malays. J. Chem., 2004, 6, 24 Search PubMed.
- T. Delair, M. H. Charles, P. Cros, A. Laayounn, B. Mandrand and C. Pichot, Polym. Adv. Technol., 1998, 9, 349 Search PubMed.
- R. Hart and A. vanDormael, Bull. Soc. Chim. Belg., 1956, 65, 517 Search PubMed.
- L. Veron, M. C. de Bignicourt, T. Delair, C. Pichot and B. Mandrand, J. Appl. Polym. Sci., 1996, 60, 235 Search PubMed.
- P. Theato and R. Zentel, Langmuir, 2000, 16, 1801 CrossRef CAS.
- P. Theato, R. Zentel and S. Schwarz, Macromol. Biosci., 2002, 2, 387 CrossRef CAS.
- A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6324 CrossRef CAS.
- G. M. Eichenbaum, P. F. Kiser, D. Shah, S. A. Simon and D. Needham, Macromolecules, 1999, 32, 8996 CrossRef CAS.
- A. E. Ivanov, J. Macromol. Sci., Part A: Pure Appl. Chem., 1999, 36, 1981 Search PubMed.
- M. Eberhardt, R. Mruk, R. Zentel and P. Theato, Eur. Polym. J., 2005, 41, 1569 CrossRef CAS.
- S. A. Robb, B. H. Lee, R. McLemore and B. L. Vernon, Biomacromolecules, 2007, 8, 2294 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155 CrossRef CAS.
- H. A. Aliyar, P. D. Hamilton and N. Ravi, Biomacromolecules, 2005, 6, 204 CrossRef CAS.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648 CrossRef CAS.
- R. Kizek, J. Vacek, L. Trnková and F. Jelen, Bioelectrochemistry, 2004, 63, 19 CrossRef CAS.
- M. Heggli, N. Tirelli, A. Zisch and J. A. Hubbell, Bioconjugate Chem., 2003, 14, 967 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713 CrossRef CAS.
- F. Cellesi, N. Tirelli and J. A. Hubbell, Biomaterials, 2003, 25, 5115 Search PubMed.
- D. F. Siquieira Petri, S. W. Choi, H. Beyer, T. Schimmel, M. Bruns and G. Wenz, Polymer, 1998, 40, 1593 Search PubMed.
- V. K. Gupta and N. L. Abbott, Science, 1997, 276, 1533 CrossRef CAS.
- T. Kim, R. M. Crooks, M. Tsen and L. Sun, J. Am. Chem. Soc., 1995, 117, 3963 CrossRef CAS.
- T. Sato, K. Terada, J. Yamauchi and T. Okaya, Makromol. Chem., 1993, 194, 175 Search PubMed.
- P. D. Mumbauer, D. H. Carey and G. S. Ferguson, Chem. Mater., 1995, 7, 1303 Search PubMed.
- J. M. Stouffer and T. J. McCarthy, Macromolecules, 1988, 21, 1204 CrossRef CAS.
- K. E. Goncalves, G. Carlson, X. Chen, J. Kumar, F. Aranda, R. Perez and M. Jose-Yacaman, J. Mater. Sci. Lett., 1996, 15, 948 CAS.
- R. Premachandran, S. Banerjee, V. T. John and G. L. McPherson, Chem. Mater., 1997, 9, 1342 CrossRef CAS.
- H. J. Yang, C.-A. Cole, N. Monji and A. S. Hoffman, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 219 CrossRef CAS.
- J. C. Han and G. J. Han, Anal. Biochem., 1994, 220, 5 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |