DOI:
10.1039/C1PY00099C
(Paper)
Polym. Chem., 2011,
2, 1737-1743
Poly(2-oxazoline) glycopolymers with tunable LCST behavior
Received
3rd March 2011
, Accepted 7th April 2011
First published on 27th May 2011
Abstract
A series of thermo-responsive glyco-poly(2-oxazoline)s based on 2-ethyl-2-oxazoline and 2-(dec-9-enyl)-2-oxazoline were prepared. To study the effect of the sugar content on the solution behavior in water, two sets of copolymers with constant monomer-to-initiator ratios of 20 and 50 and varying amounts of the hydrophobic alkene functionalized monomer were synthesized. The glycopolymers were obtained by the photoaddition of 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glycopyranose onto the double bonds followed by deacetylation of the saccharide residues. Turbidimetry measurements of the respective glycopolymers revealed a decreasing cloud point temperature with increasing amount of sugar moieties, proposed to be caused by hydrogen bonding between the sugars and the polymer amide groups, which is enabled by the flexibility of the long decyl spacer. Due to the linear relationship between cloud point temperatures and the sugar content, the cloud points can be easily tailored for specific applications.
Introduction
Thermo-responsive polymers have received major attention in the last few years as one of the main concepts for stimuli-responsive polymers.1,2 Their response to a temperature stimulus makes them perfect candidates to be used in applications where temperature variations occur depending on the location of action, such as in the human body where for instance healthy and cancer cells exhibit different temperatures. As a consequence, the design of new stimuli-responsive synthetic polymers is of great interest since they offer the possibility to generate reversible temperature response; not showing a denaturation of their structures upon heating as, for example, proteins do. In particular, the lower critical solution temperature (LCST) behavior of polymers represents an extensively exploited feature for the development of thermo-responsive materials. Commonly, an increase in temperature enhances the solubility of a polymer in (aqueous) solution. In contrast, the LCST behavior of aqueous polymer solutions represents the reverse effect upon heating resulting in a phase separation of the binary polymer/water mixture. Increasing the temperature above this phase transition temperature causes the disruption of hydrogen bonds between polar groups of the polymer and water. This cloud point temperature (Tcp) depends on the hydrophilic–hydrophobic balance of the polymer. In general, more hydrophilic polymers are better hydrated, i.e. a larger number of water molecules are hydrogen bonded to the polymer, which gives a favorable (negative) enthalpy contribution to the free energy of mixing (ΔG = ΔH − TΔS) resulting in a higher Tcp. However, H-bonding between polymer and solvent results in an enhanced ordering of the water molecules, thus, an unfavorable contribution to the entropy of mixing is accounted. The entropy term (TΔS) becomes predominant over the favorable enthalpy term with increasing temperature resulting in phase separation. This LCST effect can be tailored for special applications by varying the structure. The incorporation of hydrophobic groups, such as end groups or other more hydrophobic or hydrophilic comonomers can alter the Tcp of the system.3 In recent years, a wide range of systems were described in literature exhibiting tunable LCST behaviors based on (co-)polymerization of monomers such as N-isopropylacrylamide, ethylene glycol, (meth-)acrylates and 2-oxazolines.4–7
Glycopolymers—synthetic polymers with pendant carbohydrate moieties—appeared to be of special interest for recognition/targeting in a variety of biological processes, like immunological recognition, interaction between bacteria or viruses with cells, as well as tissue growth and repair.8–12 Due to their free hydroxyl groups sugar side-chains can have an influence on the solubility of the respective polymers and, thus, on the LCST behavior, in particular when copolymerized with a monomer that results in a LCST polymer.13–18 In combination with the ability of the carbohydrate moieties to bind to a broad range of carbohydrate-binding receptors, responsible, i.e. for cell proliferation and death,19,20 it renders thermo-responsive glycopolymers perfect candidates for biological applications, e.g. as targeted drug delivery systems, coating materials for surfaces and particles, for affinity separations, bioassays or biocapture analysis.21–26
The living cationic ring-opening polymerization (CROP) of 2-oxazolines possesses a large potential for the preparation of thermo-responsive (co-)polymers due to the possible preparation of several hydrophilic polymers and the ease in altering the hydrophilic/hydrophobic properties of the corresponding copolymers using differently substituted monomers.27,28 Poly(2-oxazoline)s themselves can be considered as analogues of poly(amino acid)s, i.e. pseudo-peptides, exhibiting in the case of 2-methyl-2-oxazoline as well as 2-ethyl-2-oxazoline a stealth behavior similar to poly(ethylene oxide) (PEO).29 Due to its biocompatibility (co-)poly(2-oxazoline)s were used in several biomedical applications.30,31 This biocompatibility in combination with the LCST behavior of poly(2-oxazoline)s is perfectly suited as a platform for the synthesis of glycopolymers with various tunable properties. Due to the living character of the polymerization, the composition and sequence of the copolymers can be tuned and different functionalities can be incorporated by the initiator, the termination agent or by the usage of suitable 2-substituted-2-oxazolines.32–34 Recently, we reported the synthesis of a copolymer system exhibiting pendant alkene functionalities in the side chains based on renewable resources,35,36 which is suitable for post-modification reactions viathiol–ene chemistry.37–39 As demonstrated by Diehl and Schlaad, this approach offers the possibility for the preparation of well-defined glycopolymers from a precursor polymer using peracetylated thio-sugars.40,41 Depending on the composition of the copolymers (2-(3-butenyl)-2-oxazoline and 2-isopropyl-2-oxazoline) the LCST behavior could be tuned showing an increase in the cloud point with increasing sugar content. Furthermore, homopolymers of glycosylated 2-(3-butenyl)-2-oxazoline revealed the formation of spherical vesicles and nanofibers upon direct dissolution in water.42 To the best of our knowledge, up to now only one additional publication exists describing the preparation of thermo-responsive glyco-poly(2-oxazoline)s based on peracetylated maltoheptanose end capped poly(2-oxazoline)s (POx).43
In this contribution, we report the systematic investigation of the LCST behavior of a series of glycopolymers derived from a precursor copolymer with a constant [M]/[I] ratio and varying ratio of 2-ethyl-2-oxazoline and 2-(dec-9-enyl)-2-oxazoline. The glycopolymers were prepared by polymer post-modification exploiting the photoaddition of thiols onto pendant alkene functionalities. The cloud points of the glycopolymers obtained showed a strong dependence on the composition of the copolymer and could be altered accordingly. Interestingly, the cloud points decreased with increasing sugar content which will be discussed in detail.
Experimental details
Materials
2-Ethyl-2-oxazoline and methyl tosylate were obtained from Acros Organics, distilled to dryness over barium oxide (BaO), and stored under argon. Acetonitrile, 2,2-dimethoxy-2-phenylacetophenone (DMPA) and sodium methoxide were purchased from Sigma Aldrich. Dialysis bags (molar mass cut-off 3.500 g mol−1) were purchased from Spectra/Por®. 2,3,4,6-Tetra-O-acetyl-1-thio-β-D-glycopyranose was prepared according to a literature procedure.44 The 2-(dec-9-enyl)-2-oxazoline (DecEnOx) was synthesized according to a recently reported method.35
General methods and instrumentation
The Initiator Sixty single-mode microwave synthesizer from Biotage, equipped with a non-invasive IR sensor (accuracy: ±2%), was used for polymerizations under microwave irradiation. Microwave vials were heated to 110 °C overnight and allowed to cool to room temperature under an argon atmosphere before usage. All polymerizations were carried out with temperature control. Size exclusion chromatography measurements were performed on an Agilent system equipped with a diode array detector and a refractive index detector. Two PSS SDV (5 μm pore size) columns were placed in series. DMA with 2.1 g L−1 of LiCl was used as eluent at 1 mL min−1 flow rate, and the column oven was set to 50 °C. Molar masses were calculated against polystyrene standards. For preparative SEC, Bio-Beads S-X1 (crosslinked polystyrene beads) from Bio-Rad were used. 1H NMR spectra were recorded on a Bruker AC 300 MHz spectrometer at room temperature, with MeOH-d4 as a solvent. The chemical shifts are given in ppm relative to the signal from residual non-deuterated solvent. Cloud points were determined in a Crystal 16 from Avantium Technologies connected to a chiller (Julabo FP 40) at a wavelength of 500 nm. The solutions were heated and cooled at a rate of 1 °C min−1 while stirring at 600 rpm. The cloud point was defined as the temperature where the transmittance decreased to 50% in the second heating run. Dynamic light scattering (DLS) measurements were carried out on a Zetasizer Nano ZS (Malvern Instrument, Malvern, UK) using a He/Ne-laser (λ = 633 nm) and a scattering angle of 173°. The mean particle size was approximated as the effective (Z average) diameter and the width of the distribution as the polydispersity index (PDI) obtained by the cumulant method assuming a spherical shape.
Microwave-assisted synthesis of the copolymers (PEtOx-stat-PDecEnOx)
A solution of initiator (methyl tosylate), monomers (EtOx and DecEnOx) and solvent (acetonitrile) was prepared. The total monomer concentration was adjusted to 2 M with a total [M]/[I] ratio of 20 and 50 with 10, 20, 30 and 40 mol% DecOx, respectively. The vial was heated to 140 °C in the microwave synthesizer. After cooling, a 15-fold excess of aqueous sodium carbonate was added and the polymerization mixture was stirred at 100 °C overnight. Subsequently, the two-phase solution was diluted with dichloromethane, the organic phase was washed three times with water, brine and dried over MgSO4. The polymer was concentrated in vacuum and precipitated into ice-cold diethyl ether or applied to preparative size exclusion chromatography.
Thiol–ene photoaddition reaction using 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glycopyranose
300 mg of the copolymer were dissolved in THF and 2,3,4,6-tetra-O-acetyl-1-thio-β-glycopyranose was added in 1.3-fold excess with respect to the double bonds. After degassing the solution for 30 min a spatula tip of 2,2-dimethoxy-2-phenylacetophenone (DMPA) was added and the solution was irradiated at 356 nm overnight. The resulting protected glycocopolymers were purified either by precipitation in ice-cold diethyl ether or by preparative size exclusion chromatography.
Deprotection of the protected glycocopolymers
200 mg of the protected glycocopolymer were dissolved in 8 mL dry methanol. 1 mL sodium methoxide solution was added and the copolymer solution was stirred for 2 h. Subsequently, the solvent was evaporated under reduced pressure. The polymer was re-dissolved in water, neutralized with diluted HCl and dialyzed against 500 mL distilled water for 3 days. The surrounding water was exchanged six times in a period of 12 h.
Cloud point measurements
4 mg of the copolymer were dissolved in deionized water applying a constant concentration of 4 mg mL−1. The turbidity of the solutions was determined in two temperature cycles ranging from 2 to 105 °C [1 K min−1]. Turbidity measurements at a wavelength of 500 nm were performed to determine the cloud point temperatures at 50% transmittance.
Dynamic light scattering measurements
The cloud point samples (4 mg mL−1) were diluted with filtered deionized water to a concentration of 1 mg mL−1 and were measured 5 times at 25 °C for 150 s.
Results and discussion
Preparation of the copolymer precursors
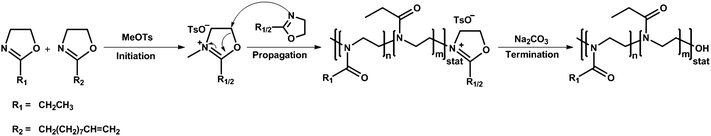
For the systematic investigation of the LCST behavior of copoly(2-oxazoline) glycopolymers two series of copolymers with a degree of polymerization of 20 and 50 were prepared, respectively. The copolymerization mechanism is schematically depicted in Scheme 1 showing the initiation with methyl tosylate, the incorporation of the monomers during the copolymerization and the subsequent termination reaction using a nucleophile. For the variation of the number of the sugar units different ratios of the hydrophilic monomer EtOx and the hydrophobic monomer DecEnOx, exhibiting pendant alkene groups, were adjusted using 10 mol% increments of DecEnOx (up to 40 mol% DecEnOx).
As can be seen in Table 1, the theoretical feed amounts of DecEnOx are in close agreement with the DecEnOx incorporated in the final copolymers as calculated by 1H NMR spectroscopy. In order to guarantee the same chemical structure of all copolymers the reaction mixture was treated with aqueous sodium carbonate at 100 °C after microwave polymerization at 140 °C to form a hydroxy end group.45 End capping of the copolymers just with water would lead to a mixture of two different end groups (hydroxy and ester) which could have a significant influence on the properties of, in particular, the shorter copolymers.46,47 As a representative example, the IR spectrum of P1b does not show the existence of an ester signal at about 1710 cm−1 (Fig. 1). Furthermore, a similar observation can be made by means of the corresponding 1H NMR spectrum (Fig. 2). In the case of the formation of an ester end group, a peak at 4.1–4.2 ppm would appear, whereas a hydroxy end group leads to a peak at lower ppm values, just left of the signals of the polymeric backbone at about 3.8 ppm. Thus, based on the absence of the peak at 4.1 to 4.2 ppm and the appearance of the signal at 3.8 ppm it can be concluded that the copolymer possesses exclusively OH end groups.
| |
n + m = 20 |
n + m = 50 |
|
P1a
|
P1b
|
P1c
|
P1d
|
P2a
|
P2b
|
P2c
|
P2d
|
| Decfeed/mol% |
10 |
20 |
30 |
40 |
10 |
20 |
30 |
40 |
| Decexp/mol% |
9 |
20 |
30 |
39 |
10 |
19 |
30 |
39 |
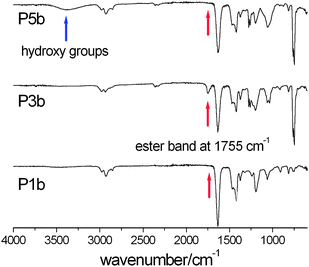 |
| | Fig. 1
IR-ATR
spectra of P1b, P3b und P5b demonstrating the successful addition of acetyl-protected glucose units (ester band at 1755 cm−1) onto the polymer precursor. The disappearance of the ester band as well as the appearance of a broad band between 3100 cm−1 and 3600 cm−1 confirms the successful deprotection of the sugar moieties. | |
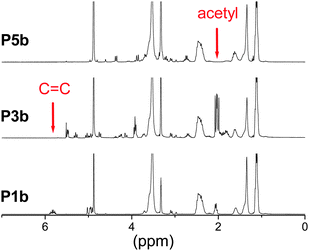 |
| | Fig. 2
1H NMR spectra (300 MHz; MeOH-d4) of the copolymers P1b, P3b (thiol–ene product) and P5b (after deacetylation). The disappearance of the signals of the double bond protons as well as the presence of signals of the acetyl protons in P3b confirms the successful thiol–ene photoaddition of Ac4GlcSH onto the copolymer P1b. The absence of the acetyl signals demonstrates the successful deprotection of the sugar units (P5b). | |
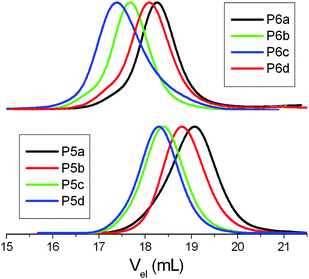
The process of the attachment of the protected sugar unit and its deprotection (Scheme 2) was monitored by IR and 1H NMR spectroscopy as well. Upon completion of the thiol–ene reaction a signal at 1740 cm−1 appeared in the IR spectrum, which originated from the acetyl protecting groups of the sugar (Fig. 1). This signal disappears again after the deprotection of the sugar moieties yielding free hydroxyl groups which cause a broad signal at about 3500 cm−1. Similar observations can be made by 1H NMR spectroscopy. The spectrum of the starting materials shows two multiplets at about 5 and 6 ppm corresponding to the terminal alkene group of the DecEnOx. These signals vanished after the reaction with Ac4GlcSH revealing quantitative conversion (>96%) of the double bonds. In addition, the signals from the protons of the sugar residues (4 to 6 ppm) and a strong signal at about 2 ppm belonging to the protons of the acetyl protecting groups appeared. Therefore, the successful deprotection of the sugar moieties by treatment with sodium methoxide could be proven by the disappearance of the signals of the acetyl groups in the final products. Applying these steps to all copolymers, two series of well-defined glycopolymers with a total degree of polymerization (DP) of 20 and 50 were obtained, respectively. They show narrow molar mass distributions in SEC (Fig. 3) and thus, possess polydispersity indices (PDI) between 1.1 and 1.2 (see Tables 2 and 3). As can be seen in Fig. 3, glycopolymers with higher sugar content eluted at shorter times than the ones with lower sugar content, which is attributed to the increase in the total molar mass of the glycopolymers. Furthermore, glycopolymers with a DP of 50 are shifted to lower elution volumes (Fig. 3, top) compared to the glycopolymers with a DP of 20 (Fig. 3, bottom).
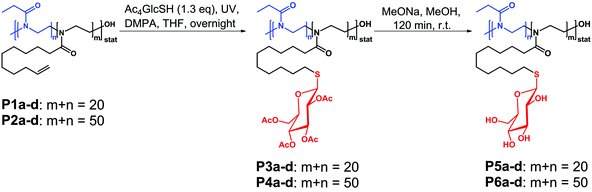 |
| | Scheme 2 Schematic representation of the preparation of glycopoly(2-oxazoline)s using the thiol–ene approach encompassing the thiol–ene photoaddition as well as the deprotection of the glucose units. | |
 |
| | Fig. 3
SEC traces of the (deprotected) glycopolymers with a DP of 20 (P5a–d; bottom) as well as with a DP of 50 (P6a–d; top) with different amounts of Dec(Glc)Ox units (a: 10 mol%, b: 20 mol%, c: 30 mol%, d: 40%); eluent: DMA/2.1 g L−1LiCl. | |
| |
M
n/g mol−1 |
PDI |
|
M
n/g mol−1 |
PDI |
|
M
n/g mol−1 |
PDI |
|
P1a
|
4100 |
1.15 |
P3a
|
5700 |
1.13 |
P5a
|
8400 |
1.20 |
|
P1b
|
4700 |
1.12 |
P3b
|
6700 |
1.10 |
P5b
|
9200 |
1.14 |
|
P1c
|
5200 |
1.12 |
P3c
|
7400 |
1.14 |
P5c
|
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
1.12 |
|
P1d
|
5200 |
1.12 |
P3d
|
7800 |
1.11 |
P5d
|
14600 |
1.12 |
| |
M
n/g mol−1 |
PDI |
|
M
n/g mol−1 |
PDI |
|
M
n/g mol−1 |
PDI |
|
P2a
|
9300 |
1.10 |
P4a
|
11200 |
1.12 |
P6a
|
14200 |
1.13 |
|
P2b
|
10500 |
1.10 |
P4b
|
12400 |
1.11 |
P6b
|
15800 |
1.15 |
|
P2c
|
9600 |
1.16 |
P4c
|
13100 |
1.17 |
P6c
|
19400 |
1.15 |
|
P2d
|
11200 |
1.13 |
P4d
|
14300 |
1.15 |
P6d
|
19900 |
1.20 |
To study the effect of the sugar content on the thermo-responsive behavior of the glycopolymers in aqueous solution, turbidimetric measurements were performed at a constant copolymer concentration of 4 mg mL−1. The water solubility of the starting copolymers with 10 to 40 mol% DecEnOx in steps of 10 mol% depends on the amount of DecEnOx incorporated in the copolymer and the total degree of polymerization (Tables 4 and 5). For a total DP of 20, copolymers with 10 mol% and 20 mol% DecEnOx revealed good water solubility, even if the cloud point of P1b is already close to room temperature. The copolymers with a higher degree of DecEnOx were insoluble in water in the investigated temperature range (2 to 105 °C). As expected, the higher the amount of the hydrophobic monomer the lower the cloud point of the copolymer. By increasing the total DP to 50, only the copolymer with the lowest DecEnOx content was still soluble in water under these conditions. A decrease in the cloud point temperature and the lower solubility upon increasing the molar mass are common observations following the Flory–Huggins model.
Table 4 Cloud point temperatures (°C; heating and cooling cycles) obtained for the statistical copolymers (P1), the acetylated glycopolymers (P3) and the corresponding glycopolymers (P5) with a total DP of 20 with different DecEnOx increments (a–d)
| |
Heating |
Cooling |
|
Heating |
Cooling |
|
Heating |
Cooling |
|
P1a
|
53.3 |
52.5 |
P3a
|
66.2 |
61.2 |
P5a
|
39.4 |
38.1 |
|
P1b
|
18.5 |
17.8 |
P3b
|
21.5 |
18.1 |
P5b
|
30.8 |
31 |
|
P1c
|
Insoluble |
Insoluble |
P3c
|
Insoluble |
Insoluble |
P5c
|
17.5 |
16.4 |
|
P1d
|
Insoluble |
Insoluble |
P3d
|
Insoluble |
Insoluble |
P5d
|
12.4 |
13.1 |
Table 5 Cloud point temperatures (°C; heating and cooling cycles) obtained for the statistical copolymers (P2), the acetylated glycopolymers (P4) and the corresponding glycopolymers (P5) with a total DP of 50 with different DecEnOx increments (a–d) [not det. = not determined]
| |
Heating |
Cooling |
|
Heating |
Cooling |
|
Heating |
Cooling |
|
P2a
|
33.7 |
32.6 |
P4a
|
Not det. |
Not det. |
P6a
|
44.6 |
42.5 |
|
P2b
|
Insoluble |
Insoluble |
P4b
|
Not det. |
Not det. |
P6b
|
34.5 |
36.8 |
|
P2c
|
Insoluble |
Insoluble |
P4c
|
Not det. |
Not det. |
P6c
|
18.3 |
21.8 |
|
P2d
|
Insoluble |
Insoluble |
P4d
|
Not det. |
Not det. |
P6d
|
14.5 |
15.3 |
Upon the photoaddition of Ac4GlcSH onto the pendant alkene groups the copolymers with 30 mol% and 40 mol% DecEnOx remained water insoluble, whereas the other ones showed a different somewhat unexpected behavior as observed for the protected glycopolymers P3a and P3b (P4a and P4b with DP of 50 were not studied in detail). Instead of showing a lower cloud point temperature as one would expect due to the increase in hydrophobicity of the complete system, the cloud point increased compared to the starting copolymers. This observation can be attributed to the formation of aggregates as revealed by dynamic light scattering (DLS) measurements of copolymer P3a (Fig. 4). The starting copolymer P1a is solubilized as individual polymer chains in aqueous solution while the attachment of the larger (hydrophobic) protected sugar moieties results in a self-assembly of the polymer chains under the formation of aggregates with a diameter of 220 nm and a low PDIparticle value. This aggregation causes a change in the thermo-responsiveness of the system, since the hydrophobic domains are shielded from the aqueous solution, thereby increasing the solubility of the hydrophilic parts that stabilize the aggregates. A similar behavior was observed for copolymer P3b possessing 20 mol% DecEnOx units. The cloud point is again slightly higher for P3b. DLS measurements revealed the existence of large ill-defined aggregates (330 nm; PDIparticle = 0.6) in the case of copolymer P3b. Similar to P3a, the formation of aggregates renders the copolymer soluble at a temperature lower than 21 °C, which would not be possible without the existence of these aggregates due to the higher hydrophobicity of the copolymer caused by the incorporation of the hydrophobic Ac4GlcSH residues. The turbidity measurements revealed a sharp decrease in transmittance consistent with a sharp phase transition for P3b in contrast to P3a (Fig. 5). The lower number of hydrophobic Dec(Ac4Glc)Ox repeating units in P3a results in a smaller hydrophobic attraction between the chains while at the same time the larger amount of hydrophilic EtOx units will stabilize initially formed aggregates by suppressing further aggregation. As a result, P3a shows a gradual collapse and assembly upon increasing the temperature resulting in a broad phase transition. Nonetheless, both polymers reveal a reversible phase transition with a similarly small hysteresis.
 |
| | Fig. 4
DLS analysis of P3a and P3b in water (4 mg mL−1) revealing the formation of large aggregates after the thiol–ene modification of the precursor copolymer P1a with Ac4GlcSH. | |
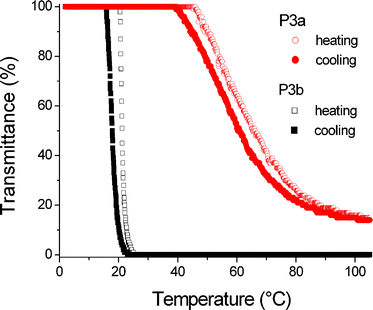 |
| | Fig. 5 Transmittance curves during heating and cooling scans (polymer concentration = 4 mg mL−1, heating/cooling rate = 1 °C min−1). | |
Upon the deprotection of the sugar moieties the corresponding glycopolymers P5a–d and P6a–d were obtained, which all exhibit cloud point temperatures. In general it can be stated that the amount of sugar moieties has a significant influence on the cloud point of the glycopolymers albeit a contrary effect, i.e. an increasing cloud point with increasing hydrophilic sugar content, was expected as observed by Schlaad and co-workers using a shorter spacer between the polymer backbone and the sugar moiety. Nevertheless, it was found in the current study that increasing the sugar content results in a decrease in the cloud point temperature of the copolymers (Fig. 6), which is in contrast to an increase in hydrophilicity of the overall system. This unexpected phenomenon is proposed to be related to the high flexibility of the DecEnOx chains in combination with hydrogen bonding. Due to the long alkyl chains acting as flexible spacers between the polymer backbone and the sugar residues, the hydroxyl groups of the sugar moieties can fold back to interact with the polymer amide groups by intramolecular hydrogen bonding. These intramolecular hydrogen bonds compete with the water hydration, thereby decreasing the enthalpy gain of the latter. As a result, the entropy term will become dominant at lower temperatures resulting in a lower cloud point. A small hysteresis is observed, in particular for the glycopolymers with a higher sugar content P5c and P5d, which might be caused by the higher number of intramolecular hydrogen bonds, which need to be disrupted during the cooling run again.1
 |
| | Fig. 6 Representative transmittance curves of the glycopolymers (P5a–d) in aqueous solution during heating (■) and cooling (□) scans (polymer concentration = 4 mg mL−1, heating/cooling rate = 1 °C min−1). | |
A similar trend could be observed for the copolymers with a total DP of 50 as shown in Fig. 7. Surprisingly, the cloud points of the longer glycopolymers are in the same range as the ones for the shorter glycopolymers. This uncommon observation might indicate that the cloud point behavior is predominantly governed by the intramolecular hydrogen bonding suppressing the Flory–Huggins chain length effect. Nevertheless, the linear dependence of the cloud point on the amount of sugar moieties allows straightforward tuning of the cloud points enabling a perfect control of the LCST behavior for specific applications.
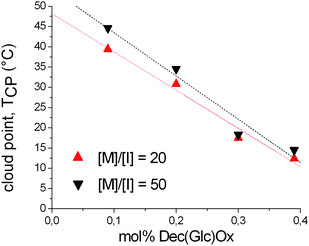 |
| | Fig. 7 Cloud point temperatures in aqueous solution of the (deprotected) glycopolymers with a total DP of 20 (P5a–d) and 50 (P6a–d) as a function of the Dec(Glc)Ox content in the copolymer. | |
Conclusions
A systematical study of the cloud points of copoly(2-oxazoline) bearing glycosylated side chains is reported based on two copolymer series of 2-ethyl-2-oxazoline and 2-(dec-9-enyl)-2-oxazoline with a total degree of polymerization of 20 and 50, respectively. All polymers were synthesized by a microwave-assisted living cationic ring-opening polymerization showing a perfect consistency between theoretical and experimentally determined molar ratios. In order to obtain the corresponding glyco-poly(2-oxazoline)s 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glycopyranose was “clicked” by thiol–ene photoaddition reactions onto the pendant double bonds of the precursor copolymers followed by the deacetylation of the sugar moieties with sodium methoxide. Turbidity measurements of the glycopolymers in aqueous solution revealed a decrease in the cloud points with increasing number of sugar moieties. This unexpected behavior is proposed to be caused by hydrogen bonding between the hydroxy sugar groups and the polymer backbone facilitated by the large flexible decyl spacer. The linear dependency of the cloud point temperatures in this systematic study allows straightforward tuning of the LCST behavior for specific applications. In the future these results will be exploited for the synthesis of more complex polymer structures to study the controlled binding of lectins by varying the type and amount of sugar moieties.
Acknowledgements
The authors thank the Dutch Polymer Institute (DPI, Technology area HTE) for financial support. K.K. is grateful to the Landesgraduiertenfoerderung Thueringen for financial support. R.H. (Veni-grant) and U.S.S. (Vici-grant) thank the Netherlands Scientific Organization (NWO) for financial support.
References
- I. Dimitrov, B. Trzebicka, A. H. E. Mueller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275–1343 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Mueller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- A. Narumi, K. Fuchise, R. Kakuchi, A. Toda, T. Satoh, S. Kawaguchi, K. Sugiyama, A. Hirao and T. Kakuchi, Macromol. Rapid Commun., 2008, 29, 1126–1133 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- C. Weber, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromolecules, 2009, 42, 2965–2971 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- N. Sharon, Biochim. Biophys. Acta, 2006, 1760, 527–537 CrossRef CAS.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS.
- D. Cunliffe, S. Pennadam and C. Alexander, Eur. Polym. J., 2004, 40, 5–25 CrossRef CAS.
- A. J. Varma, J. F. Kennedy and P. Galgali, Carbohydr. Polym., 2004, 56, 429–445 CrossRef CAS.
- S. G. Spain and N. R. Cameron, Polym. Chem., 2011, 2, 60–68 RSC.
- G. Pasparakis, A. Cockayne and C. Alexander, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS.
- M. Hetzer, G. J. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef CAS.
- L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123–129 CrossRef CAS.
- Z. Oezyuerek, H. Komber, S. Gramm, D. Schmaljohann, A. H. E. Mueller and B. Voit, Macromol. Chem. Phys., 2007, 208, 1035–1049 CrossRef.
- Z. Oezyuerek, K. Franke, M. Nitschke, R. Schulze, F. Simon, K. J. Eichhorn, T. Pompe, C. Werner and B. Voit, Biomaterials, 2009, 30, 1026–1035 CrossRef CAS.
- E. H. Min, S. R. S. Ting, L. Billon and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3440–3455.
- B. N. Stillman, D. K. Hsu, M. Pang, C. F. Brewer, P. Johnson, F. T. Liu and L. G. Baum, J. Immunol., 2006, 176, 778–789 CAS.
- H. J. Gabius, H. C. Siebert, S. Andre, J. Jimenez-Barbero and H. Ruediger, ChemBioChem, 2004, 5, 741–764.
- R. J. Pieters, Med. Res. Rev., 2007, 27, 796–816 CrossRef CAS.
- Y. Miura, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5031–5036 CrossRef CAS.
- G. Coullerez, P. H. Seeberger and M. Textor, Macromol. Biosci., 2006, 6, 634–647 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- K. Babiuch, C. R. Becer, M. Gottschaldt, J. T. Delaney, J. Weisser, B. Beer, R. Wyrwa, M. Schnabelrauch and U. S. Schubert, Macromol. Biosci., 2011, 11, 535–548 Search PubMed.
- K. Babiuch, R. Wyrwa, K. Wagner, T. Seemann, S. Hoeppener, C. R. Becer, R. Linke, M. Gottschaldt, J. Weisser, M. Schnabelrauch and U. S. Schubert, Biomacromolecules, 2011, 12, 681–691 Search PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- A. Makino and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1251–1270 Search PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS.
- H. Schlaad, C. Diehl, A. Gress, M. Meyer, A. L. Demirel, Y. Nur and A. Bertin, Macromol. Rapid Commun., 2010, 31, 511–525 CrossRef CAS.
- K. Kempe, M. Lobert, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3829–3838 CrossRef CAS.
- S. Kobayashi, S. Iijima, T. Igarashi and T. Saegusa, Macromolecules, 1987, 20, 1729–1734 CrossRef.
- S. Kobayashi, H. Uyama, N. Higuchi and T. Saegusa, Macromolecules, 1990, 23, 54–59 Search PubMed.
- K. Kempe, A. Vollrath, H. W. Schaefer, T. G. Poehlmann, C. Biskup, R. Hoogenboom, S. Hornig and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 1869–1873 Search PubMed.
- K. Kempe, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2011 DOI:10.1002/ marc.201100271.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS.
- C. Diehl and H. Schlaad, Chem.–Eur. J., 2009, 15, 11469–11472 CrossRef CAS.
- A. Gress, B. Smarsly and H. Schlaad, Macromol. Rapid Commun., 2008, 29, 304–308 CrossRef CAS.
- L. Chen, L. Zhang, J. Chen, J. Yang and R. Li, Chin. Sci. Bull., 2010, 55, 4187–4196 Search PubMed.
- D. A. Fulton and J. F. Stoddart, J. Org. Chem., 2001, 66, 8309–8319 CrossRef CAS.
- S. Kobayashi, E. Masuda, S. Shoda and Y. Shimano, Macromolecules, 1989, 22, 2878–2884 CrossRef CAS.
- O. Nuyken, G. Maier, A. Gross and H. Fischer, Macromol. Chem. Phys., 1996, 197, 83–95 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten, H. M. L. Thijs, B. M. Van Lankvelt and U. S. Schubert, Des. Monomers Polym., 2005, 8, 659–671 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.