Topological polymer chemistry: a cyclic approach toward novel polymer properties and functions
Takuya
Yamamoto
and
Yasuyuki
Tezuka
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: ytezuka@o.cc.titech.ac.jp
First published on 11th May 2011
Abstract
Recent progress observed in Topological Polymer Chemistry is outlined with particular emphasis on single-cyclic (ring) and multi-cyclic polymers having programmed chemical structures, now becoming obtainable with guaranteed purity by newly developed synthetic protocols. By making use of these topological polymers, unprecedented opportunities have now been realized to provide new insights on fundamental polymer properties either in solution or bulk, in static or dynamic states, or in self-assemblies. Moreover, unusual properties and functions for polymer materials have now been revealed based on their cyclic topologies, i.e., topology effects, unattainable either by linear or branched counterparts.
 Takuya Yamamoto | Takuya Yamamoto received his PhD in 2004 from the University of Utah where he worked with Prof. Peter J. Stang. He joined JST ERATO Aida Nanospace Project as a researcher in 2005. Since 2008, he has been an assistant professor of the Department of Organic and Polymeric Materials, Tokyo Institute of Technology. His current research interests include the synthesis and self-assembly of cyclic polymers for the development of functional materials. |
 Yasuyuki Tezuka | Yasuyuki Tezuka is a Professor of Tokyo Institute of Technology. He is a graduate of The University of Tokyo, and received his doctorate degree from Ghent University (Belgium) in 1982. He then joined Nagaoka University of Technology (Japan) as an assistant professor. In 1994, he moved to the Tokyo Institute of Technology, and has been a professor since 2003 in the Department of Organic and Polymeric Materials. He received Tokyo Tech Award of Best Teacher, 2004, and The Award of the Society of Polymer Science, Japan (2010). He has served as an Asian Editor of Reactive and Functional Polymers since 2006. His current research is focused on topological polymer chemistry, in particular on the design of topologically unique macromolecular architectures, and of novel polymer materials by their topology effects. |
1. Introduction
In the macroscopic world, the form of objects often dictates their functions and properties. Likewise in the nanoscopic world, continuing efforts have been undertaken, to fabricate extremely small objects having precisely defined structures, now opening the door to nanoscience and nanotechnology.1 In the field of synthetic polymer chemistry, the form of macromolecules has long been restricted to a linear or a randomly branched topology. In the last decade, however, a variety of precisely controlled, branched and moreover cyclic topologies has been realized along with the developments of intriguing synthetic protocols, based on living polymerization as well as on self-assembly processes.2,3 In particular, a number of breakthroughs has recently been achieved to produce a variety of single-cyclic (ring) and multi-cyclic polymers having programmed chemical structures and functionalities with guaranteed purity even by the practically viable procedures.4 Upon these developments, unprecedented opportunities have now been created not only to bring new insights in the frontier of basic polymer chemistry and physics, but also to disclose unusual properties and functions to polymer materials based on their topologies.5In this review on topological polymer chemistry,6 we describe first a brief overview on the synthetic developments for single- and multi-cyclic polymers, and subsequently on selected examples of newly prepared functional ring polymers revealing new insights on polymer properties by topology effects and on novel functions for eventual applications.
2. Synthetic developments for single-cyclic (ring) and multi-cyclic polymers
A straightforward and thus conventional synthetic protocol for ring polymers has been an end-to-end linking reaction using a linear polymer precursor having reactive groups, i.e., telechelics, and an equimolar amount of a bifunctional coupling reagent.3,4 It is notable, however, that this bimolecular reaction should be performed with strictly equimolar amounts of the telechelics and the coupling reagent. Moreover, the dilution condition should be applied to suppress the concurrent chain extension. In consequence, this bimolecular process is now recognized as a less attractive choice for the preparation of cyclic polymers in practice (Scheme 1A).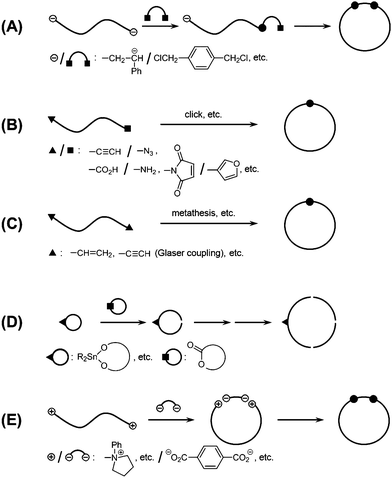 | ||
| Scheme 1 Ring polymer synthesis by (A) a bimolecular process, (B) a unimolecular process with asymmetric telechelics, (C) a unimolecular process with symmetric telechelics, (D) a ring-expansion polymerization process and (E) an electrostatic self-assembly and covalent fixation (ESA-CF) process. | ||
A significantly improved means by an alternative unimolecular process has subsequently been introduced to prepare cyclic polymers (Scheme 1B). It was first demonstrated by using an asymmetrically functional telechelic precursor, α-acetal-ω-styryl polystyrene,7 and the subsequent end-to-end polymer cyclization through the deprotection/activation treatment. This unimolecular process with asymmetrically functional telechelic precursors has been extended by employing alternative pairs of the reactive groups. Thus, an α-amino-ω-carboxyl polystyrene and poly(tert-butyl acrylate) were prepared through a living polymerization using an initiator having a protected amino group, followed by the end-capping reaction to introduce a carboxylic group.8 The resulting asymmetric telechelic precursor underwent, after the deprotection, an intramolecular amidation reaction to give a ring polymer product effectively. Alternatively, an α-(furan-protected)-maleimide-ω-cyclopentadienyl poly(methyl methacrylate) and poly(tert-butyl acrylate) were prepared through the ATRP using a protected maleimide initiator and the subsequent introduction of a cyclopentadienyl group at the living end with the nickelocene methodology.9 The obtained asymmetric telechelic precursor was subjected to the heat treatment to cause the spontaneous deprotection followed by an intramolecular Diels–Alder addition reaction to give a ring polymer product effectively.9 It is notable, however, that these processes using telechelic precursors having potentially reactive groups require multistep protection/deprotection treatments.
A versatile unimolecular process has recently been introduced by employing a click reaction using asymmetrically functional telechelic polystyrene having an alkyne and an azide group.10 The introduction of these two functional groups at the polymer chain-ends could be performed conveniently, and they can react with each other upon the addition of a Cu catalyst.11 Therefore, this click polymer cyclization has been applied to prepare such functional ring polymers, as poly(N-isopropyl acrylamide)12,13 and poly(caprolactone)14 as well as poly(valerolactone)15 to elucidate their topology-effect on phase transition, and on biodegradation, respectively, detailed in the later section. This unimolecular click process has also been employed to construct complex cyclic structures, either having multiple polymer segment components16 or having multicyclic as well as cyclic/linear hybrid polymer segments.17 On the other hand, the bimolecular click process using an azide-ended bifunctional telechelic precursor and a bifunctional alkyne compound as a linking reagent proceeds less effectively than the unimolecular counterpart.18
Another effective unimolecular process has been developed using symmetrically functional telechelic precursors, which are readily obtainable through a direct end-capping reaction of living bifunctional polymers (Scheme 1C).18–23 Thus, telechelic poly(tetrahydrofuran), poly(acrylic ester) and poly(isobutene) having olefinic end groups were prepared through the relevant living polymerization followed by the end-capping reaction. The subsequent metathesis condensation, i.e., metathesis polymer cyclization (MPC), in the presence of a Grubbs catalyst proceeded effectively to give the corresponding ring polymers in high yields even under dilution.18–20 This MPC process has further been applied for the practical synthesis of amphiphilic cyclic block copolymers21 as well as of multi-cyclic polymers.22,23 This unimolecular process using symmetrically functional telechelic precursors has been extended by alternative telechelic precursors having either alkyne groups for Glaser condensation or having bromobenzyl groups for a radical coupling process.24,25
Ring polymers are obtainable alternatively through a direct ring-expansion polymerization, in which the successive insertion of monomers into a cyclic initiator proceeds to form a ring polymer structure (Scheme 1D). This process does not require dilution in contrast to the end-to-end polymer cyclization process by linear polymer precursors, while a reactive initiator fragment is inherently included in the final ring polymer structure. Thus until recently, the ring-expansion process has been considered as a less practical means, since it is not an easy task to maintain the ring topology of the product while eliminating the initiator residues.26
A number of new processes have recently been developed to address this problem in the ring-expansion polymerization (Scheme 2). Thus, a new Ru catalyst bearing a cyclic ligand containing the Ru-alkylidene unit has been introduced and applied for the ring-expansion polymerization of cyclic olefins such as 1,5-cyclooctadiene or cyclooctene.27 During the chain-growth process, the chain transfer to the Ru-alkylidene unit took place to eliminate the initiator complex, and consequently a ring polybutadiene or a ring poly(cyclooctene) free of the catalyst fragment, respectively, is produced. The obtained ring poly(cyclooctene) was then hydrogenated to give a ring polyethylene, showing distinctive properties in comparison with commercial linear polyethylenes (Scheme 2A).27,28 It is notable, at the same time, that the random nature of this chain transfer process inherently causes the broadening of the molecular weight distribution of the product. This process has now been applied with a bulky, dendronized monomer to construct a ring nano-object directly observable by atomic force microscopy (AFM).29
 | ||
| Scheme 2 New ring-expansion polymerization processes using (A) a metathesis catalyst having cyclic ligand, (B) an N-heterocyclic carbene initiator, and (C) an cyclic stannate and subsequent intramolecular cross-linking. | ||
More recently, a new N-heterocyclic carbene initiator was introduced for the ring-expansion polymerization of lactones or lactide to form ring polyesters or polylactide, respectively (Scheme 2B).30,31 Remarkably, the obtained ring polymers possess narrow molecular weight distributions through the elimination of the initiator fragment after the quantitative consumption of the monomer, in contrast to the metathesis ring-expansion process shown before.28 By exploiting the living character, this technique has subsequently been applied for the synthesis of ring block polypeptoids and of complex topological polymers having a ring with many branches, i.e., jellyfish polymers.32,33 A further development of the ring-expansion process would provide a wider variety of chemical structures in ring polymers by broadening the choice of monomer types.
The combination of the classical ring-expansion polymerization process, in which the initiator fragment is included within the ring polymer structure, with the end-to-end polymer cyclization process could provide a new effective means for ring polymers having, in particular, large sizes (Scheme 2C).34 Thus, a ring poly(caprolactone) having short photocrosslinkable block segments was prepared through a sequential living ring-expansion polymerization using a cyclic stannate initiator. The subsequent irradiation under dilution caused the intramolecular covalent linking of the two short segments. The subsequent removal of the stannate group produced a chemically stable ring polymer product, though the exact chemical structure of the linking units is obscure.34
By employing this methodology, a high-molecular weight ring poly(chloroethyl vinyl ether) was prepared and was subjected to the grafting-onto reaction using a living polystyrene, to form a densely grafted large ring polymer product as a nanoobject, which was directly observable by the AFM.35,36 On the other hand, the AFM showed a large fraction of uncyclized, linear polymer components remained in the crude cyclization products, revealing the inherent difficulty of the end-to-end cyclization of polymer precursors of high molecular weights. It is remarkable, moreover, that a small portion of nanoobjects corresponding to complex ring polymers having tadpole, 8-shaped, catenated and knotted forms were also identified. This showed, for the first time, that not only single-cyclic but also various complex cyclic polymer topologies are indeed produced through the polymer cyclization process, as previously shown in DNA systems.37
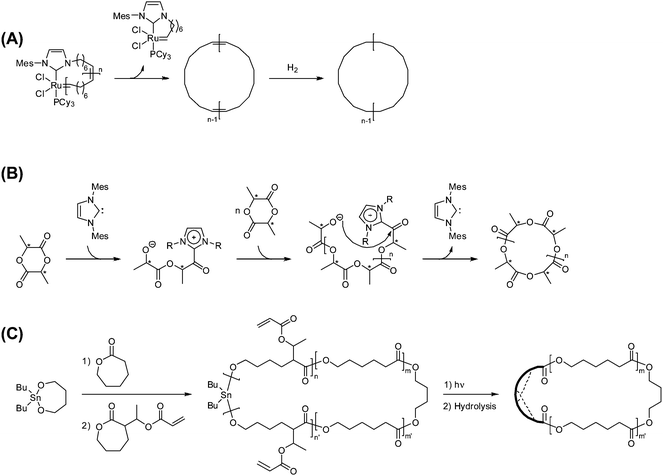
An electrostatic self-assembly and covalent fixation (ESA-CF) process has been introduced for the effective synthesis of not only cyclic but also functionalized cyclic and multicyclic polymers, where linear or star telechelic polymers having cyclic ammonium salt groups accompanying small or polymeric plurifunctional carboxylate counteranions are employed as key precursors (Scheme 1E).38–40 Various telechelic polymers have been conveniently prepared through the relevant living polymerization followed by the end-capping/end-conversion technique. The choice of such particular types of cyclic ammonium salt end groups as five- and six-membered cyclic ammonium, i.e., N-phenylpyrrolidinium and N-phenylpiperidinium salt groups, respectively, is of a crucial importance in the ESA-CF process (Scheme 3).41 The former undergoes upon heating a selective ring-opening reaction by a carboxylate counteranion to form an amino ester group through the nucleophilic attack exclusively at the N-adjacent ring (endo) position. On the other hand, the latter having unstrained six-membered cyclic ammonium salt groups gives a chemically more stable, simple ester group through the nucleophilic attack at the alternative N-adjacent (exo) position, to eliminate N-phenylpiperidine.
 | ||
| Scheme 3 The covalent conversion in the ESA-CF process by the ring-opening of N-phenyl pyrrolidinium group and by the ring-elimination of N-phenyl piperidinium group. | ||
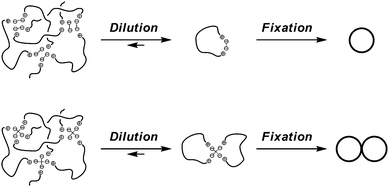
And in the electrostatic polymer self-assembly, the cations at the end of the telechelics and the anions in the carboxylates counteranions always balance the charges. Upon the dilution, the self-assembly tends to dissociate into one having the smallest number of polymer components by keeping the balance of the charges (Scheme 4). Thus, when a self-assembly comprised of a linear telechelic precursor, typically poly(THF), having cyclic ammonium salt groups accompanying either a dicarboxylate or a tetracarboxylate counteranion was heated at dilution, the selective ring-opening reaction of the cyclic ammonium groups took place to produce single-cyclic (ring) or dicyclic (8-shaped) polymer products, respectively, in high yields.38,42 Ring poly(THF)s having the molecular weights of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, corresponding to the ring size of as high as 700 atoms, were obtainable and characterized unequivocally by the MALDI-TOF mass technique. Furthermore, the ESA-CF process with dynamic equilibrium nature has been exploited to construct various multicyclic as well as cyclic-linear hybrid polymer topologies (Scheme 5).6
000, corresponding to the ring size of as high as 700 atoms, were obtainable and characterized unequivocally by the MALDI-TOF mass technique. Furthermore, the ESA-CF process with dynamic equilibrium nature has been exploited to construct various multicyclic as well as cyclic-linear hybrid polymer topologies (Scheme 5).6
 | ||
| Scheme 4 Synthesis of single-cyclic (ring) and dicyclic (8-shaped) polymersvia the ESA-CF process. | ||
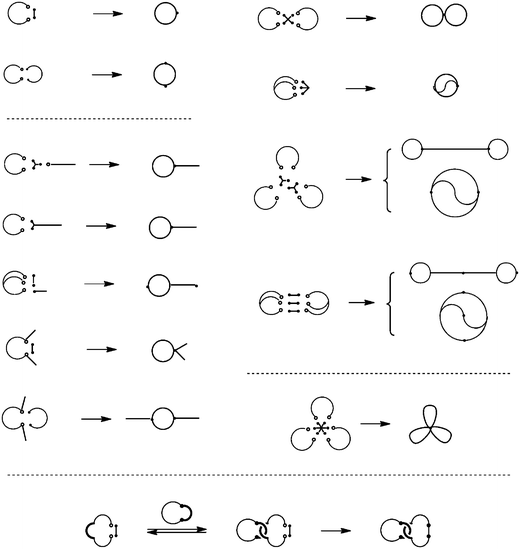 | ||
| Scheme 5 Various multicyclic and cyclic-linear hybrid polymer topologies constructed by the ESA-CF process. | ||
Moreover, ring polymers possessing the prescribed functional groups at the designated positions, i.e., kyklo-telechelics and cyclic macromonomer, have also been prepared by employing telechelic precursors possessing such functional groups as hydroxyl, olefinic, acetylenic, and azide groups at the designated, typically a center, position by using the relevant initiator, or by introducing functional linking reagents.43 They were subsequently applied to construct more complex polymer topologies either through the covalent linkage or even through the non-covalent, mechanical linkage uniquely formed by threading with ring polymer units.44,45 It was also shown that network polymers formed from cyclic polymer precursors exhibited unique gel properties compared with conventional gels obtained with linear/branched polymer precursors.46 Moreover, the topological conversion of polymers between ring/linear and even between single-cyclic/dicyclic topologies has now been achieved.45,47,48
By the combination of the ESA-CF protocol with selective covalent linking chemistry, a wide variety of multi-cyclic as well as cyclic-linear conjugated polymers have been synthesized.49 In particular, the combination of the ESA-CF process with the metathesis condensation and with the click process has now been revealed as a versatile means to construct a variety of complex multicyclic polymer topologies of fused-, spiro- and bridged forms (Scheme 6).50 Both the functional ring polymer precursors and the covalently linked multicyclic polymer product were unequivocally characterized by means of a series of spectroscopic and chromatographic techniques including MALDI-TOF mass spectroscopy and SEC. As shown in Fig. 1, for a typical example, the structure of the polymer products having complex multicyclic topologies is convincingly demonstrated. The ESA-CF process has further been applied for the effective synthesis of topologically unique polymer catenanes comprised of large polymer rings51 through the two orthogonal non-covalent interactions, namely the electrostatic and the hydrogen-bond interactions (Scheme 5).45
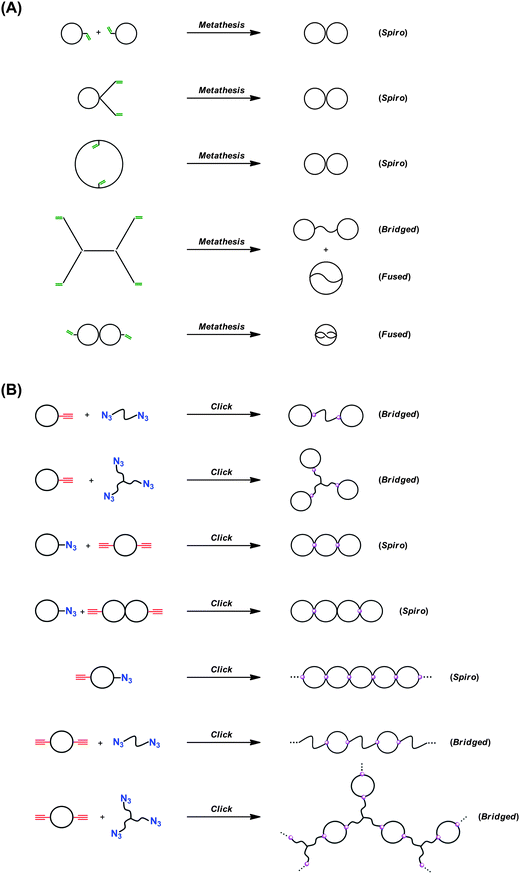 | ||
| Scheme 6 Construction of spiro-, bridged-, and fused-multicyclic polymer topologies by the ESA-CF process combined with (A) metathesis condensation and with (B) click chemistry. | ||
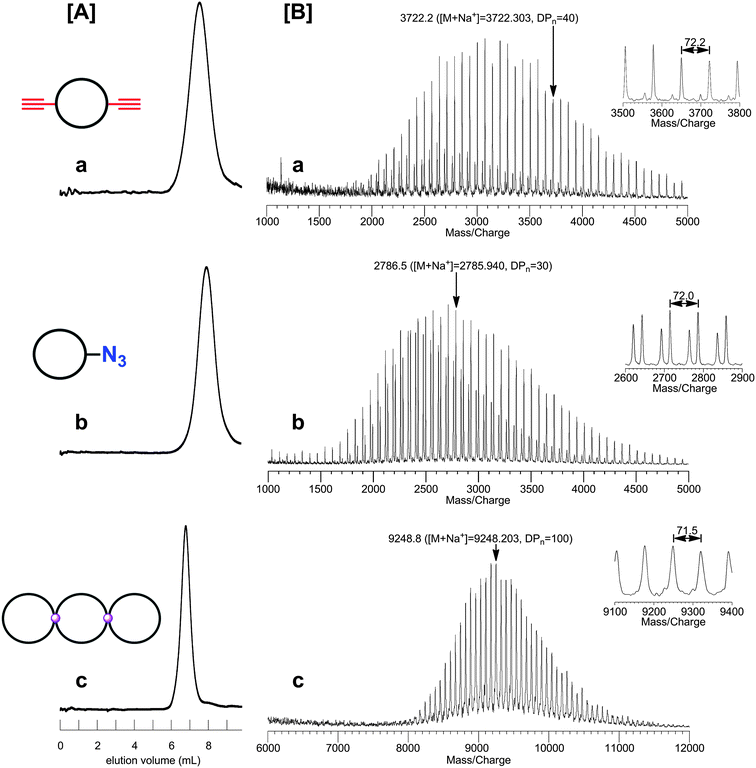 | ||
| Fig. 1 SEC (A) and MALDI-TOF mass (B) characterization of functional ring polymer precursors and of a spiro-tricyclic polymer product obtained therewith. | ||
3. Topology effects by cyclic polymers: depleting the chain-ends and beyond
In contrast to linear or branched polymer topologies, ring polymer topologies are uniquely defined by their absence of the chain ends.3,5 As recognized by the typical case of glass transition temperature (Tg), the topology effect of ring polymers is thus arisen from the depletion of the end-groups of polymer segments. Thus, the Tg of ring polymers is higher than the linear counterpart, until the polymer chain length becomes long enough to diminish the contribution of the chain end. On the contrary, the hydrodynamic volume or the melt viscosity of ring polymers is inherently smaller than their linear counterparts irrespective of their chain lengths. This indicates the distinctive conception in the topology effect by ring polymers. The latter is regarded as an inherent topology effect by ring polymers, while the former as the depletion effect of the polymer chain ends. Nevertheless, the elimination of the chain ends of polymer molecules inevitably leads to the formation of cyclic constructions to bring the topology effect, now drawing increasing interests to discover unusual properties and functions in polymer materials covering mechanical, thermal, electric, photonic and bio-related applications either in bulk or at surface. From an experimental point of view, moreover, further developments are awaited to provide ring polymers possessing high molecular weights with guaranteed purity, since the mixing of even small amount of linear counterpart might cause inconclusive or even erroneous conclusions on the topology effect by ring polymers.4. Topology effects on basic polymer properties: simulations and experiments from simple to complex cyclic polymers
Basic characteristics of single-cyclic (ring) homopolymers either in solution or bulk, in static or dynamic states, have extensively been studied and documented relevantly.3 Therefore, recent studies have elaborated either on specific polymers of practical importance like polyethylenes,52 on specific chain characteristics like semiflexible to stiff nature,53 or on multicomponent polymer systems typically block copolymers.54 Notably, moreover, theory and simulation studies have increasingly focused on multi-cyclic complex polymers, obviously promoted by synthetic developments on complex topological polymers.55–57 Unique topological polymer properties envisaged by the simulation studies are now to be verified experimentally using relevant topological polymers. Indeed, a collaborative simulation and experimental study on the liquid chromatography study has been conducted on multi-cyclic polymers, and could provide a basis for the purification and the characterization of polymers having complex topologies.58The diffusion behavior is of particular interest in view of the topology effects on polymer molecules, since the self-diffusion of linear and branched polymers follows the reptation mechanism, a snake-like translational motion, in which the polymer chain ends tend to command the motion of their in-chain segments.59 Cyclic polymers are unique, on the other hand, in the absence of chain ends, and their non-reptation dynamics has been a subject of both theory/simulation and experimental studies.60
A single-molecule fluorescence spectroscopy is a unique means to observe directly the motion of single polymer molecules.61 Thus, the ESA-CF process was applied with mono- and bifunctional telechelic poly(THF) precursors having N-phenylpiperidinium (six-membered ring) groups possessing a perylene dicarboxylate counteranion, to produce ring and linear poly(THF)s having a perylene unit applicable to the single-molecule fluorescence spectroscopy measurement (Scheme 7).62
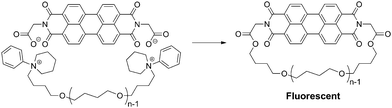 | ||
| Scheme 7 Synthesis of a fluorescent ring poly(THF) by the ESA-CF process through the elimination of N-phenyl piperidinium groups. | ||
The subsequent single-molecule imaging was conducted to observe the real-time motion of individual polymer molecules. The video-recording of the motion of single-molecule fluorescence images, followed by the computational analysis could provide the position and trajectory of the object on the x–y plane as shown in Fig. 2. The histograms of the distribution of the individual diffusion constant were subsequently obtained from large enough datasets of these consecutive images. Upon the statistical analysis of the distribution patterns, a bimodal diffusion mechanism was disclosed for ring polymers, corresponding to either threaded or non-threaded conditions in a linear molecule matrix. In contrast, the unimodal diffusion was indicated for the linear counterpart (Fig. 3).
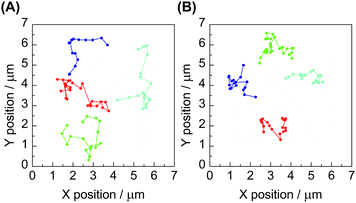 | ||
| Fig. 2 Trajectories of time-course single-molecule images of (A) linear and (B) ring poly(THF)s having a perylene unit in a linear poly(THF) matrix. | ||
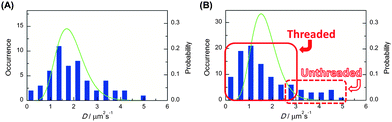 | ||
| Fig. 3 Frequency histograms (blue bars) of the diffusion coefficient determined for (A) linear and (B) ring poly(THF)s having a perylene unit. The green lines show theoretical statistical distributions corresponding to the diffusion of single molecules in homogeneous environment with diffusion coefficient given by means of the respective histograms. The measured distribution for the linear poly(THF) is reproduced reasonably well by the calculated distribution. On the other hand that for the ring poly(THF) deviates significantly from the calculated homogeneous statistical distribution. The deviation reflects the inhomogeneous nature of the system. | ||
As shown above, the significant topology effects could be envisaged in polymer properties involving their diffusion process. In this respect, the crystallization dynamics is of a considerable interest since the diffusion and the conformational transformation of polymer molecules could dictate the folding and packing of polymer segments in the crystal lattice. To elucidate the topology effect in the crystallization process, a defect-free ring poly(THF) was prepared together with the linear counterpart having the precisely relevant chemical structure.63 Thus, a living bifunctional poly(THF) was terminated with sodium allyloxide to give a telechelic precursor having allyl end groups. The subsequent metathesis polymer cyclization (MPC) with a Grubbs catalyst under dilution, followed by the hydrogenation of the unsaturated linking group afforded a defect-free ring poly(THF), consisting exclusively of oxytetramethylene units (Scheme 8). The isothermal crystallization experiments showed the slowercrystallization rate for ring poly(THF)s ascribed to their topologically constrained conformation (Fig. 4). Moreover, distinctive crystal morphologies are produced, in particular a banded structure in the spherulites exclusively for the ring poly(THF) but not for the linear counterpart. The crystallization kinetics of ring poly(ε-caprolactone)s has recently been reported by several groups.64 The fastercrystallization rate was observed for ring polymers than the linear counterpart in contrast to the case of poly(THF), and was ascribed to the higher diffusion of ring polymers than their linear counterparts.64
 | ||
| Scheme 8 Synthesis of a defect-free ring poly(THF). | ||
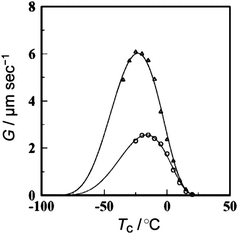 | ||
| Fig. 4 Crystallization temperature (Tc) dependence of spherulite growth rates (G) for linear (△) and ring (○) poly(THF)s. | ||
5. Topology effects by ring polymers in self-assemblies
The topology effects by ring polymers could be amplified in their assembled states rather than in their individually separated forms. Therefore, micelles formed by ring polymers should be of a remarkable interest. Thus, a series of micelles were prepared with amphiphilic cyclic AB block copolymers, comprised of either poly(butylene oxide)/poly(ethylene oxide) or poly(propylene oxide)/poly(ethylene oxide), together with their linear AB, linear ABA block copolymer counterparts.65–67 The cyclic block copolymer employed in this study was prepared by a bimolecular process using the hydroxy-terminated linear block copolymer and dichloromethane as a linking reagent in the presence of KOH, to form an acetal linking unit (Scheme 9A).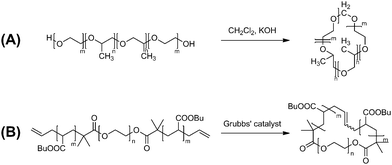 | ||
| Scheme 9 Synthesis of amphiphilic cyclic block copolymer by (A) a bimolecular and (B) a unimolecularpolymer cyclization process. | ||
The critical micelle concentration (cmc) of the micelle with the cyclic AB block copolymer was similar to or marginally smaller than those with the linear ABA counterparts. On the contrary, the corresponding micelle with the linear AB counterpart showed a significantly smaller cmc. Moreover, the association number (Nw), thermodynamic radius (rt), and hydrodynamic radius (rh) of the micelle with the cyclic AB block copolymers and with the linear ABA counterparts were comparable to each other, but distinctively smaller than those with the linear AB counterparts. These results are consistent with the geometric estimation of the size of the micelles, corresponding to the Nw, rt, and rh, which is dependent on the continuous chain length of the hydrophobic segment in the block copolymers. And, those in the cyclic AB and the linear ABA block copolymers are estimated as a half of that in the linear AB counterpart employed in this study. Moreover, the cyclic AB block copolymer tends to constitute a stiff hairpin-like conformation, resulting in a slightly larger hydrophobic core within the micelle compared to that with the linear ABA counterpart having free chain ends. The enthalpy of micellization (ΔmicH°) was also smaller with the cyclic AB block copolymer than that with the corresponding linear ABA counterpart, presumably because the segmental flip-flop is inhibited in the micelle with the former, in contrast to the motional freedom by the latter having free chain ends. This also agrees with the smaller entropy of micellization (ΔmicS°) with the cyclic AB block copolymer than with the linear counterpart.
More recently, a significant topology effect has been revealed in the thermal stability of the micelle formed with cyclic AB block copolymers in comparison with the linear ABA and even with the linear AB counterparts.68 Notably, a variety of relevant cyclic lipids, thus a type of surfactants, have been discovered in the cell membrane of thermophilic archaea, single-cell microorganisms, surviving in the hostile environment of very high-temperatures located at hot springs and submarine volcanoes.69 Thus, an amphiphilic cyclic AB block copolymer, comprised of poly(butyl acrylate)/poly(ethylene oxide) segments, was prepared. The unimolecular, metathesis polymer cyclization (MPC) process was employed using a linear ABA block copolymer precursor having olefinic end groups (Scheme 9B).21
The obtained cyclic and the relevant linear block copolymers were allowed to form the corresponding micelles, in which their cmc's and the number-average hydrodynamic volumes (Dh) were measured to be closely comparable to each other (Fig. 5).68 The size and morphology of the micelles with either block copolymers were directly observed by atomic force microscopy (AFM) (Fig. 6) and transmission electron microscopy (TEM) (Fig. 6) to show commonly flower-like spherical shapes possessing the consistent diameters determined by DLS (Scheme 10).
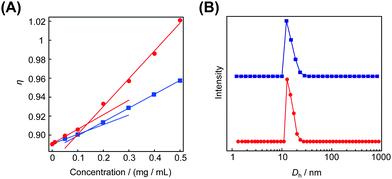 | ||
| Fig. 5 The cmc (A) and the size distribution (B) of the micelle with the cyclic block copolymer (red) and with the linear counterpart (blue). | ||
 | ||
| Fig. 6 AFM images of micelles with the linear (a) and cyclic (b) block copolymers, and TEM images of micelles with the linear (c) and cyclic (d) block copolymers. | ||
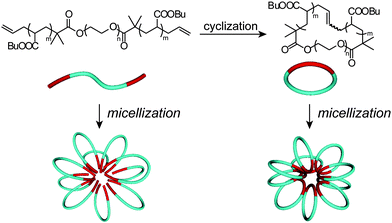 | ||
| Scheme 10 Schematic representation of the flower-like micelles with the linear (left) and cyclic (right) amphiphilic block copolymers. | ||
Remarkably, the thermal stability of the micelles was drastically enhanced using the cyclic block copolymer. Thus, the corresponding micelle solutions were heated from 20 °C in a stepwise fashion (Fig. 7), and the micelle with the linear ABA block copolymer became turbid at around 25 °C, while that with the cyclic AB counterpart was stable till above 70 °C.68
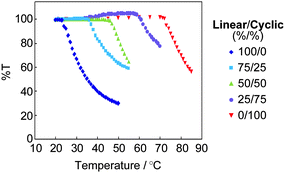 | ||
| Fig. 7 The thermal stability of the micelles solely with the linear and with the cyclic block copolymers, and also with the coassembly by both components. The total concentration of the copolymers in a solution was 0.50 mg mL−1. | ||
Based on the large deviation of the clouding point (Tc) among the micelles either solely with cyclic or with linear block copolymers, the tuning of the Tc of the micelle was achieved by the coassembly of both the linear and cyclic block copolymers.68 Thus, the Tc was systematically altered along with the relative contents of the cyclic and the linear block copolymer components in the micelle, as shown in Fig. 7.68 This topology effect will be utilized to design temperature-responsive devices encapsulating and releasing appropriate components in and from the micelle. In particular, the topology-based polymer materials design should be suitable for human body-related applications including drug delivery system, food, and cosmetics, for which the modification of chemical structure and molecular weight might induce reasonable concern over the toxicity and biocompatibility.
6. Topology effects by functional ring polymers having programmed chemical structures
Along with the introduction of new synthetic methodologies, an increasing variety of functional ring polymers and oligomers have become obtainable to exploit their topology effects. A number of recent studies are presented in this section, covering a diverse field either by conjugated cyclic oligomers,70 by photo-responsive ring polymers,71,72 by biodegradable ring polyesters,14 by temperature-responsive ring polymers,12,13or by pharmacologically functional ring polymers.73Conjugated polymers and oligomers have currently been extensively studied primarily due to their potential for diverse electronic and photonic applications.74 Ring-shaped conjugated polymers and oligomers have attracted particular interests as a prototype of the molecularly fabricated electric circuit, since the circulation of electrons/holes might be induced along the “endless” conjugated segments. A wide variety of conjugated ring oligomers possessing the increasing numbers of unsaturated groups have thus been prepared to test their electrochemical properties.70 Fully conjugated large ring polymers, on the other hand, have still been an elusive synthetic challenge. It is notable, moreover, that a twisted, Moebius stripe-like conjugated oligomer has attracted considerable interests because of their unusual non-Hückel aromaticity, in contrast to the conventional Hückel-type aromaticity which is dependent on its planar geometry by conjugated units.75
Photo-response behaviors could be influenced by polymer topologies of either ring or linear forms. Thus, a ring polymer having photo-responsive azobenzene units in the main chain was prepared through the bimolecular end-to-end cyclizationvia click chemistry.71 Upon the irradiation of UV light, both the trans-to-cis and the cis-to-transisomerization proceeded slightly faster by the ring polymer in comparison with the linear counterpart. This behavior was attributed to the higher topological constraint of the former than the latter counterpart.71 As another example, a ring polystyrene having two photoactive groups, namely fluorene and anthracene units, was prepared through the bimolecular process using a living bifunctional polystyrene produced with an initiator having a fluorene unit and a linking reagent having an anthracene unit.72 The fluorescent behavior of the obtained ring polymer was subsequently studied to show the intramolecular energy migration between the two chromophores.72
Naturally occurring ring polymers, such as cyclic DNAs, cyclic peptides as well as cyclic polysaccharides,3 and multi-cyclic counterparts like cyclotides,76 are known to exhibit higher stability against biologically induced decomposition. In this relevance, the topology effect of aliphatic biodegradable polyesters on their decomposition behavior has been investigated.14 Thus, ring poly(caprolactone) was prepared through the unimolecular click polymer cyclization using the corresponding precursor having an azide and an alkyne group. The higher hydrolytic stability was then observed in comparison with the linear counterparts.14 Notably, moreover, such biodegradable ring polyesters by poly(valerolactone) and by poly(lactide) were also prepared.15,30,31,77
Temperature-responsive properties of poly(N-isopropylacrylamide), poly(NIPAM), have extensively been studied for their eventual uses in the controlled release devices and for the cell–sheet applications.78 The two groups recently reported the topology effects by ring poly(NIPAM)s.12,13 Thus, the two groups prepared linear poly(NIPAM) precursors having alkyne and azide end groups either by a reversible addition–fragmentation chain transfer (RAFT) or by an atom transfer radical polymerization (ATRP) technique, and the unimolecular click process was commonly applied for the subsequent polymer cyclization. The cloud point (TCP) of the obtained ring poly(NIPAM) by one group was several degrees higher than that of the corresponding linear counterpart. In addition, they observed that the phase transition by the ring polymers took place distinctively slower upon heating.12 Another group, on the other hand, observed the ring poly(NIPAM) showing a few degrees lower TCP against the linear counterpart at the higher solution concentration, while higher at the lower solution concentration. Furthermore, they reported the smaller enthalpy change (ΔH) during the phase transition with the ring poly(NIPAM). These results were accounted for by the suppression of the hydrogen bond interaction between the ring poly(NIPAM) with water molecules due to their topological constraints in comparison with the flexible linear counterpart having free chain ends.13
A pharmacological application of ring polymers has recently been demonstrated by elucidating the filtration/elimination mechanism of a kidney through its nanoporous channel.73 Thus, it was anticipated that the ring polymer could remain longer in a body than the linear counterpart, since the former should be reluctant to transform its conformation into a folded shape to traverse through the nanoporous channels of the kidney.73 To test this topology effect, a series of linear and cyclic poly(caprolactone)s having poly(ethylene glycol) graft segments and having phenolic functionalities for radiolabeling by 125I were prepared. The lower secretion into urine and in turn the higher accumulation in various organs was indeed confirmed with the ring polymer having an appropriate molecular weight, corresponding to its hydrodynamic volume.73 These findings may provide new design methodologies for drug carriers and imaging agents via the topology-based control of a blood circulation time.
7. Conclusions and future perspectives
In this review, we have shown a variety of recent achievements in topological polymer chemistry, to focus on novel polymer properties and functions uniquely revealed by ring polymers. The topology effects, in particular by ring polymers have now offered unique opportunities in polymer materials design unattainable by other means. A viable promise for the practical utilization of the topology effect undoubtedly includes the modification of any existing polymeric materials dependent mostly on linear polymers, inflicting little concern on chemical toxicity and environmental pollution. Furthermore, broader opportunities of topology effects by not only single-cyclic but also multi-cyclic polymers are expected in polymer chemistry, supramolecular chemistry, and materials science.Acknowledgements
We thank Sekisui Chemical Grant Program for Research on Manufacturing Based on Learning from Nature (Y.T.), The Mitsubishi Foundation (Y.T.), Tokuyama Science Foundation (T.Y.), CASIO Science Promotion Foundation (T.Y.), The Japan Securities Scholarship Foundation (T.Y.), The Kurata Memorial Hitachi Science and Technology Foundation (T.Y.), and Yazaki Memorial Foundation for Science and Technology for financial support. This study was also supported in part by grants from the Ministry of Education, Science and Culture, Japan through the Japan Society of Promotion of Sciences (23350050, 23685022 and 23106709).References
- For example, see: (a) T. H. LaBean, Nature, 2009, 459, 331–332 Search PubMed; (b) N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- For branched polymers, see: (a) N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS; (b) A. Hirao, T. Watanabe, K. Ishizu, M. Ree, S. Jin, K. S. Jin, A. Deffieux, M. Schappacher and S. Carlott, Macromolecules, 2009, 42, 682–693 Search PubMed; (c) N. V. Tsarevsky and K. Matyjasewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- For cyclic polymers, see: (a) Cyclic Polymers, ed. J. A. Semlyen, Kluwer, Dordorecht, 2nd edn, 2000 Search PubMed; (b) K. Endo, Adv. Polym. Sci., 2008, 217, 121–184 CAS; (c) T. Yamamoto and Y. Tezuka, in Synthesis of Polymers, ed. D. Schlüter, C. Hawker and J. Sakamoto, Wiley-VCH, Weinheim, 2011, in press Search PubMed.
- (a) K. Adachi and Y. Tezuka, J. Synth. Org. Chem. Jpn., 2009, 67, 1136–1143 Search PubMed; (b) Y. Tezuka, Chem. Rec., 2005, 5, 17–26 Search PubMed; (c) S. M. Grayson, Nat. Chem., 2009, 1, 178–179 Search PubMed; (d) S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC; (e) S. Perrier, Nat. Chem., 2011, 3, 194–196 Search PubMed.
- (a) T. McLeish, Science, 2002, 297, 2005–2006 CrossRef CAS; (b) T. McLeish, Nat. Mater., 2008, 7, 933–935 Search PubMed; (c) M. E. Fox, F. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS; (d) H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS; (e) A. Deffieux and R. Borsali, Controlled Synthesis and Properties of Cyclic Polymers, Wiley-VCH, Weinheim, 2007 Search PubMed; (f) T. Yamamoto and Y. Tezuka, in Complex Macromolecular Architectures: Synthesis, Characterization, and Self-Assembly, ed. N. Hadjichristidis, A. Hirao, Y. Tezuka and F. Du Prez, Wiley, Singapore, 2011, pp. 3–20 Search PubMed.
- (a) Y. Tezuka and H. Oike, J. Am. Chem. Soc., 2001, 123, 11570–11576 CrossRef CAS; (b) Y. Tezuka and H. Oike, Prog. Polym. Sci., 2002, 27, 1069–1122 CrossRef CAS; (c) Y. Tezuka, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2905–2917 CrossRef CAS; (d) Y. Tezuka and H. Oike, Macromol. Rapid Commun., 2001, 22, 1017–1029 Search PubMed; (e) Y. Tezuka , et.al., Topology Designing (in Japanese), NTS, Tokyo, 2009, ISBN978-4-86043-162-4 Search PubMed; (f) Y. Tezuka, Kobunshi, 2008, 57, 81–85 Search PubMed.
- (a) L. Rique-Lurbet, M. Schappacher and A. Deffieux, Macromolecules, 1994, 27, 6318–6324 CrossRef CAS; (b) M. Schappacher and A. Deffieux, Macromolecules, 2001, 34, 5827–5832 CrossRef CAS.
- (a) M. Kubo, T. Hayashi, H. Kobayashi and T. Itoh, Macromolecules, 1998, 31, 1053–1057 Search PubMed; (b) M. Kubo, T. Nishigawa, T. Uno, T. Itoh and H. Sato, Macromolecules, 2003, 36, 9264–9266 Search PubMed.
- M. Glassner, J. P. Blinco and C. Barner-Kowollik, Macromol. Rapid Commun., 2011, 32, 724–728 Search PubMed.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS.
- (a) D. E. Lonsdale, C. A. Bell and M. J. Monteiro, Macromolecules, 2010, 43, 3331–3339 CrossRef CAS; (b) D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC; (c) R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- X. P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- J. Xu, J. Ye and S. Y. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- (a) J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406–6413 CrossRef CAS; (b) J. N. Hoskins and S. M. Grayson, Polym. Chem., 2011, 2, 289–299 RSC.
- H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7090–7094 Search PubMed.
- (a) G.-Y. Shi, X.-Z. Tang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2390–2401 CrossRef CAS; (b) Y.-Q. Dong, Y.-Y. Tong, B.-T. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2009, 42, 2940–2948 CrossRef CAS; (c) H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5083–5091 Search PubMed; (d) A. Touris and N. Hadjichristidis, Macromolecules, 2011, 44, 1969–1976 Search PubMed.
- (a) G.-Y. Shi and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2620–2630 Search PubMed; (b) G.-Y. Shi and C.-Y. Pan, Macromol. Rapid Commun., 2008, 29, 1672–1678 CrossRef CAS; (c) G.-Y. Shi, L.-P. Yang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6496–6508 CrossRef CAS; (d) S. Perrier, Nat. Chem., 2011, 3, 194–196 Search PubMed.
- M. Schulz, S. Tanner, H. Barqawi and W. Binder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 671–680 Search PubMed.
- Y. Tezuka and R. Komiya, Macromolecules, 2002, 35, 8667–8669 CrossRef CAS.
- S. Hayashi, K. Adachi and Y. Tezuka, Chem. Lett., 2007, 982–983 Search PubMed.
- K. Adachi, S. Honda, S. Hayashi and Y. Tezuka, Macromolecules, 2008, 41, 7898–7903 Search PubMed.
- Y. Tezuka and F. Ohashi, Macromol. Rapid Commun., 2005, 26, 608–612 Search PubMed.
- S. Hayashi, K. Adachi and Y. Tezuka, Polym. J., 2008, 40, 572–576 Search PubMed.
- Y. Zhang, G. Wang and J. Huang, Macromolecules, 2010, 43, 10343–10347 Search PubMed.
- (a) A. F. Voter and E. S. Tillman, Macromolecules, 2010, 43, 10304–10310 Search PubMed; (b) R. Nicolay and K. Matyjaszewski, Macromolecules, 2011, 44, 240–247 Search PubMed.
- (a) H. R. Kricheldorf and S. Eggerstedt, Macromol. Chem. Phys., 1999, 200, 587–593 CrossRef CAS; (b) K. J. Shea, S. Y. Lee and B. B. Busch, J. Org. Chem., 1998, 63, 5746–5747 CrossRef CAS.
- (a) C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS; (b) C. W. Bielawski, D. Benitez and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 8424–8425 CrossRef CAS.
- (a) Y. Xia, A. J. Boydston, I. A. Gorodetskaya, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 2670–2677 CrossRef CAS; (b) For the compatibility of ring and linear polymers, see also: S. Singla and H. W. Beckham, Macromolecules, 2008, 41, 9784–9792 Search PubMed.
- A. J. Boydston, T. W. Holcombe, D. A. Unruh, J. M. J. Fréchet and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 5388–5389 CrossRef CAS.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS.
- W. Jeong, E. J. Shin, D. A. Culkin, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2009, 131, 4884–4891 CrossRef CAS.
- L. Guo and D. Zhang, J. Am. Chem. Soc., 2009, 131, 18072–18074 CrossRef CAS.
- O. Coulembier, S. Moins, J. De Winter, P. Garbaux, P. Leclere, R. Lazzaroni and P. Dubois, Macromolecules, 2010, 43, 575–579 Search PubMed.
- (a) H. Li, A. Debuigne, R. Jérome and P. Lecomte, Angew. Chem., Int. Ed., 2006, 45, 2264–2267 CrossRef CAS; (b) M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2008, 130, 14684–14689 Search PubMed.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 CrossRef CAS.
- M. Schappacher and A. Deffieux, Angew. Chem., Int. Ed., 2009, 48, 5930–5933 Search PubMed.
- For recent examples, see: (a) S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS; (b) J. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. Sha, P. E. Constantinou, S. L. Ginell, C. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS; (c) C. Zhang, S. H. Ko, M. Su, Y. Leng, A. E. Ribbe, W. Jiang and C. Mao, J. Am. Chem. Soc., 2009, 131, 1413–1415 CrossRef CAS; (d) F. C. Simmel, Angew. Chem., Int. Ed., 2008, 47, 5884–5887 CrossRef CAS.
- H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592–9599 CrossRef CAS.
- H. Oike, A. Uchibori, A. Tsuchitani, H.-K. Kim and Y. Tezuka, Macromolecules, 2004, 37, 7595–7601 Search PubMed.
- (a) Y. Tezuka, N. Takahashi, T. Satoh and K. Adachi, Macromolecules, 2007, 40, 7910–7918 Search PubMed; (b) K. Adachi, H. Takasugi and Y. Tezuka, Macromolecules, 2006, 39, 5585–5588 Search PubMed.
- (a) Y. Tezuka, K. Mori and H. Oike, Macromolecules, 2002, 35, 5707–5711 Search PubMed; (b) H. Oike, M. Hamada, S. Eguchi, Y. Danda and Y. Tezuka, Macromolecules, 2001, 34, 2776–2782 CrossRef CAS.
- (a) H. Oike, S. Kobayashi, T. Mouri and Y. Tezuka, Macromolecules, 2001, 34, 2742–2744 Search PubMed; (b) Y. Peng, H. Liu, X. Zhang, S. Liu and Y. Li, Macromolecules, 2009, 42, 6457–6462 Search PubMed.
- H. Oike, T. Mouri and Y. Tezuka, Macromolecules, 2001, 34, 6229–6234 Search PubMed.
- K. Miki, Y. Inamoto, S. Inoue, T. Uno, T. Itoh and M. Kubo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5882–5890 Search PubMed.
- K. Ishikawa, T. Yamamoto, H. Harada and Y. Tezuka, Macromolecules, 2010, 43, 7062–7067 Search PubMed.
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 Search PubMed.
- M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2011, 133, 1630–1633 Search PubMed.
- For review, see: (a) T. Yamamoto and Y. Tezuka, Eur. Polym. J., 2011, 47, 535–541 Search PubMed; (b) Also see: Y. Tezuka, R. Komiya, Y. Ido and K. Adachi, React. Funct. Polym., 2007, 67, 1233–1242 Search PubMed.
- (a) Y. Tezuka and K. Fujiyama, J. Am. Chem. Soc., 2005, 127, 6266–6270 CrossRef CAS; (b) Y. Tezuka, R. Komiya and M. Washizuka, Macromolecules, 2003, 36, 12–17 Search PubMed.
- (a) N. Sugai, H. Heguri, K. Ohta, Q. Meng, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 14790–14802 CrossRef CAS; (b) See also: D. E. Lonsdale and M. J. Monteiro, Chem. Commun., 2010, 46, 7945–7947 Search PubMed; (c) B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234–238 Search PubMed.
- P. G. Clark, E. N. Guidry, W. Y. Chan, W. E. Steinmetz and R. H. Grubbs, J. Am. Chem. Soc., 2010, 132, 3405–3412 CrossRef CAS.
- (a) G. Tsolou, N. Stratikis, C. Bang, P. S. Stephanou and V. G. Mavrantzas, Macromolecules, 2010, 43, 10692–10713 Search PubMed; (b) K. Hur, C. Jeong, R. G. Winkler, N. Lacevic, R. H. Gee and D. Y. Yoon, Macromolecules, 2011, 44, 2311–2315 Search PubMed.
- D. Ida, D. Nakatomi and T. Yoshizaki, Polym. J., 2010, 42, 735–744 Search PubMed.
- A. A. Gorbunov and A. V. Vakhrushev, Polymer, 2010, 51, 3285–3292 Search PubMed.
- C. R. A. Abreu and F. A. Escobedo, Macromolecules, 2005, 38, 8532–8545 Search PubMed.
- G. Beaucage and A. S. Kulkarni, Macromolecules, 2010, 43, 532–537 Search PubMed.
- Y. Wang, I. Teraoka, F. Y. Hansen, G. H. Peters and O. Hassager, Macromolecules, 2011, 44, 403–412 Search PubMed.
- A. V. Vakhrushev, A. A. Gorbunov, Y. Tezuka, A. Tsuchitani and H. Oike, Anal. Chem., 2008, 80, 8153–8162 Search PubMed.
- (a) P.-G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, 1979 Search PubMed; (b) G. Strobl, The Physics of Polymers: Concept for Understanding Their Structures and Behavior, Springer, Berlin, 2007 Search PubMed.
- (a) T. McLeish, Adv. Phys., 2002, 51, 1379–1527 CrossRef CAS; (b) M. Kapnistos, M. Lang, D. Vlassopoulos, W. Pyckhout-Hintzen, D. Richter, D. Cho, T. Chang and M. Rubinstein, Nat. Mater., 2008, 7, 997–1002 CrossRef CAS; (c) S. Nam, J. Leisen, V. Breedveld and H. W. Beckham, Macromolecules, 2009, 42, 3121–3128 Search PubMed.
- D. Wöll, E. Braeken, A. Deres, F. C. DeSchryver and J. Hofkens, Chem. Soc. Rev., 2009, 38, 313–328 RSC.
- S. Habuchi, N. Satoh, T. Yamamoto, Y. Tezuka and M. Vacha, Angew. Chem., Int. Ed., 2010, 49, 1418–1421 CAS.
- Y. Tezuka, T. Ohtsuka, K. Adachi, R. Komiya, N. Ohno and N. Okui, Macromol. Rapid Commun., 2008, 29, 1237–1241 Search PubMed.
- (a) K. Schäler, E. Ostas, K. Schröter, T. Thurn-Albrecht, W. H. Binder and K. Saalwächter, Macromolecules, 2011, 44, 2743–2754 Search PubMed; (b) E. J. Shin, W. Jeong, H. A. Brown, B. J. Koo, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2011, 44, 2773–2779 Search PubMed; (c) M. E. Córdova, A. T. Lorenzo, A. J. Müller, J. N. Hoskins and S. M. Grayson, Macromolecules, 2011, 44, 1742–1746 Search PubMed.
- G. E. Yu, Z. Yang, D. Attwood, C. Price and C. Booth, Macromolecules, 1996, 29, 8479–8486 CrossRef CAS.
- G. E. Yu, Z. K. Zhou, D. Attwood, C. Price, C. Booth, P. C. Griffiths and P. Stilbs, J. Chem. Soc., Faraday Trans., 1996, 92, 5021–5028 RSC.
- G. E. Yu, C. A. Garrett, S. M. Mai, H. Altinok, D. Attwood, C. Price and C. Booth, Langmuir, 1998, 14, 2278–2285 CrossRef CAS.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 Search PubMed.
- (a) M. De Rosa and A. Gambacorta, Prog. Lipid Res., 1988, 27, 153–175 CrossRef CAS; (b) M. Kates, in The Biochemistry of Archaea (Archaebacteria), ed.M. Kates, et. al, Elsevier, Amsterdam, 1993, pp. 261–293 Search PubMed; (c) A. Yamagishi, Biol. Sci. Space, 2000, 14, 332–340 Search PubMed; (d) K. Arakawa, T. Eguchi and K. Kakinuma, Bull. Chem. Soc. Jpn., 2001, 74, 347–356 Search PubMed.
- For some examples of recent studies, see (a) H. Omachi, S. Matsuura, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 10202–10205 Search PubMed; (b) S.-S. Jester, N. Shabelina, S. M. Le Blanc and S. Höger, Angew. Chem., Int. Ed., 2010, 49, 6101–6105 Search PubMed; (c) M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef CAS; (d) F. Zhang, G. Götz, H. D. F. Winkler, C. A. Schalley and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 6632–6635 CrossRef CAS.
- X. Xu, N. Zhou, J. Zhu, Y. Tu, Z. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2010, 31, 1791–1797 Search PubMed.
- R. Chen, J. Ling and T. E. Hogen-Esch, Macromolecules, 2009, 42, 6015–6022 Search PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS.
- (a) Handbook of Conjugated Polymers, ed. T. A. Skotheim and J. Reynolds, CRC Press, Boca Raton, 3rd edn, 2007 Search PubMed; (b) S. S. Zade, N. Zamoshchik and M. Bendikov, Acc. Chem. Res., 2011, 44, 14–24 CrossRef CAS.
- R. Herges, Chem. Rev., 2006, 106, 4820–4842 CrossRef CAS.
- S. S. Puttamadappa, K. Jagadish, A. Shekhtman and J. A. Camarero, Angew. Chem., Int. Ed., 2010, 49, 7030–7034 Search PubMed.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538–6541 Search PubMed.
- (a) T. Tanaka, Phys. Rev. Lett., 1978, 40, 820–823 CrossRef CAS; (b) T. Okano, Adv. Polym. Sci., 1993, 110, 179–197 CAS; (c) M. Nakayama and T. Okano, J. Drug Delivery Sci. Technol., 2006, 16, 35–44 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |