Mechanistic study of hydrothermal synthesis of aromatic polyimides†
Miriam M.
Unterlass
*,
Daniel
Kopetzki
,
Markus
Antonietti
and
Jens
Weber
*
Max-Planck-Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, Am Muehlenberg 1, D-14476, Golm, Germany. E-mail: miriam.unterlass@mpikg.mpg.de; jens.weber@mpikg.mpg.de
First published on 31st May 2011
Abstract
A mechanistic study regarding the synthesis of aromatic polyimides (PIs) under hydrothermal conditions is presented. We demonstrate evidence that the first step of the reaction is the formation of a nylon type salt, which can be isolated. Upon heating the salt in the presence of water at 180 °C, polyimides are formed from the salts. Additionally performed DFT calculations substantiate the experimental findings and give evidence that the formation of imides is energetically favorable in hot water. The intermediate salts as well as the resulting PIs are characterized by diverse analytical techniques (SEC, FT-IR, WAXS, SEM). By selective fractionation, it is possible to obtain polyimides of moderate to high molecular weights that form tough films. The polymerization mechanism is discussed, and it is shown that different loci of reaction are possible, namely interfacial polycondensation, heterophase polymerization and topochemical transformations, which seem to occur in parallel. Finally, poloxamers can be used as additives for the fine-tuning of the polymer morphology.
Introduction
Hydrothermal (HT) synthesis, i.e. a chemical reaction carried out in hot water under pressure, is of growing interest, as it is inherently green, due to water being an environmentally friendly and benign solvent and the efficient process handling. Many materials are already made that way, e.g.zeolites, metal–organic frameworks (MOFs) and hybrid materials.1 Interestingly, to the best of our knowledge, the first reported HT synthesis of a pure polymer—apart from inorganic–organic hybrid materials and aqueous latices—concerns the formation of aromatic polyimides (PIs).2 This fact is remarkable since PIs are typically obtained by polycondensation reactions under the strict absence/removal of water.Given the huge academic and industrial interest for various properties such as high E-modulus, temperature stability, gas permeability, intrinsic microporosity, dielectric and optical properties and numerous others,3–13 a new and green synthesis routine towards these engineering polymers would be of high interest.
Polycondensations are step-growth processes, where each reaction step theoretically underlies the principle of micro-reversibility. At first sight, a chemical transformation with water as a side-product, carried out in water, seems to be contradictory to Le Chatelier's principle.14 The paradox of this approach is underlined by the fact that PIs could not consistently be obtained until imidization was carried out under the complete exclusion of water. This is true for both classical pathways, that is the (i) two-step procedure via polyamic acids (PAAs) and (ii) the one-step procedure in m-cresol/isoquinoline (cat.). For completeness, it should be mentioned that there are also other synthetic pathways (e.g. using molten benzoic acid as solvent), which however also require the absence of water.15,16
It has been postulated that the polyimidization under hydrothermal conditions (H2O, T > 100 °C, elevated pressure) would occur via a nylon-salt-type intermediate (Scheme 1).2
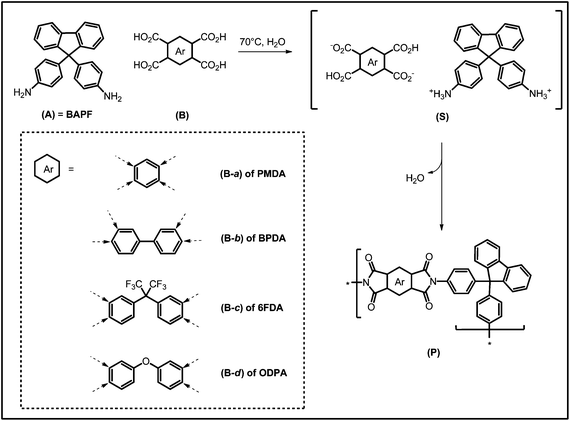 | ||
| Scheme 1 Postulated mechanism of hydrothermal polyimidization. (A) = aromatic bisamine, (B) = aromatic tetracarboxylic acid of the corresponding anhydrides PMDA, BPDA, 6FDA and ODPA, (S) = nylon-type AB-salt, (P) = polymer. (Dashed arrows indicate the position of the carboxylic acid functions.) | ||
Although the authors stated that no detailed mechanism was known up to now, the “salt-monomers” (SS) have already been synthesized by Imai,17a in order to polymerize them to different aromatic PIs via ultra-high-pressure (2800–6000 bar) solid state synthesis. This polymerization of the intermediate (SS) is indeed an applied form of the original PI synthesis pathway,18 the so-called “monomeric salt route”.19 Imai et al. also recognized that the byproduct water was present in the closed reaction vessel and concluded that H2O must have been effectively separated from the polymer product throughout the reaction.17b Moreover, the possibility of forming PIs in water has been recently exploited by Mercier et al. using microwave radiation.17c Despite the use of the hydrothermal synthesis of fully aromatic PIs, no attempts have been made up to now in order to understand this transformation and to employ it for micro- and nanostructure control.
We herein present a case study of the hydrothermal polymerization of 4 different aromatic tetracarboxylic acid anhydrides and their respective free acids (pyromellitic dianhydride (PMDA), 3,3′,4,4′-biphenyltetracarboxylic anhydride (BPDA), 4,4′-(hexafluoroisopropylidene) diphthalic anhydride (6FDA) and 4,4′-oxydiphthalic anhydride (ODPA)) with the aromatic diamine 9,9-bis(4-aminophenyl)fluorine (BAPF). For clearness of presentation, we use these abbreviations also when talking about the carboxylic acid salts (see below for details).
It will be shown that the polymerization has more than one locus of reaction and progresses most effectively along the interface of the as formed salt crystals/salt melts. This enables the formation of porous PI structures by templating processes.
Results and discussion
A clear understanding of the monomer salts (SS) (i.e.(SS)-PMDA-BAPF, (SS)-BPDA-BAPF, (SS)-ODPA-BAPF and (SS)-6FDA-BAPF) is of primary importance. To explore the sensitivity of salt formation, salts were synthesized in 3 different ways. First they were made from unpurified monomers. Secondly, recrystallized monomers and thirdly, the corresponding free acids and recrystallized diamine were employed. The salts were prepared by mixing equimolar amounts of the monomers in deionized water. The resulting mixture was heated to 70–90 °C overnight. The salt could be obtained by filtration after cooling to room temperature.As expected for the formation of salts, potential impurities (e.g. due to non-purified monomers or non-hydrolyzed anhydrides) were not incorporated in the crystalline lattice. Thus, all three approaches lead to the same salts (as indicated by FT-IR, XRD, SEM, etc., see ESI†). Further work was then restricted to the use of monomer salts obtained from the free acids and recrystallized BAPF, only.
FT-IR analysis (Fig. 1) clearly shows the appearance of ammonium modes,20i.e. the two broad arylammonium21 vibration modes at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ≈ 2830 cm−1 and ≈ 2580 cm−1 as well as the N–H deformation mode at
≈ 2830 cm−1 and ≈ 2580 cm−1 as well as the N–H deformation mode at ![[small delta, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e125.gif) = 1473 cm−1, whereas the asymmetrical and the symmetrical N–H valence modes of the primary amino groups in BAPF (i.e. ≈ 3430 cm−1, ≈ 3400 cm−1) disappear. Moreover, the bands corresponding to carboxylic acids (combinational bands due to O–H valence vibrations and intramolecular H-bonding thereof at ≈ 3000 − 2500 cm−1, C
= 1473 cm−1, whereas the asymmetrical and the symmetrical N–H valence modes of the primary amino groups in BAPF (i.e. ≈ 3430 cm−1, ≈ 3400 cm−1) disappear. Moreover, the bands corresponding to carboxylic acids (combinational bands due to O–H valence vibrations and intramolecular H-bonding thereof at ≈ 3000 − 2500 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O valence mode of arylcarboxylic acids at = 1695 cm−1) diminish strongly in intensity, whereas the carboxylate antisymmetric ( = 1600 cm−1)21 and symmetric ( = 1550 cm−1) valence modes appear.
O valence mode of arylcarboxylic acids at = 1695 cm−1) diminish strongly in intensity, whereas the carboxylate antisymmetric ( = 1600 cm−1)21 and symmetric ( = 1550 cm−1) valence modes appear.
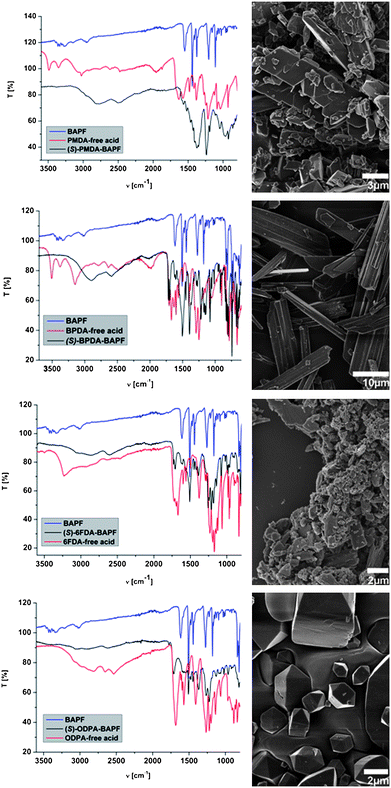 | ||
| Fig. 1 (Left side) Superimposition of FT-IR (ATR) spectra of salts (in black), the corresponding free tetracarboxylic acids (red) and BAPF (blue). T = transmittance, ν = wavenumber. (Right side) SEM micrographs of the corresponding salts. | ||
The AB-salts showed a crystalline appearance for (SS)-PMDA-BAPF, (SS)-BPDA-BAPF, partly for (SS)-ODPA-BAPF and also an organized structure for (SS)-6FDA-BAPF, as can be seen in SEM images in Fig. 1. (SS)-PMDA-BAPF shows an aligned rhombohedral morphology. Sheet-like crystals, ca. 50 μm in size, are obtained from (SS)-BPDA-BAPF. (SS)-ODPA-BAPF exhibits cubes with pyramidal caps, which are embedded in a presumably amorphous matrix. For (SS)-6FDA-BAPF, only agglomerates organized from primary nanoparticles are obtained, which are presumably nano- or liquid crystalline, as judged from their birefringence.
Due to the poor solubility of all four salts in water, it was not possible to obtain sufficiently large single crystals, in accordance with previous reports.14 Therefore, powder diffraction data were collected by wide-angle-X-ray-scattering (WAXS). The WAXS patterns (Fig. 2) show a high crystallinity for three out of four salts. Interestingly, the degree of crystallinity of the precipitate seems to decrease with the increase in geometric flexibility (i.e. increasing freedom of bending, rotation, etc.) of the aromatic dicarboxylate. This follows the order PMDA < BPDA < 6FDA < ODPA. (SS)-ODPA-BAPF and (SS)-6FDA-BAPF show indeed an amorphous background in their WAXS-patterns, indicating that these salts are only partially crystalline. The geometric complexity and the sterical demands of ODPA and 6FDA might interfere with the ability of rapid organization towards well-defined crystals, whereas PMDA and BPDA geometrically present a higher tendency to stack. The amorphous background for (SS)-6FDA-BAPF and (SS)-ODPA-BAPF may of course also arise from polydispersity in crystal size and shape, and polycrystallinity. Regarding the SEM micrographs, this may not seem very plausible, one should nonetheless keep in mind that a macroscopically uniform crystal habit does not imply monocrystallinity at all. The trend that an increasing geometric complexity results in a decreased crystallinity is well known for spiro compounds, where introducing bulky substituents into the already quite large molecules, such as tert-butyl groups, slows down the already quite unfavorable crystallization kinetics.22
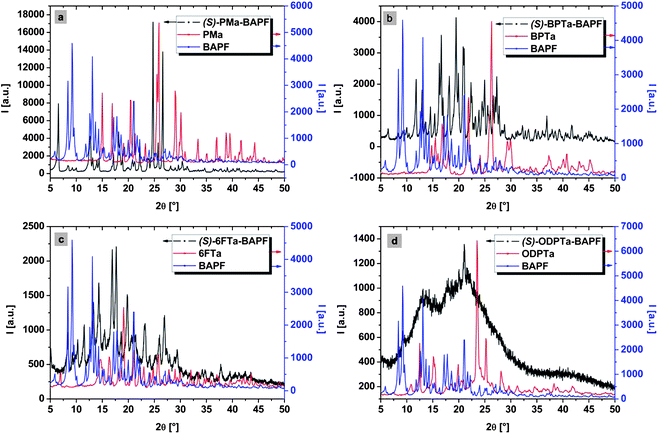 | ||
| Fig. 2 Superimposition of WAXS patterns of salts (in black), corresponding free tetracarboxylic acids (red) and BAPF (blue). (a) = (SS)-PMDA-BAPF, (b) = (SS)-BPDA-BAPF, (c) = (SS)-ODPA-BAPF and (d) = (SS)-6FDA-BAPF; the single patterns (without overlay) can be found in the ESI†. | ||
Elemental Analysis (EA) confirmed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composition of the salt. Though this is not a direct proof for an exact 1:1 cation:anion stoichiometry within the crystallites, it nevertheless implies that for polycondensations crucial equimolarity is achieved on a macroscopic scale. Neither FT-IR, nor EA or thermo gravimetric analysis (TGA) show the incorporation of water into the salt structure, in all four cases. Due to the spontaneity of salt formation, we therefore conclude that the first step in hydrothermal synthesis of aromatic PIs is the hydrolysis of the aromatic dianhydride to the corresponding free carboxylic acid, followed by the formation of an AB-type salt showing a stoichiometric composition of dicarboxylic acid, dicarboxylate and bisammonium-comonomer.
:1 composition of the salt. Though this is not a direct proof for an exact 1:1 cation:anion stoichiometry within the crystallites, it nevertheless implies that for polycondensations crucial equimolarity is achieved on a macroscopic scale. Neither FT-IR, nor EA or thermo gravimetric analysis (TGA) show the incorporation of water into the salt structure, in all four cases. Due to the spontaneity of salt formation, we therefore conclude that the first step in hydrothermal synthesis of aromatic PIs is the hydrolysis of the aromatic dianhydride to the corresponding free carboxylic acid, followed by the formation of an AB-type salt showing a stoichiometric composition of dicarboxylic acid, dicarboxylate and bisammonium-comonomer.
The isolated salts were redispersed ((SS)-PMDA-BAPF: 0.20 mol L−1, (SS)-BPDA-BAPF, (SS)-ODPA-BAPF and (SS)-6FDA-BAPF: 0.15 mol L−1) in deionized water by vigorous stirring at room temperature for 1 hour. The dispersions were then transferred to non-stirred glass vials, used as inlets of autoclaves. The autoclaves were heated to 180 °C and kept at this temperature for 12 hours. The reaction temperature has been varied in regard to the highest possible weight average molecular weights (determined viaSEC) and yield. These optimizations led to an optimal reaction temperature of 180 ± 20 °C. The autoclaves operate at autogenous pressure of water, which is at this temperature estimated to be 12–14 bar (see Fig. S7, ESI†). After cooling, the products sediment from the water phase and are slightly yellowish, very typical for aromatic polyimides. In some cases we observe oxidation of the educts, presumably of the diamine, above the waterline and at the interface (see Fig. 3). In principle this could be avoided by purging with nitrogen, this was however not applied.
 | ||
| Fig. 3 Optical appearance of the reaction products. (a) (PP)-PMDA-BAPF, (b) (PP)-BPDA-BAPF, (c) (PP)-ODPA-BAPF and (d) (PP)-6FDA-BAPF. | ||
We restricted analysis to the well separable sediment. In all cases, imides were formed ( = 1775 cm−1, = 1720 cm−1 and = 1365 cm−1) whereas no carboxylic acid, carboxylic acid anhydride, carboxylate, amine or ammonium bands are found left in FT-IR (spectra displayed in the ESI†) of the crude products.
As the validity of the PS universal calibration23 in Size Exclusion Chromatography (SEC) has been investigated for aromatic PIs based on BAPF24 and proven to be valid, SEC has been carried out on the four products in order to analyze their molecular weight distribution (MWD). The elugrams of (PP)-PMDA-BAPF, (PP)-BPDA-BAPF, (PP)-6FDA-BAPF and (PP)-ODPA-BAPF are shown in Fig. 4.
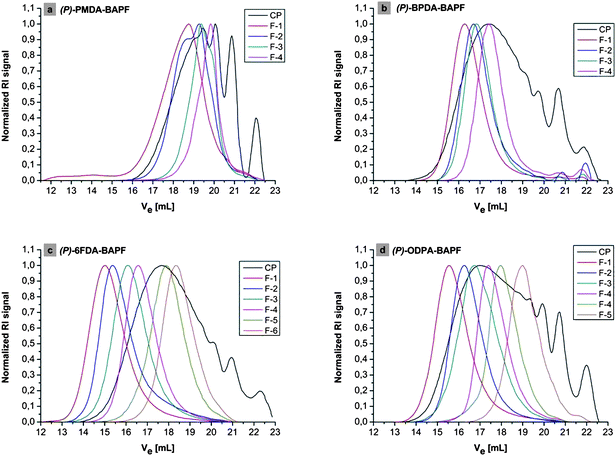 | ||
| Fig. 4 Elugrams of the four polymeric products (a) (PP)-PMDA-BAPF, (b) (PP)-BPDA-BAPF, (c) (PP)-6FDA-BAPF, (d) (PP)-ODPA-BAPF after HT polymerization, designated as CP for the crude product, and of the fractions after fractionate precipitation (F-1, F-2, …,F-n). Normalized RI signal is plotted vs. the elution volume in mL. | ||
Interestingly, the elugrams of the washed and dried crude product show distinct separate peaks, for all four PIs (see black curves in Fig. 4). The following weight average molecular weights and polydispersity indices D were determined (Table 1) (complete SEC data are depicted in Table S1†).
| Compound |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w
(RI)c/g mol−1
w
(RI)c/g mol−1 |
D(RI)c | Yield [%] |
w
(RI)F−1/g mol−1 |
D(RI)F−1 | Yield [%] |
|---|---|---|---|---|---|---|
| a Index c: crude product; index F-1: 1st fraction, highest molecular weight; yield is given in weight percent with respect to the monomer weight. | ||||||
| (PP)-PMDA-BAPF | 4400 | 1.64 | 86 | 24100 |
4.69 | 22 |
| (PP)-BPDA-BAPF | 10000 |
4.49 | 91 | 22000 |
1.40 | 15 |
| (PP)-6FDA-BAPF | 14000 |
3.32 | 77 | 48000 |
1.66 | 2 |
| (PP)-ODPA-BAPF | 13000 |
4.19 | 85 | 33700 |
1.60 | 16 |
The last peak at an elution volume of ∼22 mL corresponds to pure BAPF. It is not surprising that the excess of non-reacted dicarboxylic acid-dicarboxylate is not visible in SEC, since it is dissolved in the aqueous phase. The second lowest peak at ∼21 mL might be related to cyclic or oligomeric byproducts which are typical dilute solution products.5,26 It should furthermore be noted that in the elugram of (PP)-PMDA-BAPF, the highest fraction (F-1, red curve) in Fig. 4a shows traces of high molecular weights (Ve between ∼12 and 14 mL). This finding points towards the possibility of solid state transformations, i.e.polymers whose molecular weight is controlled by the crystal size. A discussion of the possible pathways can be found below.
The average molecular weight is—for a polycondensation—rather acceptable, where fractions up to Mw ≈ 50000 g mol−1 can be isolated. This combination, in agreement with the rather high polydispersity index of up to 5, speaks for more than one locus of reaction, where the polymer species of different weights are generated.
The fact that non-reacted diamine monomers were found after cooling points towards solution polycondensation as one of the loci of reaction, i.e. monomers get dissolved and react while being dissolved. Indeed, it is known that phthalimides can be synthesized under hydrothermal conditions in solution.27 High molecular weights however cannot be formed at such low concentrations, and it will be reasoned below that the crystal interface is the second place where high polymerization takes place.
An examination of the reaction products by scanning electron microscopy (SEM) gives strong indications for those reaction pathways. Fig. 5 shows micrographs of the polyimides obtained after washing with aqueous HCl to remove the residual BAPF.
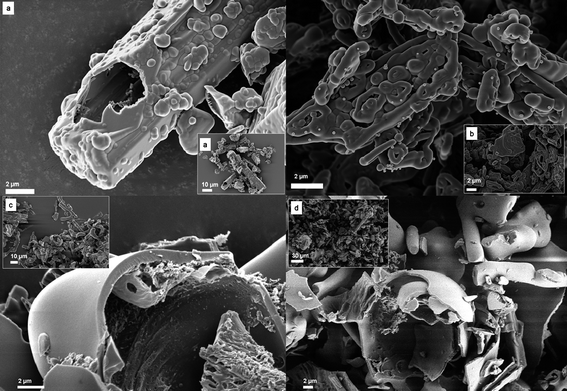 | ||
| Fig. 5 SEM images of (a) (PP)-PMDA-BAPF, (b) (PP)-BPDA-BAPF, (c) (PP)-6FDA-BAPF and (d) (PP)-ODPA-BAPF. Images in the corner show an overview. | ||
In the case of (PP)-PMDA-BAPF (Fig. 5a), the observed morphology is a hollow copy of the original salt crystals, i.e. the polymer is formed or precipitated onto the original monomer salt crystal which is being consumed at the same time. We can only speculate that the crystal slowly melts/dissolves while the polymer forms. Moving along for the still stiff BPDA based PI, the provenience from the salt-precursors is equally still visible: trapeze-like shaped particles with an interior that one could describe like a trumpet's piston valves (Fig. 5b). Interestingly, here the surface layer of the crystals breaks in a sequence of about 500 nm width ribbons, which however as a group remold the previous crystal shape. (PP)-ODPA-BAPF and (PP)-6FDA-BAPF, however, lead always to roundish, molten-like structures that are hollow. This suggests a molten-like, monomer-rich liquid state as starting situation.
Additionally performed TGA measurements of the salts showed a loss in mass, which can most probably be related to the condensation side-product water. The temperature, at which the mass loss was observed, showed a dependence on the monomer type, i.e. the mass loss was observed at 261 °C, 246 °C, 220 °C and 160 °C for (SS)-PMDA-BAPF, (SS)-BPDA-BAPF, (SS)-6FDA-BAPF and (SS)-ODPA-BAPF, respectively (see Fig. S2† for TGA data). This dependency is in accordance with the previously discussed flexibility issue and potential melting/pre-melting points of the isolated solids. In order to verify a potential melt polycondensation pathway, the isolated salts were checked by heating the salts in a capillary and monitoring the capillary by eye. No melting transitions were visible up to 200 °C in all four cases. However, these findings only relate to the dry salts. We believe that the observed molten-like morphologies could arise either from premelting or from a decrease in the melting temperatures under HT conditions, but are not comparable to classical melt-polycondensation. As it is however not easily possible to measure the phase transition under HT conditions, melting processes cannot be excluded fully at the current state. Finally, the fact that the monomer salts can be polymerized in a TGA capsule without the addition of solvent or pressure points towards polymerization as a solid-state transformation.
Bell reported that his “salt monomers” did not show any melting point, meaning that they were polymerizing roughly around their supposed melting point.21 As in our studies, hydrothermal polycondensations were carried out at 180 °C, i.e. at a temperature lower than the ones observed in TGA for the pure salt crystals (except (SS)-ODPA-BAPF), we think that two reaction pathways are effective.
Firstly, it is possible that ions get transferred into solution and react in a solution polycondensation pathway to cycles and oligomers (that might precipitate out of solution onto the crystals). Secondly, the monomers might undergo polycondensation reactions at the interface of the crystals from premelting surface layers (note that the surface of a crystal is always more disorganized than the bulk, especially after adhering “impurities”, such as formed oligomers). This could lead to high molecular weight fractions as the monomers are already well aligned for reaction. A scheme illustrating both potential pathways is depicted in Scheme 2.
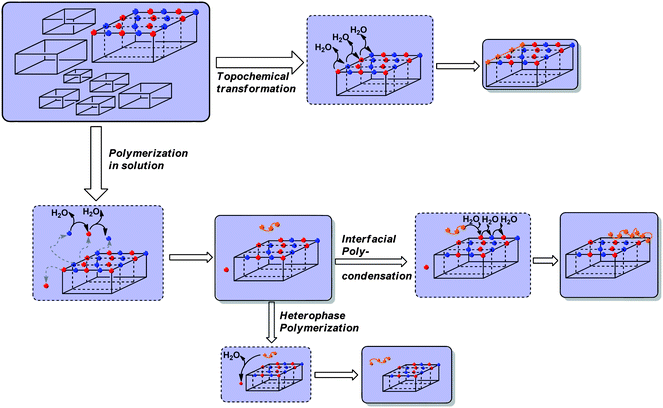 | ||
| Scheme 2 Possible transformations from monomer salt crystallites to PI. Red spheres = diacid-dicarboxylate ions, blue spheres = diammonium ions, orange = oligo-/polyimide chains. | ||
A combination of the pathways could explain both the structured molecular weight distributions as well as the formation of hollow replicas of the original crystals, as observed in the case of PMDA-BAPF and BPDA-BAPF.
The actual reaction pathway for each system is certainly a mixture of the different possible transformations with a participation of each varying for the different monomer combinations and the temperature.
A final experiment was performed to clarify the solution pathway by varying the stoichiometry in the synthesis of (PP)-PMDA-BAPF. To do so, a dispersion of (SS)-PMDA-BAPF was hydrothermally polymerized as obtained (a), with the addition of 0.2 equivalents of pyromellitic acid (b) and with additional 0.2 molar equivalents of BAPF (c). The pH of the aqueous phase after reaction was between 1.5 and 2 for all reactions, indicating that unreacted pyromellitic acid was the main solute in all cases. SEC analysis of the crude products (Fig. S6, ESI†) clearly shows that the highest weight average molecular weights were obtained in the presence of a slight excess of PMDA (case b), while an excess of BAPF resulted in the smallest Mw (case c). We think that these findings substantiate the theory that carboxylic acid monomers can pass into solution. As the dissolved amount consequently misses in the salt crystal—the stoichiometry is not of the ideal 1:1 composition anymore—the presence of additional pyromellitic acid yields higher molecular weights. However, it should be kept in mind that BAPF is visible in SEC, whereas PMDA is not. Since the arylamine is of low molecular weight, its addition already decreases the overall average molecular weight in itself.
Computational studies
Moreover, we were interested in the question, which reaction product (imide, amide and anhydride) from which starting product would be possible, energywise, and how temperature influences thermodynamic equilibria. To the best of our knowledge, no computational calculations have been performed up to date concerning the imide formation via the monomer salt pathway, with the exception of Artem'eva et al.,28 who performed semiempirical AM129 simulations investigating the formation of polyimide from diester–dicarboxylic-acid and diamine monomers. Interestingly, a minimum for the salt structure of the simulated compound was found on the surface of the potential energy. Therefore, model reactions (see Scheme 3) were analyzed using the DFT approach. The free energy of each of the educts/products of the model reactions was calculated using the B3LYP functional with a 6-31G(d,p) basis set. The Gibbs free energy of the reaction ΔG and the equilibrium constant K could then be calculated from these values.30
The free energy was calculated for different temperatures; solvent influences were not considered. Fig. 6 shows a plot of the Gibb's free reaction energy versus the temperature and the corresponding equilibrium constants K, also plotted vs. the temperature.
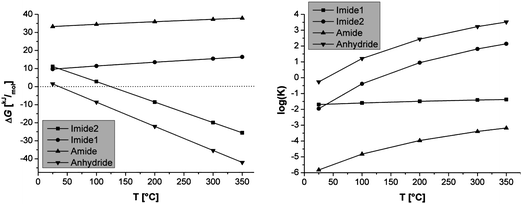 | ||
| Fig. 6 (Left side) Temperature dependence of ΔG. (Right side) Temperature dependence of equilibrium constants K. | ||
It is seen that both the formation of the imide from the free diacid and the amine and the ring closure from the free ortho-diacid to the anhydride are thermodynamically favored at the reaction temperature (180 °C), i.e. indeed imide formation drives the polycondensation in the presence of an excess of water. The latter is not surprising, as it is well known that ortho-dicarboxylic acid (e.g. phthalic acids) forms intramolecular anhydrides upon heating.31 Both the anhydride and the Imide2 formation are clearly entropy driven, as the calculated ΔrS values (see Tables S3a–S3e, ESI†) point out. This result is intuitively understandable for the anhydride formation, as one reactant gives rise to two molecules. Interestingly, the difference in entropies ΔΔrS between Imide2- and anhydride formation decreases from 24.12 kJ mol−1 to 18.73 kJ mol−1 with the increase in temperature from 298 K to 623 K. The increase in the temperature strongly shifts equilibria towards the products throughout the whole probed temperature range. The equilibrium constant increases roughly three orders of magnitude from room temperature to 200 °C. Although the formation of the amide increases with temperature, too, the dependence is less pronounced, and overall thermodynamics are still highly unfavorable. At room temperature however the hydrolysis of the anhydride is favored. Thus no matter if anhydride or diacid is employed and mixed with the amine, the salt is formed in the presence of water at moderate temperature. In the subsequent hydrothermal treatment however the free Gibbs energy is altered so that here the imide formation takes place, whenever kinetically possible. These results substantiate our experimental findings of the imide formation from a monomeric salt and furthermore point out a considerable overall thermodynamic driving force (ΔG < 0) at the chosen hydrothermal reaction conditions.
Finally, we have to admit that this computational procedure opens up another potential promoting bypass for the reaction. If the formation of the anhydride is favorable at elevated temperatures, it can be imagined that it forms first in all liquid or dissolved situations. This anhydride not only further lowers the melting point of the surface layer, but also accelerates reaction with the nucleophilic amine monomer to form the amic acid intermediate, which then might consecutively react towards the imide.
Morphology control
Finally, the possibility of synthesizing fully aromatic PIs in molten surface layers in water offers some prospects for material design. Addition of a ternary compound miscible with the monomer melt will at the one hand lower the melting point, but on the other hand enable stabilization and template routes thus giving rise to nanostructured engineering plastics.As an example, we used water-soluble polymeric surfactants of the poloxamer type (polyethylene oxide-block-propylene oxide) block-copolymers, also commonly known as Pluronics as potential templating agents. The result is quite a drastic change in the original morphology: the formed polymer structures are now compact spherical PI beads upon the usual reaction conditions (180 °C, 12 h, autogenous pressure), here depicted for (PP)-PMDA-BAPF in the SEM micrograph below (Fig. 7). This indicates that the addition of F127 indeed enables a liquid monomer dispersion, which then polycondenses towards PI latex spheres. The template obviously demixed at later stages of polycondensation, leaving pronounced surface porosity within the PI as easily seen from higher magnification SEM images. To our disappointment, the porosity is not preserved throughout the spheres, as also indicated by only moderate specific BET surface areas, but rather remains in a thin surface layer, only.
 | ||
| Fig. 7 SEM images of (PP)-PMDA-BAPF templated with Pluronic® F127. | ||
We attribute this to the insufficient compatibility of template and polycondensing oligomers, which however could be addressed by alternative choices of the template. As such porous PI spheres of a diameter of around 10 microns (depending on used reaction conditions) seem to be quite interesting for separation techniques32,33 (given the potential microporosity of some of the investigated polyimides),34 such template optimizations are currently ongoing.
Conclusion
The present study shed for the first time some light on the hydrothermal synthesis of aromatic polyimides. We could show that the key intermediate is a nylon-type AB salt of the hydrolyzed acid anhydrides and the aromatic amine. The salts could be isolated, and it was shown that no water is incorporated in the salt structure. The organic salts show a defined structure with an apparently high crystallinity, which was however dependent on the flexibility of the organic molecules. The presence of amorphous phases was found with increasing flexibility of the anhydride monomer. The mere existence as well as the type of the monomer salt has an impact on the morphology and the molecular mass distribution of the final polymers. The molecular weights of the crude products were only moderate as a high number of oligomers was still present. Upon fractionation, polymers with weight-average molecular weights of up to 50000 g mol−1 could be isolated. Such molecular weights are already high enough for the formation of stable, freestanding films. Hence, the here presented green chemistry pathway offers an attractive alternative to classic synthesis routes.
A thorough analysis of the presented experimental data brought us to the conclusion that the transformation of the salt precursor to the polymer concedes via 3 routes: (i) solution polycondensation, which gives most probable only oligomeric species, (ii) reaction at the interface of the salt crystals and (iii) a direct solid-state transformation within the salt crystal. As it is experimentally challenging to follow hydrothermal synthesisin situ, future studies will concentrate first on the solid-state transformation in the absence of water, including a detailed analysis of the crystallinity of the organic salts.
Finally, it should be stated that the morphology of high-performance plastics can be controlled by a templating process with water-soluble block copolymers. This finding itself is noteworthy, as morphology control of such polymers is typically not easy to achieve.35 We believe that the hydrothermal pathway opens up a lot of new possibilities in material design.
Experimental part
Materials and synthesis
4,4′-Oxydiphthalic tetracarboxylic acid, 3,3′,4,4′-biphenyl tetracarboxylic acid and 4,4′-(hexafluoroisopropylidene) diphthalic tetracarboxylic acid were synthesized via the same procedure.
(SS)-PMDA-BAPF: C35H26O8N2, calculated: w(C): 69.76%, w(H): 4.35%, w(N): 4.65%, obtained: w(C): 68.95%, w(H): 4.49%, w(N): 5.29%. (SS)-BPDA-BAPF: C41H32O10N2, calculated: w(C): 70.89%, w(H): 4.35%, w(N): 4.30%, obtained: w(C): 72.82%, w(H): 4.46%, w(N): 4.85%. (SS)-6FDA-BAPF: calculated: w(C): 62.17%, w(H): 3.64%, w(N): 5.06%, obtained: w(C): 62.59%, w(H): 3.79%, w(N): 4.11%. (SS)-ODPA-BAPF: C41H30O9N2, calculated: w(C): 70.89%, w(H): 4.35%, w(N): 4.03%, obtained: w(C): 69.24%, w(H): 4.46%, w(N): 4.62%. All obtained values were calculated via arithmetic mean of repeat determination.
Methods
FT-IR spectra were collected with a Varian 1000 FT-IR (scimitar series) spectrometer, equipped with an attenuated total reflection (ATR) setup. Size Exclusion Chromatography (SEC) in 0.05 mol L−1LiBr in NMP as the eluent and methyl benzoate as internal standard was performed with a system from Thermo Separation Products containing PSS GRAM 100 Å/1000 Å to 7 μm (particle size) columns from PSS-Polymer Standards Service. GRAM is a polyester column material without reactive functional groups. The column dimensions are of 8 × 50 mm for the pre-column and 8 × 300 mm for the main columns, respectively. It provides simultaneous UV and RI detection. 100 μL of sample with a concentration of 1.5 g L−1 were injected and separated at 70 °C and a flow of 0.8 mL min−1. Samples were filtered through 0.45 μm syringe filters prior to use. Polystyrene calibration standards of different molecular weights from PSS-Polymer Systems Service were used. Data treatment was performed with the software package NTeqGPCv6.4 from hs GmbH.Thermogravimetric analysis on the decomposition temperature was carried out on a Netzsch TG 209 F1 at a heating rate of 10 K min−1 under nitrogen atmosphere. Elemental analysis was measured with a Micro from Vario.
Scanning Electron Microscopy was carried out with a Gemini Leo 1550microscope at 3 kV. The samples were loaded on carbon coated stubs and coated by sputtering with Au/Pd alloy prior to imaging.
Wide Angle X-ray Scattering measurements were performed with a D8 Advance machine from Bruker Instruments. The radiation was of the wavelength of CuKα (0.154 nm). Measurements were undertaken in reflection geometry. Samples were measured as fine powders on a silicon sample holder.
Computational details
The herein presented thermochemical data were computed using the B3LYP36 density functional method as implemented in the Gaussian03 software package.37 The 6-31G(d,p) basis set was used to perform geometry optimizations and single point energy calculations in the gas phase at the desired temperature and a pressure of 1 atm.Acknowledgements
The authors would like to thank Marlies Gräwert for SEC measurements and Jekaterina Jeromenok for TGA and DSC measurements. Helmut Schlaad and Klaus Tauer are thanked for helpful discussions. Funding from the Max Planck Society is acknowledged. This work was supported by the ERC Advanced Grant “HYDRAChem”, project no. 227639.References
- E. R. Parnham and R. E. Morris, Acc. Chem. Res., 2007, 40(10), 1005 CrossRef CAS.
- (a) B. Laycock, D. Hawthorne, J. Hodgkin and T. C. Morton, WO Pat., 99/06470 1999; (b) B. Dao, J. Hodgkin and T. C. Morton, High Perform. Polym., 1999, 11, 205 Search PubMed.
- (a) H. Ohya, V. V. Krudyavtsev and S. I. Semenova, Polyimide Membranes—Applications, Fabrications and Properties, Gordon and Breach Science Publishers, SA, 1996 Search PubMed; (b) W. Volksen, R. D. Miller and G. Dubois, Chem. Rev., 2010, 110, 56 CrossRef CAS.
- J. Weber, O. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- N. Ritter, M. Antonietti, A. Thomas, I. Senkovska, S. Kaskel and J. Weber, Macromolecules, 2009, 42, 8017 CrossRef CAS.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730 RSC.
- B. S. Ghanem, N. B. McKeown, P. M. Budd, N. M. Al-Harbi, D. Fritsch, K. Heinrich, L. Starannikova, A. Tokarev and Y. Yampolskii, Macromolecules, 2009, 42, 7881–7888 CrossRef CAS.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. Van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254–258 CrossRef CAS.
- J. D. Wind, D. R. Paul and W. J. Koros, J. Membr. Sci., 2004, 228, 227–236 CrossRef CAS.
- D. M. Kim, S. Park, T. J. Lee, S. G. Hahm, K. Kim, J. C. Kim, W. Kwon and M. Ree, Langmuir, 2009, 25, 11713–11719 CrossRef CAS.
- J. Wakita, H. Sekino, K. Sakai, Y. Urano and S. Ando, J. Phys. Chem. B, 2009, 113, 15212–15224 Search PubMed.
- D. P. Erhard, R. Giesa, V. Altstädt and H. Schmidt, Macromol. Chem. Phys., 2007, 208, 1522–1529 Search PubMed.
- J. H. Hodgkin and T. C. Morton, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2000, 41, 2008 Search PubMed.
- A. A. Kuznetsov, High Perform. Polym., 2000, 12, 445–460 Search PubMed.
- J. K. Fink, High Performance Polymers, William Andrew Inc., Norwich, NY, 2008 Search PubMed.
- (a) Y. Imai, J. Photopolym. Sci. Technol., 1994, 7(2), 251 Search PubMed; (b) Y. Imai, et al., J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 39 Search PubMed; (c) R. Brunel, C. Marestin, V. Martin and R. Mercier, High Perform. Polym., 2010, 22, 82 Search PubMed.
- W. M. Edwards and I. M. Robinson, US Pat., 2,710,583, 1955.
- Synthetic Methods in Step-Growth Polymers, ed. M. E. Rogers and T. E. Long, John Wiley Sons, Inc., Hoboken, New Jersey, 2003 Search PubMed.
- D. Dimov, E. Spassova, I. Karamancheva, I. Zhivkov and G. Danev, Vacuum, 2004, 76, 223 Search PubMed.
- V. L. Bell, Polym. Lett., 1967, 5, 941 Search PubMed.
- (a) R. Pudzich and J. Salbeck, Synth. Met., 2003, 138, 21 CrossRef CAS; (b) T. P. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS.
- J. Moore, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 835 Search PubMed.
- (a) M. Konáš, T. M. Moy, M. E. Rogers, A. R. Schultz, T. C. Ward and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1429 Search PubMed; (b) M. Konáš, T. M. Moy, M. E. Rogers, A. R. Schultz, T. C. Ward and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1441 Search PubMed.
- (a) A. Wexler, J. Res. Natl. Bur. Stand., Sect. A, 1976, 80, 775 Search PubMed; (b) B. Hardy, ITS-90 Formulations for Vapor Pressure, Frostpoint Temperature, Dewpoint Temperature, and Enhancement Factors in the Range −100 to +100 °C, in Proceedings of Third International Symposium on Humidity & Moisture, Teddington, London, England, 1998 Search PubMed.
- H. R. Kricheldorf, D. Fritsch, L. Vaklangishili and G. Schwarz, Makromol. Chem. Phys., 2005, 206, 2239 Search PubMed.
- L. M. Dudd, E. Venardou, E. Garcia-Verdugo, P. Licence, A. J. Blake, C. Wilson and M. Poliakoff, Green Chem., 2003, 5, 187 RSC.
- V. N. Artem'eva, V. V. Kudryavtsev, P. I. Chupans, A. V. Yakimansky and G. V. Lubimova, Russ. Chem. Bull., 1995, 44(6), 1021 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107(13), 3902 CrossRef.
- J. W. Ochterski, Thermochemistry in Gaussian, Gaussian, Inc., Pittsburgh, PA, 2000 Search PubMed.
- B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical, Harlow, UK, 5th edn, 1989 Search PubMed.
- F. Svec, J. Chromatogr., A, 2010, 1217, 902 CrossRef CAS.
- M. M. Titirici, A. J. Hall and B. Sellergren, Chem. Mater., 2003, 15, 822 CrossRef.
- N. Ritter, I. Senkovska, S. Kaskel and J. Weber, Macromolecules, 2011, 44, 2025 Search PubMed.
- K. Kimura, S. Kohama and S. Yamazaki, Polym. J., 2006, 38(10), 1005 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98 (Revision A. x), Gaussian, Inc., Pittsburgh PA, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00109d |
| This journal is © The Royal Society of Chemistry 2011 |