Clickable poly(ester amine) dendrimer-grafted Fe3O4 nanoparticles prepared via successive Michael addition and alkyne–azide click chemistry
Min
Li
a,
Li Qun
Xu
a,
Liang
Wang
a,
Y. P.
Wu
b,
Jun
Li
c,
Koon-Gee
Neoh
a and
En-Tang
Kang
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Kent Ridge, Singapore 119260. E-mail: cheket@nus.edu.sg; Fax: +65-67791936; Tel: +65-6516-2189
bTemasek Laboratories, National University of Singapore, Kent Ridge, Singapore 119260
cDivision of Bioengineering, National University of Singapore, Kent Ridge, Singapore 119260
First published on 14th April 2011
Abstract
Nanospheres, consisting of a Fe3O4 core, a silica inner shell and dendritic poly(ester amine) (PEA) outer shell, were synthesized via the sol–gel reaction, and successive Michael addition and alkyne–azide click chemistry. The resultant PEA dendrimer-grafted magnetic nanoparticles (Fe33O44-gg-PEA) were characterized by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared (FT-IR) spectroscopy, and thermogravimetric (TGA) analysis. Transmission electron microscopy (TEM) images reveal that the Fe33O44-gg-PEA magnetic nanoparticles (MNPs) have a uniform size distribution of about 30 nm in diameter. The hydrodynamic size of Fe33O44-gg-PEAMNPs changes reversibly with pH of the aqueous medium in the pH range of 3 to 9. The clickable alkyne groups on the surface of Fe33O44-gg-PEAMNPs endow these nanoparticles with a functionalizable surface viaalkyne–azide click chemistry. Assays with 3T3 fibroblasts and RAW macrophage cells indicate that the Fe33O44-gg-PEAMNPs possess very low cytotoxicity. Hence, the PEA dendrimer-grafted magnetic nanospheres have potential applications as biomedical materials.
1. Introduction
Advances in polymerization technology in the past decades have permitted the synthesis of macromolecules with tailored compositions, novel architectures and unique biological and physicochemical properties.1–6 Synthetic polymers can assume linear, cross-linked, branched and dendritic microstructures. Among them, dendritic polymers, also known as dendrimers, have received considerable attentions in recent years. The highly branched and well-defined nanostructure of dendrimers makes them attractive candidates for applications in biosensing and drug delivery, protein separation and purification, and gene therapy.7–9 The two most widely studied dendrimer families are the Fréchet-type poly(benzyl ether) and the Tomalia-type poly(amidoamine) (PAMAM) dendrimers.10–12 Poly(benzyl ether) dendrimers are mainly prepared by a convergent approach introduced by Fréchet and co-workers.13 The core is installed in the final step and provides great structural control. On the other hand, the PAMAM dendrimers are synthesized by the divergent method, which involves preparing the dendrimers from the core by an iterative synthetic procedure.11,14Dendrimers are well-suited for encapsulation and functionalization of metal nanoparticles because of their fairly uniform composition and structure, and the presence of abundant terminal functional groups on the dendrimer periphery. The general preparation method for dendrimer protected metal nanoparticles depends on complexation of the metal cations with amine groups in the dendrimer, followed by reduction with a chemical reductant.15,16 Development of nanoparticles with specific functional properties has attracted considerable attention in recent years due to their promising applications in optics, drug release, biomedicine, catalysis, electrochemistry and as sensing materials.17–21 A widely used method for designing and fabrication of nanoparticles with tailored surface properties is surface modification by covalently tethered functional polymers.22–26 Recently, a versatile “grafting-to” method based on alkyne–azide “click chemistry”, developed by Sharpless et al., has gained significant attention due to its fidelity, high efficiency and mild reaction conditions.27,28 Diblock and triblock copolymers, branched and star-like polymers, bioconjugates, as well as gels and networks, have been prepared viaalkyne–azide click chemistry.29–32 In recent years, the alkyne–azide click chemistry has been widely used for the synthesis of biopolymers and biomaterials due to specificity of the reaction and the absence of byproducts.33,34
In this work, poly(ester amine) (PEA) dendrimer-grafted Fe3O4 magnetic nanoparticles (MNPs) were synthesized via combined inorganic sol–gel reaction, and successive Michael addition and alkyne–azide click chemistry. The resultant core–shell hybrid MNP consists of a magnetic core, a silica inner shell and an organic outer shell of well-defined PEA dendrimer. The surface properties, cytotoxicity, and further surface functionalization viaalkyne–azide click reaction of the PEA dendrimer-grafted MNPs were investigated.
2. Experimental section
2.1 Materials
Iron(III) chloride (98%), sodium oleate (95%), oleic acid (90%), 1-octadecene (90%), copper(I) bromide (CuBr, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), Igepal® CO-520 (average Mn ≈ 441), propargyl acrylate (98%), 11-azido-3,6,9-trioxaundecan-1-amine (ATXDA, ≥90%), tetraethyl orthosilicate (TEOS, 98%) and 3-aminopropyltriethoxysilane (APS, 99%) were purchased from Sigma-Aldrich Chem. Co. and were used as received. All solvents (analytical grade) were also obtained from Sigma-Aldrich Chem. Co. and were used as received. Purified argon was used in all reactions. 1-(2-(2-Azidoethoxy)ethyl)-3-(3′,6′-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9′-xanthene]-4-yl)thiourea (azide-terminated fluorescein) was synthesized according to the method reported in the literature.35 Spectra-Por dialysis membranes were obtained from Spectrum Laboratories, Inc. (molecular weight cut-off: 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Mouse macrophages (RAW 264.7) and 3T3 fibroblasts were purchased from the American Type Culture Collection (ATCC).
000). Mouse macrophages (RAW 264.7) and 3T3 fibroblasts were purchased from the American Type Culture Collection (ATCC).
2.2 Synthesis of the amine-functionalized Fe3O4-silica core–shell nanoparticles (Fe33O44-gg-NH22, step 1)
The magnetic nanoparticles (MNPs) were prepared according to the method described in the literature.36 Initially, 10.8 g of iron chloride (40 mmol) and 36.5 g of sodium oleate (120 mmol) were dissolved in a solvent mixture comprising 80 mL ethanol, 60 mL distilled water and 140 mL hexane. The resulting mixture was heated to 70 °C and kept at that temperature for 6 h. Upon completion of the reaction, the upper organic layer containing the iron–oleate complex was washed three times with 30 mL of doubly distilled water in a separatory funnel. After washing, the iron–oleate complex in a waxy solid form was obtained by evaporating the hexane. In the next step, 36 g (40 mmol) of the iron–oleate complex synthesized as described above and 5.7 g (20 mmol) of oleic acid were dissolved in 200 g of 1-octadecene at room temperature. The reaction mixture was heated to 320 °C with a constant heating rate of 3.3 °C min−1, and then kept at that temperature for 40 min. The resulting solution containing the MNPs was then cooled to room temperature, and 500 mL of ethanol was added to the solution. The black precipitate was separated viacentrifugation, and then dissolved in 40 mL of hexane in the presence of oleic acid (1 mL) and oleylamine (1 mL). Centrifugation was applied to remove any undispersed residue. The product, oleic acid-stabilized Fe3O4 nanoparticles, was precipitated with ethanol, and collected by centrifugation. The nanoparticles were then dried under reduced pressure and stored at 0–4 °C.The amine-terminated silica shell on the magnetic (Fe3O4) core Fe33O44-gg-NH22 was prepared via inorganic sol–gel reaction. Approximately 30 mg of Fe3O4 nanoparticles, 160 mL of hexane, 8 mL of Igepal® CO-520 and 140 μL of ammonia solution (25%) were introduced into a 250 mL flask equipped with a magnetic stirrer. After stirring at 1000 rpm for 30 min, 200 μL of TEOS was added dropwise into the reaction mixture. The flask was sealed with a rubber stopper under an argon atmosphere and the reaction was allowed to proceed under continuous stirring at room temperature for 24 h. Another 200 μL of APS was then added dropwise into the flask. The flask was sealed again and the reaction was allowed to proceed under continuous stirring at room temperature for another 24 h. After the reaction, the Fe33O44-gg-NH22nanoparticles were purified by washing thoroughly with methanol.
2.3 Synthesis of the alkyne-functionalized Fe3O4 nanoparticles (first generation, Fe33O44-gg-G1, step 2)
For the synthesis of Fe33O44-gg-G1nanoparticles, 30 mg of the Fe33O44-gg-NH22nanoparticles was first dispersed in 60 mL of anhydrous methanol in a 150 mL flask. After purging with argon for 30 min, about 0.1 mL (0.9 mmol) of propargyl acrylate was added dropwise into the reaction mixture. The flask was sealed under an argon atmosphere and the reaction was allowed to proceed under continuous stirring at room temperature for 48 h. The resulting Fe33O44-gg-G1nanoparticles were centrifuged and redispersed in methanol for purification. The purification process was repeated thrice to ensure the complete removal of residual propargyl acrylate.2.4 Synthesis of the amine-terminated Fe33O44-gg-G1nanoparticles (Fe3O4-g-G1–NH2, step 3)
The Fe3O4-g-G1–NH2nanoparticles were prepared viaalkyne-azide click chemistry. 30 mg of the Fe33O44-gg–G1 were first dispersed in 20 mL of anhydrous N,N-dimethylformamide (DMF). After ultrasonication for 10 min, about 3.8 mg (26.4 μmol) of CuBr and 5.8 μL (0.26 mmol) of ATXDA were added into the solution. The reaction mixture was degassed by bubbling argon through the solution for 30 min. 6 μL of PMDETA was then added into the reaction mixture. The reaction mixture was purged with argon for an additional 10 min. The flask was sealed tightly with a rubber stopper under an argon atmosphere. The reaction was allowed to proceed under continuous stirring at 60 °C for 48 h. The reaction was terminated by exposing the reaction mixture to air. The resultant Fe3O4-g-G1–NH2nanoparticles was centrifuged and redispersed in DMF. The dispersion was subject to dialysis in doubly distilled water for 72 h. The Fe3O4-g-G1–NH2 solution was freeze-dried after dialysis.2.5 Synthesis of more generations of dendritic poly(ester amine) (PEA)-grafted Fe3O4 nanoparticles
As illustrated in Scheme 1, Fe3O4 nanoparticles grafted with more generations of PEA dendrimers (Fe33O44-gg-G2, Fe3O4-g-G2–NH2, and Fe33O44-gg-G3 in Table 1) can be obtained simply by repeating steps 2 and 3 in that sequence. | ||
| Scheme 1 Schematic illustration of the synthesis of PEA dendrimer-grafted magnetic nanoparticlesvia sol–gel reaction (step (1)) and successive Michael addition (step (2)) and alkyne–azide click chemistry (step (3)). | ||
| MNP | [N]:[C]a |
[N]:[C]b |
[C–Si]:[C–C]:[C–N]:[C–O]:[O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]c O]c |
[C–Si]:[C–C]:[C–N]:[C–O]:[O–CO]d |
[–N]:[–N–]e |
[–N]:[–N–]f |
|---|---|---|---|---|---|---|
|
a Theoretical [N]:[C] molar ratio calculated from the chemical structure of grafted dendrimer.
b [N]:[C] molar ratio calculated from the sensitivity factor-corrected XPS N 1s and C 1s core-level spectral area ratio.
c Theoretical [C–Si]:[C–C]:[C–N]:[C–O]:[O–CO] species molar ratio calculated from the chemical structure of grafted dendrimer.
d [C–Si]:[C–C]:[C–N]:[C–O]:[O–CO] species molar ratio calculated from peak component area ratio of XPS C 1 s core-level spectra.
e Theoretical [–N]:[–N–] species molar ratio calculated from the chemical structure of grafted dendrimer.
f [–N]:[–N–] species molar ratio calculated from peak component area ratio of XPS N 1s core-level spectra.
g The primary amine terminated Fe3O4–SiO2 core–shell nanoparticles synthesized by sol–gel reaction.
h The first generation of PEA dendrimer-grafted MNPs.
i The primary amine terminated Fe33O44-gg-G1.
j The second generation of PEA dendrimer-grafted MNPs.
k The primary amine terminated Fe33O44-gg-G2.
l The third generation of PEA dendrimer-grafted MNPs.
|
||||||
| Fe33O44-g-g-NH22 g | 0.33:1 |
0.31:1 |
1:1:1:-:- |
0.98:1:0.95:-:- |
-:1 |
-:1 |
| Fe33O44-gg-G1 h | 0.067:1 |
0.063:1 |
0.14:1:0.43:0.29:0.29 |
0.14:1:0.48:0.3:0.23 |
-:1 |
-:1 |
| Fe3O4-g-G1–NH2i | 0.29:1 |
0.27:1 |
0.33:1:3.67:4.67:0.67 |
0.3:1:3.76:4.89:0.75 |
0.8:1 |
0.82:1 |
| Fe33O44-gg-G2 j | 0.16:1 |
0.15:1 |
0.07:1:1:1.2:0.4 |
0.1:1:1.8:1.9:0.62 |
0.8:1 |
0.76:1 |
| Fe3O4-g-G2–NH2k | 0.29:1 |
0.26:1 |
0.14:1:4.43:6:0.86 |
0.11:1:29.6:40.5:5.6 |
0.92:1 |
0.83:1 |
| Fe33O44-gg-G3 l | 0.18:1 |
0.17:1 |
0.03:1:1.26:1.61:0.45 |
-:1:1.35:1.76:0.55 |
0.92:1 |
0.89:1 |
2.6 Grafting of azide-terminated fluorescein onto the Fe33O44-gg-G3MNPs
One of the promising characteristics of the resulting dendritic PEA-grafted MNPs is the preservation of active alkyne end groups of grafted PEA dendrimers (G1, G2 and G3). The active chain ends of the grafted dendrimers are available for reaction with azide-terminated molecules for further functionalization viaalkyne–azide click chemistry. Thus, for the grafting of azide-terminated fluorescein on the Fe33O44-gg-G3MNPs, 30 mg of the Fe33O44-gg-G3 were first dispersed in 15 mL of anhydrous N,N-dimethylformamide (DMF) in a 25 mL flask equipped with a magnetic stirrer followed by ultrasonication for 10 min. 1.6 mg (11 μmol) of CuBr and 2.3 μL (0.11 mmol) of azide-ended fluorescein were then introduced into the solution. The reaction mixture was degassed by bubbling argon through the solution for 30 min. Then, 5 μL of PMDETA was added into the reaction mixture. The reaction mixture was purged with argon for an additional 10 min and the reaction was allowed to proceed under continuous stirring at 60 °C for 48 h. After the reaction, the resultant fluorescein-grafted Fe33O44-gg-G3MNPs (Fe33O44-gg-G3–fluorescein) were centrifuged and redispersed in ethanol. The dispersion solution was subject to dialysis in doubly distilled water for 72 h.2.7 Cytotoxicity of Fe33O44-gg-G3MNPs
The cytotoxicity of Fe33O44-gg-G3MNPs was evaluated by determining the viability of mouse macrophages and 3T3 fibroblasts after incubation in media containing Fe33O44-gg-G3 at concentrations of 0.1, 0.2, 0.5 and 1.0 mg mL−1. Control experiments were carried out using the complete growth culture media without the MNPs. Cell viability testing was carried out via the reduction of the MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide) reagent. The MTT assay was performed in a 96-well plate following the standard procedure with minor modifications. These cells were seeded at a density of 1 × 104cells per well and incubated at 37 °C for 24 h before the medium was replaced with one containing the Fe33O44-gg-G3MNPs. The cells were incubated at 37 °C for another 24 h in the medium containing the Fe33O44-gg-G3MNPs. The culture medium in each well was then removed and 90 μL of media and 10 μL MTT solution (5 mg mL−1 in PBS) were then added to each well. After 4 h of incubation at 37 °C, the medium was removed and the formazan crystals were dissolved with 100 μL of DMSO for 15 min. The optical absorbance was then measured at 560 nm on a microplate reader (Tecan GENios). The results were expressed as percentages relative to that obtained in the control experiments. The difference in the results obtained from Fe33O44-gg-G3MNPs and the controls was analyzed statistically using the two-sample t-test. The difference observed between samples was considered significant for P < 0.05.2.8 Cell uptake of Fe33O44-gg-G3MNPs
Mouse macrophage cells were employed to assess the intracellular uptake of the MNPs. The macrophage cells were routinely cultured in their medium and seeded at a density of 1 × 105cells per well in a 24-well culture plate for 24 h before the medium was replaced with those containing the MNPs at 0.1, 0.2, 0.5, and 1.0 mg mL−1. In the control experiment, the cells were seeded and cultured in the same manner but without MNPs. After incubation at 37 °C for 24 h, the cells were washed three times with PBS to remove the nanoparticles in the medium and then were detached from the well walls with trypsin–EDTA (EDTA = ethylenediaminetetraacetic acid) solution. After counting with a hemocytometer, the cells were collected by centrifugation, and the cell pellets were dissolved in 37% HCl at 60 °C for 4 h. The iron concentration was determined using a Thermal Jarrell Ash Duo Iris inductively coupled plasma-mass spectrometer (ICP-MS). At each condition, four runs were made and the mean ± SD was reported.2.9 Characterization
The X-ray photoelectron spectroscopy (XPS) provides a versatile tool for the analysis of surface chemical compositions.37,38XPS measurements were performed on a Kratos AXIS HSi spectrometer equipped with a monochromatized Al Kα X-ray source (1486.6 eV photons), at a constant dwelling time of 100 ms and pass energy of 40 eV. The core-level signals were obtained at the photoelectron take-off angle (α, with respect to the sample surface) of 90°. All binding energies (BEs) were referenced to the C 1s hydrocarbon peak at 284.6 eV. In peak analysis, the line width (full width at half-maximum or FWHM) for the Gaussian peaks was maintained constant for all peak components in a particular spectrum. Surface elemental stoichiometries were determined from peak-area ratios, after correcting with the experimentally determined sensitivity factors, and were reliable to ±10%. Fourier transform infrared (FT-IR) spectra were obtained in transmission mode on a Bio-Rad FT-IR spectrophotometer (Model FTS135) under a nitrogen atmosphere. Transmission electron microscopy (TEM) images were obtained on a JEOL JEM-2100F field emission transmission electron microscope. The nanoparticle solution was spread onto the surface of a carbon coated copper grid and dried under reduced pressure. Thermogravimetric (TGA) measurements were carried out on TA instruments TGA 2090 analyzer in air in the temperature range of 50 °C to 700 °C at a heating rate of 15 °C min−1. Dynamic light scattering (DLS) was performed on a Brookhaven 90 plus laser light scattering spectrometer at the scattering angle of 90°. ζ Potentials of the hybrid magnetic nanospheres were measured using a Zetasizer Nano ZS (Malvern Instruments, Southborough, MA) with a laser wavelength of 633 nm at a scattering angle of 173°, using a capillary ζ potential cell in automatic mode. Measurement of magnetization was carried out with a vibrating sample magnetometer (Model 1600, DMS). Fluorescence spectra were measured on a Shimadzu RF-5031 fluorescence spectrophotometer.3. Results and discussion
3.1 Characterization of the dendritic poly(ester amine) (PEA)-grafted Fe3O4 nanoparticles
The synthetic procedures for the PEA dendrimer-grafted Fe3O4 magnetic nanoparticles (MNPs) are shown in Scheme 1. The oleic acid stabilized magnetic nanoparticles, as shown by the transmission electron microscopy (TEM) image in Fig. 1(a), have an average diameter of about 15 nm. After the sol–gel reaction, the average diameter of resultant spherical Fe3O4–SiO2 core–shell hybrid MNPs (Fe33O44-gg-NH22 in Table 1) increases to about 30 nm, as shown by the TEM image in Fig. 1(b). Thus, the thickness of the silica shell is about 7 nm. The TEM image of the third generation of PEA dendrimer-grafted MNPs (Fe33O44-gg-G3 in Table 1) is shown in Fig. 1(c). The hybrid core–shell Fe33O44-gg-G3MNPs disperse readily in water, mostly as segregated particles. | ||
| Fig. 1 TEM images of (a) oleic acid stabilized Fe3O4 MNPs, (b) Fe33O44-gg-NH22MNPs, and (c) Fe33O44-gg-G3MNPs. | ||
The chemical structure and composition of the PEA dendrimer-grafted MNPs at each step of preparation were analyzed by X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FT-IR) spectroscopy. The XPS characterization results are summarized in Table 1. The XPS widescan spectra of Fe33O44-gg-NH22, Fe33O44-gg-G1, Fe3O4-g-G1–NH2 and Fe33O44-gg-G3 in Table 1 are shown in Fig. 2(a), (d), (g) and (j), respectively. In comparison to the widescan spectrum of the oleic acid-stabilized MNPs in the inset of Fig. 2(a), (a′), the presence of Si 2s and Si 2p signals in Fig. 2(a), (d), (g) and (j) indicates that a silica shell which gelated from tetraethyl orthosilicate (TEOS) and 3-aminopropyltriethoxysilane (APS) was successfully coated on the MNPs. The N 1s to C 1s peak component intensity ratio varies significantly in the widescan spectra of Fig. 2(a)–(j), reflecting the changing [N]/[C] ratio with each step of dendritic PEA growth. The C 1s core-level spectrum of Fe33O44-gg-NH22 (Fig. 2(b)) can be curve-fitted into three peak components with binding energies (BEs) at about 284.1, 284.6 and 285.7 eV, attributable to the C–Si, C–C (C–H) and C–N species, respectively.39 The area ratio of C–Si, C–C (C–H) and C–N is calculated to be 0.98:1:0.95, which is close to the theoretical ratio of 1:1:1 calculated from the chemical structure of the grafted APS. In the N 1s core-level spectrum of Fe33O44-gg-NH22 (Fig. 2(c)), the only peak component at 399.4 eV can be attributed to the –NH2group introduced by APS. The [N]/[C] ratio, as determined from the sensitivity factor-corrected XPS N 1s and C 1s core-level spectral area ratio, is about 0.31:1, which is close to the theoretical value of 0.33:1 calculated from the grafted APS. The primary amino groups on the surface of MNPs play an important role in the subsequent fabrication of dendritic PEA-grafted MNPs.
 | ||
| Fig. 2 XPS widescan, C 1s core-level and N 1s core-level spectra of (a–c) Fe33O44-gg-NH22, (d–f) Fe33O44-gg-G1, (g–i) Fe3O4-g-G1–NH2 and (j–l) Fe33O44-gg-G3MNPs in Table 1. (a′) The inset shows the widescan spectrum of oleic acid stabilized Fe3O4 MNPs. | ||
Fig. 3 shows the FT-IR spectra of the oleic acid-stabilized Fe3O4, Fe33O44-gg-NH22, Fe33O44-gg-G1, Fe3O4-g-G1–NH2 and Fe33O44-gg-G3MNPs. From a comparison of the spectra of oleic acid-stabilized Fe3O4 MNPs (Fig. 3(a)) and Fe33O44-gg-NH22MNPs (Fig. 3(b)), it is apparent that after the sol–gel reaction, the characteristic absorption peak of Si–O at 1110 cm−1 has appeared, and the characteristic symmetric (2851 cm−1) and asymmetric (2929 cm−1) stretches arising from the methylene groups have become weaker. At the same time, the characteristic absorption band of methyl groups at 2958 cm−1 and the asymmetric vibration of carboxyl groups at 1711 cm−1, which only exist in oleic acid and not in TEOS or APS, have disappeared almost completely after the sol–gel reaction. These results indicate that the oleic acid on the surface of the MNPs has been successfully replaced by the primary amine-terminated silica shell.
 | ||
| Fig. 3 FT-IR spectra of (a) oleic acid stabilized Fe3O4 MNPs, and (b) Fe33O44-gg-NH22, (c) Fe33O44-gg-G1, (d) Fe3O4-g-G1–NH2 and (e) Fe33O44-gg-G3MNPs. | ||
The Fe33O44-gg-G1MNPs were synthesized via the Michael addition reaction between the surface primary amino groups of Fe33O44-gg-NH22 and the vinyl groups of propargyl acrylate. In the XPS widescan spectrum of Fe33O44-gg-G1 (Fig. 2(d)), the Fe 2p and Fe 3p signals were much weaker in comparison to those in the widescan spectrum of Fig. 2(a), indicating that the thickness of inorganic–organic hybrid grafting layer on MNPs has almost reached the XPS probing depth in an organic matrix (about 8 nm).40 The C 1s core-level spectrum of Fe33O44-gg-G1 (Fig. 2(e)) can be curve-fitted into five peak components with BEs at about 284.1, 284.6, 285.7, 286.2 and 288.7 eV, attributable to the C–Si, C–C (C–H), C–N, C–O and O–CO species, respectively.41 The area ratio of the C–Si, C–C (C–H) C–N, C–O and O–CO species is calculated to be 0.14:1:0.48:0.3:0.23, which is comparable to the theoretical ratio of 0.14:1:0.43:0.29:0.29 calculated from the chemical structure of grafted PEA (first generation, G1). There was only one peak component at about 399.4 eV in the N 1s core-level spectrum of Fe33O44-gg-G1 (Fig. 2(d)), which is associated with the tertiary amino group of grafted PEA (G1). The [N]/[C] ratio of about 0.063:1 is close to the theoretical value of 0.067:1 calculated from the chemical composition of the G1 dendrimer. Furthermore, in the FT-IR spectrum of Fe33O44-gg-G1 (Fig. 3(c)), the appearance of characteristic absorption band of alkyne group28 at 2121 cm−1 and the asymmetric vibration of ester group at 1731 cm−1 provide additional evidence on the grafting of the G1 dendrimer on the MNPs.
For the preparation of Fe3O4-g-G1–NH2MNPs, the alkyne-functionalized Fe33O44-gg-G1MNPs were reacted with the active azide-containing 11-azido-3,6,9-trioxaundecan-1-amine (ATXDA) viaalkyne–azide click reaction in N,N-dimethylformamide (DMF) using CuBr/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) complex as the catalyst system. PMDETA is a widely used ligand to accelerate the alkyne–azide click reaction in the presence of copper catalyst in organic solvent. The XPS C 1s core-level spectrum of Fe3O4-g-G1–NH2 (Fig. 2(h)) can be curve-fitted into five peak components with BEs at about 284.1, 284.6, 285.7, 286.2 and 288.7 eV, attributable to the C–Si, C–C (C–H), C–N, C–O and O–C O species, respectively, with a corresponding area ratio of 0.3:1:3.76:4.89:0.75, which is comparable with the theoretical value of 0.33:1:3.67:4.67:0.67 calculated from the chemical structure of NH2-terminated PEA (G1–NH2). The N 1s core-level spectrum of Fe3O4-g-G1–NH2 (Fig. 2(i)) can be curve-fitted with two peak components. The peak component at BE of 398.2 eV was attributed to the imine groups in the 1,4-triazole rings resulted from the click reaction. The peak component at 399.4 eV was assigned to the tertiary amino group and the primary amino group. The peak component area ratio of N– to –N– (–NH2) is 0.82:1, which is close to the theoretical value of 0.80:1 calculated from the chemical composition of grafted G1–NH2 on the MNPs. In the FT-IR spectrum of Fe3O4-g-G1–NH2 (Fig. 3(d)), the characteristic absorption band of alkyne group at 2121 cm−1 was no longer present after the alkyne–azide click reaction.
Finally, the Fe 2p and Fe 3p photoelectron lines in the widescan spectrum of Fe33O44-gg-G3 (Fig. 2(j)) have disappeared completely, indicating that the thickness of inorganic–organic hybrid graft layer on the MNPs has exceeded the XPS probing depth of about 8 nm. The C 1s core-level spectrum of Fe33O44-gg-G3 (Fig. 2(k)) can be curve-fitted into four peak components with BEs at about 284.6, 285.7, 286.2 and 288.7 eV, attributable to the C–C (C–H), C–N, C–O and O–CO species, respectively, and with a corresponding area ratio of 1:1.35:1.76:0.55. This ratio is comparable to the theoretical value of 1:1.26:1.61:0.45 calculated from the chemical structure of PEA dendrimer (third generation, G3). In the N 1s core-level spectrum of Fe33O44-gg-G3 (Fig. 2(l)), two peak components were discernible: the imine peak component at 398.2 eV and the peak component for tertiary and primary amino group at 399.4 eV. The imine to amine peak component area ratio is about 0.89:1, which is close to the theoretical ratio of 0.92:1 calculated from the composition of grafted PEA G3 on the surface of MNPs. Furthermore, in the FT-IR spectrum of Fe33O44-gg-G3 (Fig. 3(e)), the presence of characteristic absorption peak of alkyne group at 2121 cm−1 suggests that the Fe33O44-gg-G3MNPs have the active alkyne-terminated groups, which can serve as coupling sites for the subsequent alkyne–azide click reaction to prepare further generations of the dendrimers or to introduce other functionalities.
3.2 TGA measurements of the dendritic PEA-grafted MNPs
The thermogravimetric analysis (TGA) curves of Fe33O44-gg-NH22, Fe33O44-gg-G1, Fe3O4-g-G1–NH2 and Fe33O44-gg-G3MNPs are shown in Fig. 4. The weight loss curves of MNPs after each step of modification indicate the amount of organic compound on the MNPs. In Fig. 4(a), a 9% weight loss is observed for the Fe33O44-gg-NH22MNPs after heating to 700 °C in air. The weight loss can be attributed to the evaporation of adsorbed water, the decomposition of organic fraction and the oxidation of Fe3O4 to Fe2O3.42 The TGA curve of Fe33O44-gg-G1MNPs in Fig. 4(b) shows a higher weight loss of about 21% in comparison to that of the Fe33O44-gg-NH22MNPs in Fig. 4(a). For the TGA curves of Fe3O4-g-G1–NH2 (Fig. 4(c)) and (Fig. 4(d)) MNPs, the grafted dendritic PEA degraded to low molecular weight units and then decomposed with increasing temperature, as indicated by the two major weight loss stages at about 275 and 430 °C. The two major weight loss stages can be attributed to the decomposition of C–N bond and the thermal cleavage of ester groups, respectively.43,44 The weight losses for the two types of MNPs are about 30% and 48%, respectively. The TGA results are thus in agreement with the increase in organic content for the increasing generation of grafted PEA dendrimers. The grafting density of the poly(ester amine) dendrimer on Fe33O44-gg-G3MNPs is calculated to be about 2.6 chain nm−2, using the method proposed by Ohno et al.45 | ||
| Fig. 4 TGA curves of (a) Fe33O44-gg-NH22, (b) Fe33O44-gg-G1, (c) Fe3O4-g-G1–NH2 and (d) Fe33O44-gg-G3MNPs. | ||
3.3 Analysis of magnetic properties
The magnetic properties of Fe33O44-gg-NH22, Fe33O44-gg-G1 and Fe33O44-gg-G3MNPs, determined by vibrating sample magnetometry (VSM) at room temperature, are shown in Fig. 5. The saturation magnetization (Ms) of Fe33O44-gg-NH22MNPs was about 17.7 emu g−1. After grafting of the PEA dendrimer, the Ms values of Fe33O44-gg-G1 and Fe33O44-gg-G3MNPs decreased to 15.8 and 8.1 emu g−1, respectively. The decrease in Ms of the PEA dendrimer-grafted MNPs can be attributed to the substantial decrease in the relative Fe3O4 magnetic core content in the MNPs, which is consistent with the TGA analysis. The Ms of PEA dendrimer-grafted MNPs is acceptable for bioapplications, such as magnetic resonance imaging (MRI) agents, where Ms of 7–22 emu g−1 is usually needed.46 The nano-scaled size and the absence of hysteresis in their magnetic profile suggest that these MNPs are superparamagnetic.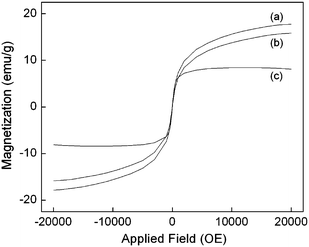 | ||
| Fig. 5 Field dependent magnetization at 25 °C of (a) Fe33O44-gg-NH22, (b) Fe33O44-gg-G1, and (c) Fe33O44-gg-G3MNPs. | ||
3.4 pH-sensitivity of the PEA dendrimer-grafted MNPs
In an aqueous dispersion, the average size of PEA dendrimer-grafted MNPs is sensitive to the pH of the environment. The pH-dependent MNP size was evaluated by dynamic light scattering (DLS) studies. Fig. 6 shows the pH dependence of hydrodynamic diameter (Dh) of Fe33O44-gg-G3. At pH 9, the Fe33O44-gg-G3MNPs have a smaller Dh of about 55 nm. The Dh of the MNPs increases to about 77 nm at pH 3. The diameter of the hydrophilic dendrimer-grafted MNPs observed by TEM (about 30 nm) in the dry state was about 2–3 times smaller than that measured by DLS for the swollen nanoparticles in aqueous media. The results are in agreement with earlier studies on different hydrophilic polymer-based nanoparticles.47,48 These results also indicate the large water absorption ability of the hydrophilic dendritic PEA-grafted MNPs. The pH dependence of Dh can be attributed to the presence of tertiary amino groups and the triazole rings in the grafted PEA dendrimer on the MNPs. Tertiary amine and triazole groups are both weak bases and are well-known pH-sensitive groups commonly found in stimuli-responsive materials.49,50 At low pH (ca. pH = 3), the tertiary amino groups and triazole rings of grafted PEA dendrimer are protonated, giving rise to the fully extended PEA chains. As the solution pH is increased to basic pH, the amino groups and triazole rings become deprotonated, leading to the association of PEA chains and a smaller diameter of the MNPs.51 | ||
| Fig. 6 Hydrodynamic diameter (Dh) distribution of Fe33O44-gg-G3MNPs in aqueous media of (a) pH = 9 and (b) pH = 3 at 25 °C. | ||
ζ Potential measurements were further employed to investigate the effect of pH on surface charges of the Fe33O44-gg-G3MNPs in aqueous media. As shown in Fig. 7, the ζ potential decreases with the increase in pH of the medium, from 25.7 mV at pH 3 to 3.1 mV at pH 9, consistent with the pH-responsive properties of the tertiary amino groups and triazole rings. The pH-dependent surface potential further confirms the pH-sensitivity of the PEA dendrimer-grafted MNPs.
 | ||
| Fig. 7 Effect of pH on ζ potential of Fe33O44-gg-G3MNPs in aqueous media. | ||
3.5 Cytotoxicity and cell uptake of the PEA dendrimer-grafted MNPs
Cytotoxicity of the PEA dendrimer-grafted MNPs was evaluated by incubation with mouse macrophage (RAW 264.7, RAW macrophage) cells and 3T3 fibroblasts. The 3T3 fibroblasts cell line is an important fibroblast culture and widely used in biomedical and biochemical applications, such as in vitro carcinogenic analysis.52,53Fig. 8 shows the dose-dependent viability of 3T3 fibroblasts and RAW macrophage cells after 24 h of incubation with Fe33O44-gg-G3MNPs of 0.1, 0.2, 0.5 and 1.0 mg mL−1 in the culture medium. At pH 7, the ζ potential of the hybrid MNPs is about +8.2 mV due to partial protonation of the tertiary amino groups of the grafted dendrimers. The weakly positively charged MNPs may be responsible for the increasing cytotoxicity of the hybrid materials when the concentration of the MNPs increases. Nevertheless, for both cell lines, the viabilities were above 90% for the MNP concentration tested and were not significantly different from those in the control experiment (P > 0.05). These results show that the PEA dendrimer-grafted MNPs have very low toxicity towards both the RAW macrophages and the 3T3 fibroblasts, and thus can be potentially suitable for biomedical applications. | ||
| Fig. 8 Effect of Fe33O44-gg-G3MNPs on the viability of 3T3 fibroblasts and RAW macrophage cells. | ||
Fig. 9 shows the uptake of Fe33O44-gg-G3MNPs by RAW macrophage cells after 24 h of incubation in medium containing 0.1, 0.2, 0.5 and 1.0 mg mL−1 of the nanoparticles. The iron uptake increases with the concentration of Fe33O44-gg-G3MNPs, reaching about 1.1, 2.1, 2.7 and 3.3 pg Fe per cell at the Fe33O44-gg-G3 MNP concentrations of 0.1, 0.2, 0.5, and 1.0 mg mL−1, respectively. For the incubation conditions and doses, the uptake of Fe33O44-gg-G3 by the macrophages remained at a relatively low level, suggesting good biocompatibility of the Fe33O44-gg-G3MNPs.
 | ||
| Fig. 9 Uptake of Fe33O44-gg-G3MNPs by RAW macrophage cells. | ||
3.6 Grafting of 1-(2-(2-azidoethoxy)ethyl)-3-(3′,6′-dihydroxy-3-oxo-3H-spiro[isobenzofuran-1,9′-xanthene]-4-yl)thiourea (azide-ended fluorescein) onto the Fe33O44-gg-G3MNPs
Another important issue for the as-synthesized PEA dendrimer-grafted MNPs is that the nanoparticles can be further functionalized for multipurpose applications.54 For each generation of the PEA dendrimer-grafted MNPs (Fe33O44-gg-G1, Fe33O44-gg-G2 and Fe33O44-gg-G3), the PEA chains still retain the terminal alkyne groups, which can be readily utilized for the subsequent alkyne–azide click reaction to further enhance the functionality of the MNPs. In the present work, an azide end-functionalized fluorescein was “clicked” onto the Fe33O44-gg-G3 to further modify the surface properties of MNPs. Fluorescein is currently the most commonly used fluorescent dye and is used in a wide-ranging applications, including fluorescence activated cell sorter (FACS) analysis and flow cytometry.55,56Fig. 10 shows the emission spectra of Fe33O44-gg-G3 and fluorescein-functionalized Fe33O44-gg-G3 (Fe33O44-gg-G3–fluorescein) MNPs in the aqueous medium excited at a wavelength of 490 nm. The Fe33O44-gg-G3–fluorescein MNPs exhibit a characteristic emission at the wavelength of about 527 nm, which can be attributed to the clicked fluorescein species. On the other hand, the Fe33O44-gg-G3 shows negligible emission in the wavelength range of 500 to 650 nm. These results suggest that the alkyne-terminated groups of the PEA dendrimer-grafted MNPs are preserved, and further tailoring of the MNP surface functionalities viaalkyne–azide click chemistry is feasible. | ||
| Fig. 10 Emission spectra of (excitation wavelength at 490 nm) of (a) fluorescein-functionalized Fe33O44-gg-G3 (Fe33O44-gg-G3–fluorescein) MNPs and (b) Fe33O44-gg-G3MNPs. | ||
4. Conclusions
Inorganic–organic hybrid magnetic nanoparticles (MNPs), comprising a Fe3O4 core, a silica inner shell and dendritic poly(ester amine) (PEA) outer shell, have been synthesized via the inorganic sol–gel reaction, and successive Michael addition reaction and alkyne–azide click chemistry. After surface grafting of the inorganic–organic hybrid shell, the so-obtained PEA dendrimer-functionalized MNPs exhibit good solubility in an aqueous medium and are superparamagnetic with a saturation magnetization (Ms) of 8.1 emu g−1. The PEA dendrimer-grafted MNPs are pH-sensitive, resulting in a pH-dependent hydrodynamic size in an aqueous medium. The PEA dendrimer-functionalized MNPs do not exhibit significant cytotoxicity towards 3T3 fibroblasts and RAW macrophage cells after 24 h of incubation. The uptake of PEA dedrimer-functionalized MNPs by macrophage cells is low, even in the presence of a relatively high concentration of the MNPs (e.g. 1.0 mg mL−1) in the culture medium, suggesting good biocompatibility of the MNPs and their potential biomaterial applications. In addition, the preservation of alkyne-terminated groups in the grafted PEA dendrimers allows further functionalization of the MNPsviaalkyne–azide click reaction for multipurpose applications. This synthesis method of successive Michael addition reaction and alkyne–azide click chemistry proceeds with quantitative yields and without the need for protection and/or deprotection of functional groups. It can be readily extended to the functionalization of other substrates with PEA dendrimers and will in turn provide an insight into the preparation of various functional dendrimers under mild conditions.Acknowledgements
This work was supported under the SMF-NUS Research Horizon Award grant no. 270-000-303-592/646.References
- H. Kakwere and S. Perrier, Polym. Chem., 2011, 2, 270–288 RSC.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889–1895 CrossRef CAS.
- I. Dierking, Polym. Chem., 2010, 1, 1153–1159 RSC.
- P. Archer and I. Dierking, Soft Matter, 2009, 5, 835–841 RSC.
- C. T. Adkins and E. Harth, Macromolecules, 2008, 41, 3472–3480 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- P. K. Lo, K. L. Metera and H. F. Sleiman, Curr. Opin. Chem. Biol., 2010, 14, 597–607 CrossRef CAS.
- J. P. Gunning, J. W. Levell, M. F. Wyatt, P. L. Burn, J. Robertson and I. D. W. Samuel, Polym. Chem., 2010, 1, 730–738 RSC.
- K. C. F. Leung and K. N. Lau, Polym. Chem., 2010, 1, 988–1000 RSC.
- J. W. Lee, H. J. Kim, S. C. Han, J. H. Kim and S. H. Jin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1083–1097 CrossRef CAS.
- J. Zheng, M. S. Stevenson, R. S. Hikida and P. G. Van Patten, J. Phys. Chem. B, 2002, 106, 1252–1255 CrossRef CAS.
- Y. Liu, V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Am. Chem. Soc., 2009, 131, 2798–2799 CrossRef CAS.
- M. Kawa and J. M. J. Fréchet, Chem. Mater., 1998, 10, 286–296 CrossRef CAS.
- G. Franc and A. Kakkar, Chem. Commun., 2008, 5267–5276 RSC.
- A. T. Gates, E. G. Nettleton, V. S. Myers and R. M. Crooks, Langmuir, 2010, 26, 12994–12999 CrossRef CAS.
- E. Murugan and R. Rangasamy, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2525–2532 CrossRef CAS.
- C. C. Corten and M. W. Urban, Polym. Chem., 2011, 2, 244–250 RSC.
- A. Misra and M. W. Urban, Macromolecules, 2009, 42, 7828–7835 CrossRef CAS.
- M. L. Vargo, C. P. Gulka, J. K. Gerig, C. M. Manieri, J. D. Dattelbaum, C. B. Marks, N. T. Lawrence, M. L. Trawick and M. C. Leopold, Langmuir, 2010, 26, 560–569 CrossRef CAS.
- C. E. Dowdy and M. C. Leopold, Thin Solid Films, 2010, 519, 790–796 CrossRef CAS.
- Y. Q. Wen, C. K. McLaughlin, P. K. Lo, H. Yang and H. F. Sleiman, Bioconjugate Chem., 2010, 21, 1413–1416 CrossRef CAS.
- B. Mu, P. Liu, Y. Dong, C. Y. Lu and X. L. Wu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3135–3144 CrossRef CAS.
- D. Serrano-Ruiz, S. Rangou, A. Avgeropoulos, N. E. Zafeiropoulos, E. Lopez-Cabarcos and J. Rubio-Retama, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1668–1675 CrossRef CAS.
- C. Chevigny, J. Jestin, D. Gigmes, R. Schweins, E. Di-Cola, F. Dalmas, D. Bertin and F. Boue, Macromolecules, 2010, 43, 4833–4837 CrossRef CAS.
- S. E. Harton, S. K. Kumar, H. C. Yang, T. Koga, K. Hicks, E. Lee, J. Mijovic, M. Liu, R. S. Vallery and D. W. Gidley, Macromolecules, 2010, 43, 3415–3421 CrossRef CAS.
- M. D. Rowe, D. H. Thamm, S. L. Kraft and S. G. Boyes, Biomacromolecules, 2009, 10, 983–993 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217–6223 CrossRef CAS.
- J. Huang, G. Habraken, F. Audouin and A. Heise, Macromolecules, 2010, 43, 6050–6057 CrossRef CAS.
- X. Y. Wei, W. Chen, X. J. Chen and T. P. Russell, Macromolecules, 2010, 43, 6234–6236 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- D. J. Siegwart, J. K. Oh, H. F. Gao, S. A. Bencherif, F. Perineau, A. K. Bohaty, J. O. Hollinger and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 2180–2193.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS.
- P. Lundberg, M. V. Walter, M. I. Montanez, D. Hult, A. Hult, A. Nystrom and M. Malkoch, Polym. Chem., 2011, 2, 394–402 RSC.
- C. M. Santos, A. Kumar, W. Zhang and C. Z. Cai, Chem. Commun., 2009, 2854–2856 RSC.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS.
- E. T. Kang, K. G. Neoh, Y. K. Ong, K. L. Tan and B. T. G. Tan, Synth. Met., 1990, 39, 69–80 CAS.
- L. Wang, K. G. Neoh, E. T. Kang and B. Shuter, Biomaterials, 2011, 32, 2166–2173 CrossRef CAS.
- F. J. Xu, E. T. Kang and K. G. Neoh, Macromolecules, 2005, 38, 1573–1580 CrossRef CAS.
- K. L. Tan, L. L. Woon, H. K. Wong, E. T. Kang and K. G. Neoh, Macromolecules, 1993, 26, 2832–2836 CrossRef CAS.
- F. J. Xu, Z. L. Yuan, E. T. Kang and K. G. Neoh, Langmuir, 2004, 20, 8200–8208 CrossRef CAS.
- L. Wang, K. G. Neoh, E. T. Kang, B. Shuter and S. C. Wang, Adv. Funct. Mater., 2009, 19, 2615–2622 CrossRef CAS.
- Z. J. Zheng, C. Y. Pan, D. Wang and Y. Liu, Macromol. Chem. Phys., 2005, 206, 2182–2189 CrossRef CAS.
- O. Ozturk, T. J. Black, K. Perrine, K. Pizzolato, C. T. Williams, F. W. Parsons, J. S. Ratliff, J. Gao, C. J. Murphy, H. Xie, H. J. Ploehn and D. A. Chen, Langmuir, 2005, 21, 3998–4006 CrossRef CAS.
- K. Ohno, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 8989–8993 CrossRef CAS.
- P. Tartaj and C. J. Serna, J. Am. Chem. Soc., 2003, 125, 15754–15755 CrossRef CAS.
- D. Donghi, D. Maggioni, G. D'Alfonso, F. Amigoni, E. Ranucci, P. Ferruti, A. Manfredi, F. Fenili, A. Bisazza and R. Cavalli, Biomacromolecules, 2009, 10, 3273–3282 CrossRef CAS.
- E. Ranucci, M. A. Suardi, R. Annunziata, P. Ferruti, F. Chiellini and C. Bartoli, Biomacromolecules, 2008, 9, 2693–2704 CrossRef CAS.
- P. Bertrand and J. P. Gesson, J. Org. Chem., 2007, 72, 3596–3599 CrossRef CAS.
- Y. Diaz-Fernandez, F. Foti, C. Mangano, P. Pallavicini, S. Patroni, A. Perez-Gramatges and S. Rodriguez-Calvo, Chem.–Eur. J., 2006, 12, 921–930 CrossRef CAS.
- G. L. Li, G. Liu, E. T. Kang, K. G. Neoh and X. L. Yang, Langmuir, 2008, 24, 9050–9055 CrossRef CAS.
- K. Glunde and N. J. Serkova, Pharmacogenomics, 2006, 7, 1109–1123 CrossRef CAS.
- A. Martin, G. Romito, I. Pepe, G. De Vivo, M. R. Merola, A. Limatola and V. Gentile, Curr. Med. Chem., 2006, 13, 1895–1902 CrossRef CAS.
- L. Zhang, H. Xue, C. L. Gao, L. Carr, J. N. Wang, B. C. Chu and S. Y. Jiang, Biomaterials, 2010, 31, 6582–6588 CrossRef CAS.
- R. Cavalli, A. Bisazza, R. Sessa, L. Primo, F. Fenili, A. Manfredi, E. Ranucci and P. Ferruti, Biomacromolecules, 2010, 11, 2667–2674 CrossRef CAS.
- T. Vermonden, S. S. Jena, D. Barriet, R. Censi, J. van der Gucht, W. E. Hennink and R. A. Siegel, Macromolecules, 2010, 43, 782–789 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |