Fluorene-containing cardo polymers as ion conductive membranes for fuel cells
Kenji
Miyatake
*ab,
Byungchan
Bae
b and
Masahiro
Watanabe
*b
aClean Energy Research Center, University of Yamanashi, 4 Takeda, Kofu, Yamanashi 400-8510, Japan. E-mail: miyatake@yamanashi.ac.jp; Fax: +81 552208707; Tel: +81 552208707
bFuel Cells Nanomaterials Center, University of Yamanashi, 4 Takeda, Kofu, Yamanashi 400-8510, Japan. E-mail: m-watanabe@yamanashi.ac.jp; Fax: +81 552547091; Tel: +81 552547091
First published on 13th May 2011
Abstract
For polymer electrolyte fuel cells, proton conducting electrolyte membranes are key materials. This review highlights aromatic polymers containing cardo groups and ionic functions as emerging electrolyte materials. First, the role of biphenyl fluorene groups on sulfonated polyimides is discussed. Discussion then focuses on how to balance proton conductivity and membrane stability with sulfonated poly(arylene ether)s making the most of the bulky cardo groups. Block copolymer structure containing fully sulfonated biphenyl fluorene groups in its hydrophilic component and compact hydrophobic component is proposed. Effect of superacid groups onto the properties of fluorene-containing polymers is discussed. Replacing sulfonic acid groups with ammonio groups afforded the cardo polymers highly anion conducting properties, which make them potentially applicable to alkaline fuel cells.
 Kenji Miyatake | Kenji Miyatake received his PhD degree in polymer chemistry from Waseda University under the supervision of the late Prof. Eishun Tsuchida in 1996. He joined Prof. Allan S. Hay's group in 1999 as a postdoctoral fellow, in the department of chemistry at McGill University. In 2001, he was offered an associate professor position in Clean Energy Research Center at the University of Yamanashi, where he currently serves as a professor. His research interest involves design, synthesis, and characterization of new functional polymers especially for polymer electrolyte fuel cell applications. He is a member of Royal Society of Chemistry, American Chemical Society, and Chemical Society of Japan. |
 Byungchan Bae | Byungchan Bae received his PhD degree in chemical engineering from Sungkyunkwan University under the supervision of Prof. Dukjoon Kim in 2005. In 2006, he joined Profs. Miyatake and Watanabe's group at Clean Energy Research Center of the University of Yamanashi as a postdoctoral fellow. He is currently an assistant professor of Polymer Research Division of Fuel Cell Nanomaterials Center at the University of Yamanashi. He is interested in design, synthesis, and characterization of polymer electrolyte membranes as well as proton and water transport in PEMFC system. |
 Masahiro Watanabe | Masahiro Watanabe received his PhD degree in physical chemistry from the University of Tokyo under the supervision of the late Prof. Kazuo Fueki in 1976. He was a postdoctoral fellow at the University of Florida with Prof. Herbert A. Laitinen. He has been a professor since 1989 and served as the director of Clean Energy Research Center at the University of Yamanashi. He is currently the director of Fuel Cell Nanomaterials Center at the University of Yamanashi. He serves as a member of the editorial board of Fuel Cells and a Fellow of the International Society of Electrochemistry. |
1. Introduction
Ion conductive polymers are of interest for a wide variety of applications, such as batteries, sensors, and actuators.1 Among them, proton conductive polymers have attracted considerable attention in the last decade for fuel cells.2Fuel cells are energy conversion devices that produce electricity via chemical reaction of fuels (e.g., hydrogen) and oxidants (e.g., oxygen). Polymer electrolyte fuel cells (PEFCs) or proton exchange membrane fuel cells (PEMFCs) employ proton conductive polymer membranes as electrolyte and are operated at moderate temperatures (typically at <80 °C).3 They are potentially, highly efficient, light-weighted, and low-cost, and are therefore expected as a possible power source for electric vehicles of the next generation. Some of the big car companies have announced that the fuel cell driven vehicles will come onto the market in or maybe before the year of 2015.The state-of-the-art proton conductive polymers for PEFCs are perfluorinated ionomers (or perfluorosulfonic acid ionomers; PFSAs), which are copolymers composed of poly(tetrafluoroethylene) and poly(trifluoroethylene) with pendant sulfonic acid groups on perfluoroalkylether side chains.4 The perfluorinated ionomers are often recognized as Nafion, which is a commercial product by du pont. Due to their highly proton conductive properties and excellent chemical and physical stabilities, it is no doubt that the perfluorinated ionomers retain their status as the most used electrolyte for PEFCs in their initial stage of commercialization. In a long range, there is a strong demand for alternative membranes based on unfluorinated materials that are less expensive and environmentally more compatible.
Requirements for PEFC membranes include:
(1) high proton conductivity (>10 mS cm−1) and its little dependency on temperature and humidity;
(2) chemical, thermal, and mechanical stability for 5000 hours under fuel cell operation (for applications in electric vehicles);
(3) gas (hydrogen and oxygen) impermeability;
(4) high water transport capability (from the cathode to the anode) and
(5) film forming capability, easy processability, and compatibility with gas diffusion electrodes.
There have been a number of approaches to develop alternative membranes. One of the promising candidates is hydrocarbon polymers with acidic functions. Aromatic polymers have been extensively studied due to their high stability, high susceptibility toward acid functionalization reaction such as sulfonation and phosphonation, and availability in structural modification.5–8 For example, poly(ether ether ketone)s, poly(arylene ether sulfone)s, polyimides, polybenzimidazoles, polyphenylenes, and others have been sulfonated or doped with mineral acids. Some of them are claimed to show high proton conductivity, very low gas permeability, and reasonable stability. However, none of them can compete with PFSAs. The most critical issues of the aromatic ionomer membranes are insufficient durability and significant dependence of the proton conductivity upon humidity. Challenge is to achieve these two conflicting properties with a single ionomer membrane.
Introducing cardo groups, i.e., pendant rings in which a carbon of the ring is also a member of the polymer main chain, provides aromatic polymers some characteristic features such as improved solubility in organic solvents, enhanced thermal stability (high glass transition temperature) and mechanical properties, high transparency and high refraction index, and low dielectric constant.9–11 Since these properties are attractive for fuel cell membranes as well, we have focused on proton conductive aromatic polymers containing cardo groups in the last decade. Especially, bulky biphenyl fluorene groups (see Fig. 1) attracted us because, in addition to the above,
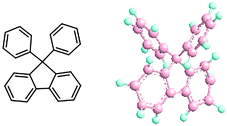 | ||
| Fig. 1 Chemical structure and ball-stick model of 9,9-diphenyl-fluorene. | ||
(1) a number of hydroxy and amino substituted diphenyl fluorene monomer compounds are readily available (commercial products or can be readily synthesized as summarized in Table 1);

(2) these functional monomers are highly reactive in polycondensation reactions to provide high molecular weight fluorene-containing cardo polymers;
(3) ionic groups (sulfonic acid groups in most cases) can be substituted on the fluorene groups by simple electrophilic substitution reaction and
(4) the introduction of ionic groups is controllable (in terms of the degree and position of ionization).
In the present review, we describe synthesis, characterization, properties, and fuel cell performance of our fluorene-containing cardo polymers as ion conductive membranes. Polyimides and poly(arylene ether)s are the main chain structures for the purpose. The review begins with the effect of fluorene cardo groups on the hydrophilic and proton conductive properties of sulfonated polyimides. Then, molecular tuning of sulfonated poly(arylene ether)s is presented for high temperature and low humidity operable ionomer membranes. The approaches contain substitution of methyl groups onto the main chain or superacid groups in the side chain, and sequenced multiblock copolymer architecture. Possibility of fluorene-containing cardo polymers as anion conductive membranes for alkaline fuel cells is also discussed briefly in the last part.
2. Sulfonated polyimides
2.1 Synthesis and properties
Sulfonated polyimides (SPIs) are one of the most studied aromatic ionomers in terms of synthesis, properties, chemical and physical structures, and fuel cell performance. Since pre-sulfonated diamine monomers, such as 2,2′-benzidine disulfonic acid (BDSA), are readily available and reactive in typical polycondensation reaction, a number of SPIs have been reported in the literature. In the early study on SPIs, major interests of their applications were electrodialysis and gas separation.12–21 In the late 1990s–early 2000s, they found new applications in fuel cells as possibly alternative proton conductive membranes to PFSAs.22 While the possibility of some SPIs was initially investigated by French groups,23,24 it was probably Prof. Okamoto's group who first reported fluorene-containing SPIs for the purpose.25–27Amino-containing fluorene monomer, 4,4′-(9-fluorenylidene)dianiline (entry 1 in Table 1), could be copolymerized with BDSA and naphthalene tetracarboxylic dianhydride (NTDA) to provide SPI copolymers. Prof. Litt et al. also synthesized the similar SPIs nearly at the same time.28 They have proved that the fluorene-containing SPIs showed some attractive properties (water absorbing capability and proton conductivity) as fuel cell membranes. However, the composition of fluorene groups relative to BDSA was rather low (ca. <10 mol%) so that the SPI copolymer membranes were deliquescent when exposed to high activity of water at high temperature.We have investigated in more detail the effect of fluorene groups onto the properties of the SPI copolymer membranes.29–36 We assumed that introducing more fluorene groups could provide more hydrophobicity and more free volume in the membranes, resulting in the improved stability at little expense of proton conducting properties. According to Scheme 1, a series of fluorene-containing SPI-1s were synthesized, in which composition of fluorene-containing units (x) ranged from 0 to 60 mol%. Regardless of the composition, the SPI-1 copolymers were obtained as soluble high molecular weight polymers (Mn > 60 kDa and Mw > 140 kDa relative to polystyrene standards) to give brown ductile membranes by solution casting. In Fig. 2 is plotted water absorbability of SPI-1 membranes at 85 °C and 93% RH (relative humidity) as a function of the fluorene composition. The SPI-1 (x = 0), which carried no fluorene groups, showed highest water uptake (103 wt%) due to its highest ion exchange capacity value (IEC = 3.47 meq g−1). The water uptake decreased to 51 wt% for x = 10, and 20 wt% for x = 20 with increasing the fluorene content. It was surprising to note that the water uptake showed maximum (57 wt%) at x = 30 in spite of increased hydrophobicity or lowered IEC (2.42 meq g−1). It is considered that the bulky and rigid fluorene groups force each polymer chain apart to produce large interchain separations (free volume), in which water molecules could be confined (Fig. 3). Such effect was not observed in another series of SPI-2 copolymer (Fig. 4) membranes, which contained naphthalene groups in place of fluorene groups as hydrophobic components.37 The water uptake decreased rather monotonously with increasing naphthalene content. The planar and compact naphthalene groups were unlikely to build up rigid spaces for confining water molecules.
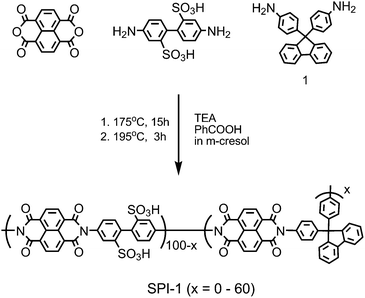 | ||
| Scheme 1 Synthesis of SPI-1 copolymers. | ||
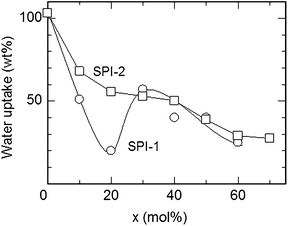 | ||
| Fig. 2 Water uptake of SPI-1, 2 membranes at 85 °C and 93% RH. | ||
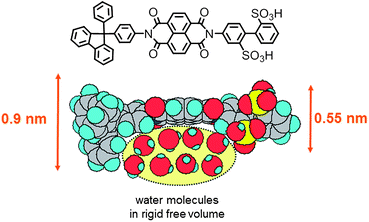 | ||
| Fig. 3 Concept of water confinement in rigid free volume produced by fluorene-containing sulfonated polyimides. | ||
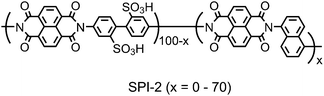 | ||
| Fig. 4 Structure of SPI-2 copolymers. | ||
The SPI-1 membranes showed high proton conductivity. The conductivity of hydrated samples (measured under 100% RH conditions) was higher than 0.1 S cm−1 when x was higher than 40 mol%, and increased with the temperature (Fig. 5). At 120 °C, SPI-1 (x = 30) showed the highest proton conductivity of 1.67 S cm−1, which was ca. 9 times higher than that of PFSA (Nafion 112) membrane (0.19 S cm−1) under the same conditions. This value of proton conductivity is extremely high for a solid polymer membrane, and nearly comparable to that of an aqueous solution of mineral acids. The conductivities of SPI-1 membranes (x = 30–60) did not decrease even above boiling temperature of water, indicating water holding capability. This behavior supported our claim that the water molecules were located in a rigid space produced by the fluorene groups. It is assumed that such water would not evaporate easily from the membranes so that the conductivity kept increasing even above 100 °C (note that the proton conductivity of Nafion membrane leveled off at >100 °C). This was not the case for SPI-1 membrane with x = 10 and 20, of which the content of fluorene units was probably insufficient to provide such space and water confinement effect. The conductivity showed an approximate Arrhenius-type temperature dependence. The activation energy estimated from the slope in Fig. 5 was ca. 21 kJ mol−1 for the SPI-1s and seemed independent on the content of fluorene units. This value was comparable to that of PFSA membranes, indicating that, despite of considerable differences in the main chain and side chain structures, SPI-1 and PFSA share the similar proton conduction mechanism involving hydronium ions.
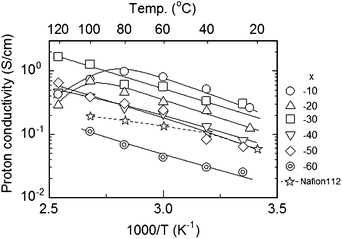 | ||
| Fig. 5 Temperature dependence of the proton conductivity of SPI-1 (x = 10–60) and Nafion 112 membranes under fully hydrated (100% RH) conditions. | ||
The SPI-1 membrane (x = 30) showed considerably high proton conductivity even under low hydrated conditions. In Fig. 6 is shown humidity dependence of the proton conductivity of SPI-1 (x = 30) membrane at 80, 100, and 120 °C. While the conductivities did depend on the humidity, the dependency was similar to that of Nafion and much less compared to the other nonfluorinated aromatic ionomer membranes (see below). It is noticeable that the conductivity seemed practically independent of the temperature at low humidity (<30% RH), since the carrier (hydronium ions) concentration would be more crucial than its mobility.
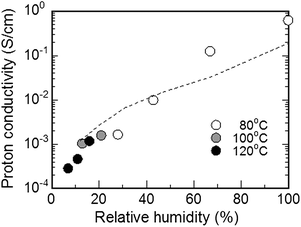 | ||
| Fig. 6 Humidity dependence of the proton conductivity of SPI-1 (x = 30) membrane at 80, 100, and 120 °C. | ||
2.2 Stabilization
One of the disadvantages of using polyimides as the main chain of hydrocarbon ionomers is their susceptibility to hydrolytic degradationvia a depolymerization mechanism under fuel cell operating (high temperature and humidified) conditions.38–40 It has been reported in the literature that electronic modification by introducing electron-donating substituents could effectively improve hydrolytic stability of SPIs.41,42 We have investigated the effect of chemical branching on the properties of SPI-1 membranes.30,43 By replacing a small amount (3 mol%) of fluorene-containing monomer (1) with triflunctional cross-linkable monomer (melamine), the obtained branched SPI-1B (Fig. 7) membrane showed much improved stability in hot Fenton's reagent (aqueous hydrogen peroxide solution containing iron sulfate). The branched membrane, however, showed lower water uptake and proton conductivity especially at high temperature than those of the original SPI-1 membrane, probably because such structural irregularity impeded preferable water holding capability in the free volume.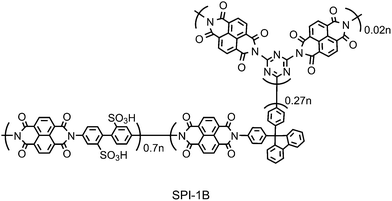 | ||
| Fig. 7 Chemical structure of chemically cross-linked SPI-1B. | ||
SPI-1 membrane was cross-linkable by electron beam irradiation. The obtained radiochemically cross-linked membrane showed similar water uptake properties to the chemically branched one. When irradiated only on the surface of the membrane, loss in the proton conductivity was minor with the highest conductivity of 1.16 S cm−1 at 120 °C. The thoroughly irradiated membrane, however, was less conductive with a rather lower conductivity of 0.36 S cm−1. As an another effective cross-linking method, phosphorus pentoxide-catalyzed condensation reaction was reported, in which sulfone cross-linking occurred via dehydration condensation of sulfonic acid groups and aromatic rings. It was claimed that the covalently cross-linked SPI membranes displayed good balance of the conductivity and oxidative stability. While these cross-linked SPIs showed improved oxidative stability, hydrolysis still remains an issue.
3. Sulfonated poly(arylene ether)s
3.1 Synthesis and properties
The above results on SPIs stimulated us to produce different cardo-polymers with sulfonic acid groups to confirm whether the fluorene groups could function similarly in the other series of ionic polymers. We focused our interest on poly(arylene ether)s as the main chain structure since the ether bonds are more robust to hydrolysis than the imide bonds.44–47 The monomer, 9,9-bis(4-hydroxyphenyl)fluorene (entry 5 in Table 1), which is readily available as a commercial product, was polymerized with bis(4-fluorophenyl)sulfone under nucleophilic substitution conditions to obtain corresponding poly(arylene ether) (PE-1). The precursor polymer was sulfonated with chlorosulfonic acid to give the sulfonated poly(arylene ether) (SPE-1) (Scheme 2). It was confirmed by 1H and 13C NMR analyses that the sulfonation reaction was selective and quantitative when the reaction conditions were carefully chosen. The sulfonic acid groups were substituted only at 2,7-positions of the fluorene groups and the degree of sulfonation was controllable up to x = 2.0 per repeating unit, which corresponded to IEC = 2.76 meq g−1.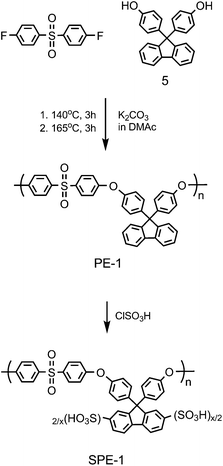 | ||
| Scheme 2 Synthesis of SPE-1. | ||
Proton conductivity of SPE-1 membranes was reasonably high under fully hydrated (or 100% RH) conditions. For example, SPE-1 with IEC = 1.48 meq g−1 showed 0.1–0.2 S cm−1 of the conductivity at a wide range of temperature (from r.t. to 150 °C). Proton conductivity of SPE-1 membranes, however, was dependent on the humidity (Fig. 8). It dropped by several orders of magnitude by decreasing the humidity from 100 to 40% RH. The major reason for this is that aromatic ionomers are less likely to have well-developed and interconnected hydrophilic domain as ionic channels. The ionic connectivity becomes even worse under less hydrated conditions resulting in the loss of proton conductivity. Humidity dependence of the proton conductivity was more significant for SPE-1 membranes than the SPI-1 membranes. Because of the possibly smaller molecular interaction between polymer main chains of poly(arylene ether)s containing flexible ether bonds compared to the rigid polyimides, the water holding capability provided by the fluorene groups would be less effective in SPE-1 than in SPI membranes.
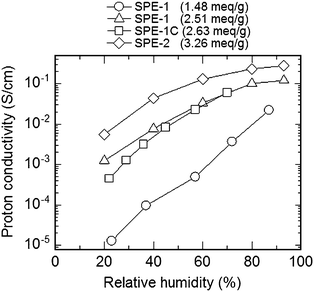 | ||
| Fig. 8 Humidity dependence of the proton conductivity of SPE-1, 1C, and 2 membranes at 80 °C. | ||
In order for SPE-1 to have high proton conductivity at low RH, much higher IEC was required. Humidity dependence of SPE-1 membrane with higher IEC = 2.51 meq g−1 was mitigated compared to the above lower IEC equivalent membrane. Its proton conductivity was ca. 10−3 S cm−1 at 20% RH and 80 °C and higher two orders of magnitude than that of the lower IEC membrane. High IEC membranes, however, suffer from over-hydrophilicity. Water uptake of SPE-1 (2.51 meq g−1) at 80 °C was 35 wt% (90% RH) and 10 wt% (20% RH), respectively, which was approximately two times higher than that of SPE-1 (1.48 meq g−1) membrane under the same conditions. Such high water absorbability often causes mechanical failure under fuel cell operating conditions, where typical 10 kgf cm−2 of pressure is applied to membranes to ensure gas sealing. Similar to the above mentioned SPI-1 membranes, chemical branching and cross-linking were used for high IEC SPE membranes to alleviate the hydrophilic properties.48 A typical cross-linking agent was bis(2,4-dihydroxy)diphenyl sulfide as a tetrafunctional comonomer, 2 mol% of which was incorporated into the highly sulfonated polymer (Fig. 9). The cross-linked SPE-1C with IEC = 2.63 meq g−1 showed lower water absorbability, better oxidative stability and mechanical strength, at minor expense of proton conducting properties.
 | ||
| Fig. 9 Chemical structure of SPE-1C. | ||
Another effective approach included introducing methyl groups onto the polymer structure, expecting increased rigidity of the main chains.49–51 Among a number of SPEs with different numbers and/or positions of methyl groups, SPE-2 (Fig. 10) turned to be an appropriate chemical structure for high IEC membrane. The methylation on the hydrophobic component, e.g., isopropylidene biphenylene (bisphenol-A) unit, provided high IEC SPE-2 membranes with high mechanical strength and good dimensional stability. Methyl group substitution on fluorene units was rather less effective, due to the co-substitution with sulfonic acid groups. SPE-2 membrane with the highest IEC = 3.26 meq g−1 was obtained, in which x and y + z (degree of sulfonation) were 1.46 and 1.80, respectively. The membrane showed comparable proton conductivity to that of Nafion under a wide range of conditions (80–120 °C and 20–93% RH). Its proton conductivities at 80 °C reached as high as 6 × 10−3 S cm−1 at 20% RH and 0.3 S cm−1 at 93% RH. Durability of the conductivities was confirmed under severe conditions (120 °C and 40% RH) for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h without a significant decline. Scanning transmission electron microscopic (STEM) images clearly showed that ionic clusters were developed and inter-connected more than that of the lower IEC SPE membranes. The high IEC SPE-2 membrane proved its availability in operating fuel cells with low ohmic resistance at a high temperature of 90 °C.
000 h without a significant decline. Scanning transmission electron microscopic (STEM) images clearly showed that ionic clusters were developed and inter-connected more than that of the lower IEC SPE membranes. The high IEC SPE-2 membrane proved its availability in operating fuel cells with low ohmic resistance at a high temperature of 90 °C.
 | ||
| Fig. 10 Chemical structure of SPE-2. | ||
3.2 Block copolymers
For improving further properties of SPE membranes, molecular structures needed more careful investigation. Especially, molecular design should be devoted to produce well-developed hydrophilic/hydrophobic phase separation.52 Such morphology seemed one of the most important parameters in determining the membrane properties. Hydrophilic domains play a role as ionic pathway, while hydrophobic domains are responsible for mechanical strength and gas permeation. Compared to typical randomly sulfonated ionomers (Fig. 11a), block copolymers tend to construct phase-separated morphology in hydrocarbon ionomer membranes (Fig. 11b) since the sequenced blocks induce self-aggregation of ionic and nonionic components. The strategy was confirmed in many ionomers of different chemical structures.53–58Block copolymer membranes show considerably higher proton conductivity than the random copolymer membranes with similar IEC value. Another approach recently proposed was introducing highly sulfonated groups into polymer structure. Locally dense sulfonic acid groups tethered at the end of polymer chains or contained in the main chain functioned similarly to the sequenced block copolymers.59–67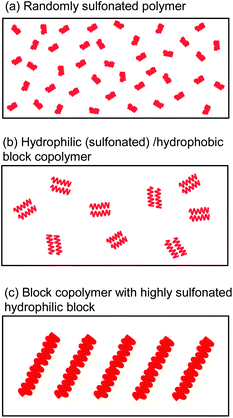 | ||
| Fig. 11 Schematic concept of hydrophilic/hydrophobic phase separation in (a) randomly sulfonated polymer, (b) sulfonated block copolymer, and (c) block copolymer with highly sulfonated hydrophilic block. Red parts represent hydrophilic domains composed of sulfonic acid cluster. | ||
It was our idea to combine both strategies into a single polymer architecture (multiblock copolymers containing sulfonic acid clusters in their hydrophilic blocks) for more pronounced and ordered phase separated morphology (Fig. 11c). We have designed and synthesized two series of multiblock poly(arylene ether sulfone)s (Fig. 12).68–72 Both block copolymers contained highly sulfonated fluorene biphenylene groups in the hydrophilic block. Differences between the two lie in the hydrophobic block, in which tetramethyl bisphenol-A units were contained in B-SPE-1 (similar to SPE-2) and benzophenone units were contained in B-SPE-2. Due to the insufficient stability of tetramethyl bisphenol-A groups in the sulfonation reaction conditions, the degree of sulfonation was 64–85% in B-SPE-1. The B-SPE-2 was more robust, and well-controlled post-sulfonation reaction enabled preferential sulfonation on each aromatic ring of the fluorene biphenylene groups in 100% degree of sulfonation. More recently, we have proposed a new synthetic route for the sulfonated block poly(arylene ether)s involving oligomeric sulfonation. In this method, we have more choices in the hydrophobic components.
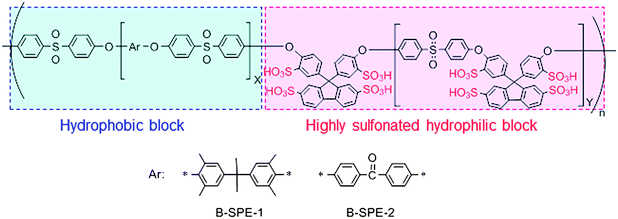 | ||
| Fig. 12 Chemical structure of multiblock poly(arylene ether sulfone)s, B-SPE-1, and 2, with highly sulfonated diphenylene fluorene groups. | ||
As expected, sulfonated block copolymer membranes showed morphologies with well-developed hydrophilic/hydrophobic phase separation. Compared to the randomly sulfonated SPE-1 membranes, B-SPE membranes showed distinct and unique nanophase separation. In the STEM images of lead-ion stained samples, ionic clusters (represented as black domains) were small and separated for SPE-1 membrane. In contrary, interconnected ionic clusters were confirmed for B-SPE-2 membrane despite of its lower IEC value (Fig. 13). The degree of sulfonation also had an impact on the morphology. B-SPE-2 membrane showed more enhanced phase separation than that of B-SPE-1 due to higher local concentration of sulfonic acid groups in the former's hydrophilic blocks. Longer block length and/or higher IEC resulted in larger and better-connected ionic clusters under dry conditions, while the morphology seemed less dependent on these factors under fully hydrated conditions.
 | ||
| Fig. 13 Scanning transmission electron microscopic (STEM) images of (a) SPE-1 (IEC = 2.0 meq g−1) and (b) B-SPE-2 (IEC = 1.62 meq g−1) membranes stained with lead ions. | ||
The water absorbing capability was in the order of Nafion NRE 212 < B-SPE-2 < B-SPE-1 membranes, according to the IEC values (Fig. 14a). Effect of block length on the water uptake was minor. B-SPE-2 membrane with IEC = 1.86 meq g−1 showed much higher proton conductivity than that of the random copolymer membrane with similar chemical structure and IEC. The proton conductivities were similar or even higher compared to those of Nafion NRE212 over a wide humidity range (Fig. 14b). The B-SPE-2 membrane retained high proton conductivity at 110 °C. It should be noted that proton conductivities were comparable for B-SPE-1 (2.20 meq g−1) and B-SPE-2 (1.86 meq g−1) membranes despite the former's lower IEC. The high conductivity resulted from the high proton diffusion coefficient. Longer block length was effective in increasing proton diffusion coefficient, which coincided with the above morphological observations.
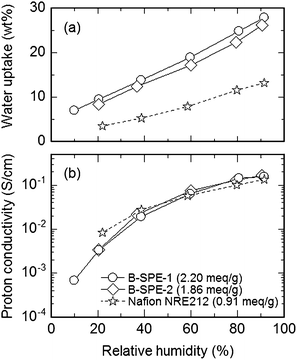 | ||
| Fig. 14 Humidity dependence of (a) water uptake and (b) proton conductivity of B-SPE-1, 2, and Nafion NRE 212 membranes at 80 °C. | ||
B-SPE-2 membranes were stable to hydrolysis in hot water at 140 °C for 24 h or at 100 °C for 1000 h. The membranes degraded to some extent under harsh oxidative conditions (in Fenton's reagent), which is still an issue for hydrocarbon ionomer membranes. Oxidative degradation is likely to occur at 9-positional fluorene carbon atoms and phenylene carbon atoms ortho to the ether bonds by the attack of highly oxidative hydroxyl radicals.
A fuel cell was successfully operated with B-SPE-2 membrane at 100 °C and 30 and 53% RH (Fig. 15). The current density was 250 mA cm−2 at 30% RH and 410 mA cm−2 at 53% RH at a cell voltage of 0.6 V. The high proton conductivity of the membrane at high temperature and low RH was confirmed as low ohmic resistance in practical fuel cell operation.
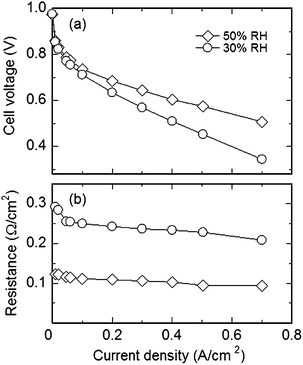 | ||
| Fig. 15 Fuel cell performance of B-SPE-2 membrane at 100 °C, 30 and 53% RH. (a) Cell voltage and (b) ohmic resistance. | ||
3.3 Superacid-modification
One of the significant differences between PFSAs and aromatic ionomers is the acidity. The pKa value of benzenesulfonic acid (PhSO3H) is ca. −2.5 and that of trifluoromethanesulfonic acid (CF3SO3H) is ca. −13. For ionomer membranes, the pKa value was estimated to be ca. −1 (sulfonated polyether ketone) and ca. −6 (Nafion), respectively.52 The large difference in pKa values implies that the effective proton concentration should be lower in the aromatic ionomer membranes compared to that of Nafion when insufficiently hydrated. This is partly responsible for low proton conductivity of the aromatic ionomer membranes at low RH. Recently, it has been reported that poly(arylene ether sulfone)s containing perfluorosulfonic acid side chains showed several times higher proton conductivity than those of conventional sulfonated poly(arylene ether sulfone)s with similar IEC value.73–75We have investigated the effect of superacid groups onto the properties of the poly(arylene ether) ionomers containing fluorene groups.76–79 The synthesis of superacid-modified poly(arylene ether)s (F-SPE-1, 2, 3) is summarized in Scheme 3. Fluorene biphenol monomers with dibromo (10) or diiodo (11) substituents were polymerized with decafluorobiphenyl, equimolar mixture of bis(4-fluorophenyl)sulfone and difluorobenzophenone, or difluorobenzophenone to obtain the corresponding halogenated (co)polymers (Br-PEs and I-PEs). The halo substituents on these precursors were converted to superacid groupsvia Ullmann coupling reaction with copper powder to obtain the title ionomers F-SPE-1–3. As expected, the iodo groups were more reactive than the bromo groups and yielded a higher degree of superacidification. The degree of superacidification was controllable, by simply changing the reaction conditions, up to x = 1.84 per repeating unit, which corresponded to IEC = 1.52 meq g−1.
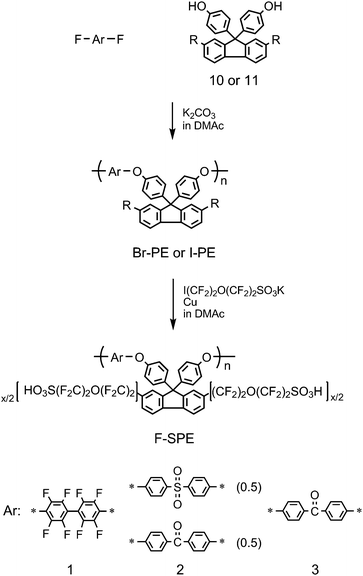 | ||
| Scheme 3 Synthesis of superacid-modified F-SPE-1, 2, and 3. | ||
F-SPE membranes showed similar thermal and gas permeation properties to those of the conventional sulfonated SPE-1 ionomers. In contrast, their morphology was more similar to that of Nafion. Well-developed hydrophilic/hydrophobic phase separation was observed, while the hydrophilic clusters were ca. 2–3 nm in diameter and smaller than those of Nafion (5–6 nm). More significant differences were observed in water uptake and proton conductivity. For example, proton conductivity of F-SPE-1 membrane (IEC = 1.40 meq g−1) was ca. 2 mS cm−1 at 80 °C and 20% RH, which was considerably higher than that (0.02 mS cm−1) of SPE-1 with higher IEC (1.58 meq g−1) under the same conditions. The two membranes showed very similar water uptake behavior at wide range of humidity. The results revealed that the F-SPEs showed properties between Nafion and SPE-1; water uptake and proton conductivity similar to Nafion and thermal and mechanical stabilities similar to SPE-1. It was also confirmed that the sequenced block structure further improved the proton conducting properties of F-SPE membranes. The effect of main chain structure has not yet been well-understood and needs further investigation.
4. Poly(arylene ether)s containing ammonium groups
4.1 Synthesis and properties
The above mentioned research on aromatic PEMs containing cardo-fluorene structure prompted us to investigate the effect of the fluorene groups on the other ion conducting ionomers. We focused on hydroxide-ion conducting ionomers since the conduction mechanism of hydroxide ions, based on molecular diffusion (vehicle mechanism) and/or structure reorientation (Grotthuss mechanism), is similar to that of protons while the electric charge of carrier ions is opposite. Hydroxide ion exchange membranes are useful for alkaline fuel cells (AFCs), which have potential advantages over PEMFCs in terms of cost and performance.It was advantageous for us to use the same precursor polymer (PE) and the similar electrophilic substitution reaction to produce ammonio-groups substituted (quaternized) poly(arylene ether)s (QPEs).80,81 Two series of QPEs, containing perfluorobiphenylene (QPE-1) or diphenyl sulfone and benzophenone (QPE-2) in the main chain, were obtained as high-molecular-weight polymersvia Friedel-Crafts chloromethylation of poly(arylene ether)s followed by quaternization (Scheme 4). By carefully optimizing the reaction conditions, the chloromethylation reaction (which often accompanies undesirable cross-linking) was controllable. For QPE-2, the degree of chloromethylation per fluorene unit (x) was up to 1.80, which corresponded to high IEC of 2.54 meq g−1 since the quaternization with tertiary amine and the subsequent ion exchange reaction were quantitative.
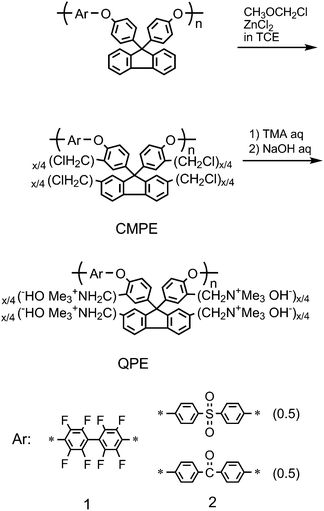 | ||
| Scheme 4 Synthesis of ammonio-groups substituted (quaternized) poly(arylene ether)s, QPE-1 and 2. | ||
The thermal and mechanical properties of the QPE membranes were reasonable. The decomposition temperature was ca. 180 °C under dry nitrogen and maximum stress at break and Young's modulus at 80 °C and 60% RH were 48 MPa and 0.7 GPa, respectively, for a QPE-1 membrane with IEC = 1.23 meq g−1. The QPE membranes showed high hydroxide ion conductivity in water (Fig. 16). Hydroxide ion conductivity of QPE-2 membrane with the highest IEC of 2.54 meq g−1 was 50 mS cm−1 at 30 °C and increased to 78 mS cm−1 at 60 °C. Compared to the other quaternized aromatic ionomer membranes, of which hydroxide ion conductivity ranges from 2.3 to 35 mS cm−1 under the similar conditions, QPE membranes achieved much higher conductivity probably due to their high molecular weights and high IECs. The random sulfone-co-ketone structure in the main chain with ammonio-functionalized fluorene groups would be responsible to realize such high IEC membranes with insolubility and less swellability in water. The conductivity of the QPE membranes showed approximate Arrhenius-type temperature dependency. The apparent activation energy estimated from the slope was ca. 10 kJ mol−1 and seemed independent of the IEC. The activation energy of QPEs was similar to or somewhat lower than those of the other reported materials (10–23 kJ mol−1). QPE-2 membranes were durable in hot water for 1000 h. During the stability test some of the ammonio groups were lost, while the conductivities increased.
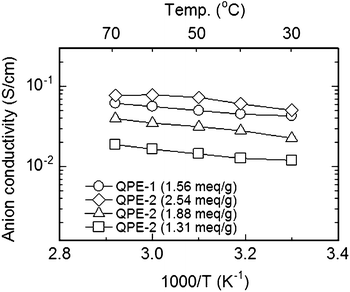
The main chain structure affected the properties of QPE membranes. QPE-1 membrane (IEC = 1.56 meq g−1) showed higher hydroxide ion conductivity than that of the QPE-2 membrane with higher IEC (1.88 meq g−1). The perfluorinated biphenylene structure may induce nanophase separation between the hydrophobic domains (composed of main chain) and the hydrophilic domains (composed of ammonio groups). While these issues need further investigation, the preliminary results on the QPE membranes seem promising for alkaline fuel cell applications.
Conclusions and perspectives
In addition to the above mentioned research works of ours, there have been a number of ion-functionalized polymers containing cardo biphenylene fluorene groups in the last decade. A simple literature search with the keywords ‘fluorene’ and ‘ionic polymers’ gives more than 100 hits in the years after 2001. While these studies are yet in the stage of basic research, they mostly focus on fuel cells as the main applications. Taking the many advantages of fluorene groups such as high reactivity toward polycondensation and ionization reactions, good solubility, high thermal and chemical stability, and film forming capability of the resulting polymers, these ionic polymers would attract more and more attentions for the applications to electrochemical sensors, batteries, and solar cells.There are some concerns for fluorene groups. The sp3carbon connecting four phenylene rings is more likely to be attacked than the aromatic rings under specific conditions, resulting in the polymer degradations. Fluorene-containing monomers are rather expensive compared to the conventional monomeric compounds used for functional aromatic polymers. These issues need careful attention. However, since the general concept of using cardo groups should be effective for ionic polymers, it can be applicable to the other aromatic components with bulky, rigid, and twisted chemical structure, in which we have a considerable number of options.
Notes and references
- M. R. Tant, K. A. Mauritz and G. L. Wilkes, Ionomers: Synthesis, Structure, Properties and Applications, 1997 Search PubMed.
- P. Colomban, Proton Conductors: Solids, Membranes and Gels—Materials and Devices, 1992 Search PubMed.
- D. P. Wilkinson, J. Zhang, R. Hui, J. Fergus and X. Li, Proton Exchange Membrane Fuel Cells: Materials Properties and Performance, 2010 Search PubMed.
- ACS Symp. Ser., No. 180: Perfluorinated Ionomer Membranes, ed. A. Eisenberg and H. L. Yeager, 1982 Search PubMed.
- M. Rikukawa and K. Sanui, Prog. Polym. Sci., 2000, 25, 1463–1502 CrossRef CAS.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4611 CrossRef CAS.
- K. Miyatake and M. Watanabe, Electrochemistry, 2005, 73, 12–19 CAS.
- J. Peckham Timothy and S. Holdcroft, Adv. Mater., 2010, 22, 4667–4690 Search PubMed.
- V. V. Korshak, S. V. Vinogradova and Y. S. Vygodski, J. Macromol. Sci., Rev. Macromol. Chem., 1974, 11, 45–142 Search PubMed.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- M. Sauviat and R. Salle, FR Pat., 2050251, 1971.
- C. Giori and V. A. Adamaitis, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1974, 15, 676–681 Search PubMed.
- Y. F. Ozcayir, G. Goetz and B. Bikson, EP Pat., 750939, 1997.
- Y. F. Ozcayir, G. Goetz and B. Bikson, EP Pat., 811421, 1997.
- Y. F. Ozcayir, G. Goetz and B. Bikson, US Pat., 5725633, 1998.
- E. Vallejo, G. Pourcelly, C. Gavach, R. Mercier and M. Pineri, J. Membr. Sci., 1999, 160, 127–137 CrossRef CAS.
- M. Pineri, G. Pourcelly and R. Mercier, WO Pat., 2000048718, 2000.
- V. Detallante, D. Langevin, C. Chappey, M. Metayer, R. Mercier and M. Pineri, J. Membr. Sci., 2001, 190, 227–241 Search PubMed.
- V. Detallante, D. Langevin, C. Chappey, M. Metayer, R. Mercier and M. Pineri, Desalination, 2002, 148, 333–339 CrossRef CAS.
- F. Piroux, E. Espuche, R. Mercier, M. Pineri and G. Gebel, J. Membr. Sci., 2002, 209, 241–253 Search PubMed.
- N. Gunduz and J. E. McGrath, Abstracts of Papers, 220th ACS National Meeting, Washington, DC, United States, August 20–24, 2000, POLY-355.
- S. Faure, M. Pineri, P. Aldebert, R. Mercier and B. Sillion, WO Pat., 9742253, 1997.
- S. Faure, N. Cornet, G. Gebel, R. Mercier, M. Pineri and B. Sillion, New Materials for Fuel Cell and Modern Battery Systems II, Proceedings of the International Symposium on New Materials for Fuel Cell and Modern Battery Systems, 2nd, Montreal, July 6–10, 1997, 1997, pp. 818–827.
- T. Watari, J. Fang, K. Tanaka, H. Kita and K.-I. Okamoto, Polym. Mater. Sci. Eng., 2001, 85, 334 Search PubMed.
- X. Guo, J. Fang, T. Watari, K. Tanaka, H. Kita and K. Okamoto, Macromolecules, 2002, 35, 6707–6713 CrossRef CAS.
- J. Fang, X. Guo, T. Watari, K. Tanaka, H. Kita and K.-i. Okamoto, Polyimides Other High Temp. Polym., 2003, 2, 137–154 Search PubMed.
- H.-J. Kim and M. Litt, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2001, 42, 486–487 CAS.
- K. Miyatake, H. Zhou, H. Uchida and M. Watanabe, Chem. Commun., 2003, 368–369 RSC.
- K. Miyatake, H. Zhou and M. Watanabe, Macromolecules, 2004, 37, 4956–4960 CrossRef CAS.
- K. Miyatake, H. Uchida and M. Watanabe, JP Pat., 2003277501, 2003.
- T. Yasuda, K. Miyatake, M. Hirai, M. Nanasawa and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4439–4445 Search PubMed.
- H. Zhou, K. Miyatake and M. Watanabe, Fuel Cells, 2005, 5, 296–301 Search PubMed.
- K. Miyatake, T. Yasuda, M. Hirai, M. Nanasawa and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2006, 45, 157–163 Search PubMed.
- T. Yasuda, Y. Li, K. Miyatake, M. Hirai, M. Nanasawa and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3995–4005 Search PubMed.
- K. Miyatake, T. Yasuda and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4469–4478 Search PubMed.
- K. Miyatake, N. Asano and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3901–3907 CrossRef CAS.
- C. Lee, S. Sundar, J. Kwon and H. Han, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3612–3620 Search PubMed.
- C. Lee, S. Sundar, J. Kwon and H. Han, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3621–3630 Search PubMed.
- W. Jang, C. Lee, S. Sundar, Y. G. Shul and H. Han, Polym. Degrad. Stab., 2005, 90, 431–440 Search PubMed.
- Y. Yin, S. Chen, X. Guo, J. Fang, K. Tanaka, H. Kita and K.-I. Okamoto, High Perform. Polym., 2006, 18, 617–635 Search PubMed.
- Y. Yin, Y. Suto, T. Sakabe, S. Chen, S. Hayashi, T. Mishima, O. Yamada, K. Tanaka, H. Kita and K.-I. Okamoto, Macromolecules, 2006, 39, 1189–1198 CrossRef CAS.
- K. Miyatake, H. Uchida and M. Watanabe, JP Pat., 2004269658, 2004.
- K. Miyatake, Y. Chikashige and M. Watanabe, Macromolecules, 2003, 36, 9691–9693 CrossRef CAS.
- Y. Chikashige, Y. Chikyu, K. Miyatake and M. Watanabe, Macromolecules, 2005, 38, 7121–7126 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, J. Membr. Sci., 2008, 310, 110–118 CrossRef CAS.
- T. Shimura, K. Miyatake and M. Watanabe, Eur. Polym. J., 2008, 44, 4054–4062 CrossRef CAS.
- Y. Chikashige, Y. Chikyu, K. Miyatake and M. Watanabe, Macromol. Chem. Phys., 2006, 207, 1334–1343 CrossRef CAS.
- K. Miyatake, Y. Chikashige, E. Higuchi and M. Watanabe, J. Am. Chem. Soc., 2007, 129, 3879–3887 CrossRef CAS.
- T. Yoda, T. Shimura, B. Bae, K. Miyatake, M. Uchida, H. Uchida and M. Watanabe, Electrochim. Acta, 2009, 54, 4328–4333 CrossRef CAS.
- T. Yoda, T. Shimura, B. Bae, K. Miyatake, M. Uchida, H. Uchida and M. Watanabe, Electrochim. Acta, 2010, 55, 3464–3470 CrossRef CAS.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- T. Nakano, S. Nagaoka and H. Kawakami, Polym. Adv. Technol., 2005, 16, 753–757 CrossRef CAS.
- N. Asano, K. Miyatake and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2744–2748 CrossRef CAS.
- X. Yu, A. Roy, S. Dunn, J. Yang and J. E. McGrath, Macromol. Symp., 2006, 245–246, 439–449 Search PubMed.
- H.-S. Lee, A. Roy, A. S. Badami and J. E. McGrath, Macromol. Res., 2007, 15, 160–166 Search PubMed.
- A. Roy, H.-S. Lee and J. E. McGrath, Polymer, 2008, 49, 5037–5044 CrossRef CAS.
- N. Li, J. Liu, Z. Cui, S. Zhang and W. Xing, Polymer, 2009, 50, 4505–4511 Search PubMed.
- S. Matsumura, A. R. Hlil and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6365–6375 CrossRef CAS.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay and A. S. Hay, Macromolecules, 2008, 41, 277–280 CrossRef CAS.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, Macromolecules, 2008, 41, 281–284 CrossRef CAS.
- S. Matsumura, A. R. Hlil, M. A. K. Al-Souz, J. Gaudet, D. Guay and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5461–5473 Search PubMed.
- S. Tian, Y. Meng and A. S. Hay, Macromolecules, 2009, 42, 1153–1160 CrossRef CAS.
- S. Tian, Y. Meng and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4762–4773 Search PubMed.
- T. Higashihara, K. Matsumoto and M. Ueda, Polymer, 2009, 50, 5341–5357 CrossRef CAS.
- K. Matsumoto, T. Higashihara and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3444–3453 Search PubMed.
- K. Matsumoto, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 1161–1166 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, ACS Appl. Mater. Interfaces, 2009, 1, 1279–1286 Search PubMed.
- B. Bae, K. Miyatake and M. Watanabe, ECS Trans., 2009, 25, 415–422 Search PubMed.
- B. Bae, T. Yoda, K. Miyatake, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 317–320 CAS , S317/311-S317/311.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2684–2691 Search PubMed.
- B. Bae, T. Yoda, K. Miyatake, M. Uchida, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2010, 114, 10481–10487 CrossRef CAS.
- K. Yoshimura, JP Pat., 2008208193, 2008.
- K. Yoshimura and K. Iwasaki, Macromolecules, 2009, 42, 9302–9306 CrossRef CAS.
- K. Nakabayashi, T. Higashihara and M. Ueda, Macromolecules, 2011, 44, 1603–1609 Search PubMed.
- K. Miyatake, T. Shimura, T. Mikami and M. Watanabe, Chem. Commun., 2009, 6403–6405 RSC.
- T. Mikami, K. Miyatake and M. Watanabe, ACS Appl. Mater. Interfaces, 2010, 2, 1714–1721 Search PubMed.
- T. Shimura, K. Miyatake and M. Watanabe, Bull. Chem. Soc. Jpn., 2010, 83, 960–968 Search PubMed.
- T. Mikami, K. Miyatake and M. Watanabe, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 452–464 Search PubMed.
- M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2657–2659 CrossRef CAS.
- M. Tanaka, M. Koike, K. Miyatake and M. Watanabe, Polym. Chem., 2011, 2, 99–106 RSC.
- Z. Y. Wang and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 1045–1052 Search PubMed.
- A. S. Hay and Z. Y. Wang, US Pat., 5110993, 1992.
- Y. Yamakawa, M. Higami and T. Kadota, JP Pat., 2006342242, 2006.
| This journal is © The Royal Society of Chemistry 2011 |