Photodecarbonylation and photoinitiated polymerization from a monomer and polymer based on the α-keto ester methacryloyl phenylglyoxylate
Kamel
Omrane
,
Jin-Jin
Feng
,
Richard E.
Partch
* and
Devon A.
Shipp
*
Department of Chemistry and Biomolecular Science, and Center for Advanced Materials Processing, Clarkson University, Potsdam, New York, USA 13699-5810. E-mail: dshipp@clarkson.edu; partch@clarkson.edu
First published on 12th April 2011
Abstract
The polymerizable α-keto ester methacryloylethyl phenylglyoxylate (MEPG), and its homopolymer, were synthesized and tested for their photoinitiation capabilities in a crosslinking monomer resin system containing bis-phenol A-glycidyl methacrylate (BisGMA) and triethylene glycol dimethacrylate (TEGDMA). Monomer conversion kinetic data were collected using both the MEPG and poly(MEPG) as initiator and/or comonomer. Furthermore, the evolution of CO resulting from the photoinduced decarbonylation led to a significant reduction in the volume shrinkage of the resin upon photocuring with a UV light. The shrinkage of the resin was reduced by 63% when 45% of MEPG was added to the BisGMA/TEGDMA monomer mixture of the resin. However, dispersion of the CO voids was uncontrolled, resulting in large voids which are likely to be detrimental to material properties. Addition of inorganic filler (SiO2) to the resin mixture did not affect CO generation and produced even less shrinkage compared to systems without SiO2 at given MEPG concentrations.
Introduction
One of the major causes for the failure of polymer resins and resin composites is volume change, usually a contraction or shrinkage, which occurs during polymerization (curing). This shrinkage is the result of the change in the intermolecular distance between the monomer units (0.3–0.4 nm) when covalently bonded (∼0.15 nm) in polymer chains. Polymerization shrinkage has significant negative effects on a variety of properties of the material and, if used in a medical application such as dental resins, on the patients' health as a result of microcracking and microleakage. For instance, Kleverlaan and Feilzer1 showed that the polymerization shrinkage causes the failure of the bond between the resin and the tooth structure. Failure of the material occurs when the values of the polymerization stress (shrinkage) exceed the strength of the adhesive bond to the surrounding mold.2The aim of this work is to evaluate the possibility that photodecarbonylation, which occurs during photocuring when an α-keto acid or an α-keto ester functionality is present, might lead to a reduction in polymerization shrinkage. The approach is to utilize a monomer with an α-keto acid or an α-keto ester functionality that upon irradiation with the UV and/or visible light undergoes photodecarboxylation (carbon dioxide release) or photodecarbonylation (carbon monoxide release). Incorporation of this into a resin formulation is expected to generate gas bubbles in the resin and as a result form a porous material, thus reducing or perhaps eliminating the shrinkage caused by the conversion of the carbon–carbon double bonds in monomers into covalent bonds of the polymer. We also examine the possibility of using a polymer derived from the α-keto ester monomer as an additive to the monomer mixture, in addition to determining if a common filler (SiO2) has any effect on photodecarbonylation. While the resin used as a model system is based on a formulation often used in dental composites, we expect this approach to be more general and have potential use in many of the polymerizing systems; thus this paper aims to provide a proof of concept—more details regarding specific applications, including physical and mechanical evaluations, will follow in subsequent publications.
Experimental section
Materials
Anhydrous ether and pyridine were both purchased from J.T.Baker Corp. Ether was used as received, and pyridine was distilled and stored over molecular sieves 4 Å. Bis-phenol A-glycidyl methacrylate (BisGMA), triethylene glycol dimethacrylate (TEGDMA), 2-hydroxyethyl methacrylate, azobisisobutyronitrile (AIBN) and thionyl chloride were purchased from Aldrich and used as received. The reversible addition–fragmentation chain transfer (RAFT) agent, cyanoisopropyl dithiobenzoate (CPDTB), was synthesized according to the literature3 by reaction of phosphorus pentasulfide (P4S10) (Aldrich) with benzoic acid, followed by reaction with AIBN.Synthesis of methacryloylethyl phenylglyoxylate (MEPG)
The phenylglyoxylyl chloride was synthesized according to the following method.4 A solution of 2.9 g of thionyl chloride (24 mmol) and 30 mL of anhydrous ether was added drop-wise to a mixture of benzoylformic acid (3 g, 20 mmol) and pyridine (1.58 g) in 25 mL of anhydrous ether at room temperature. After the addition, the reaction mixture was stirred for 1.5 hours. After reducing its volume by rotoevaporation, 20 mL of hexane was added to the ethereal solution and the volume was reduced using a rotary evaporator and dried under vacuum to obtain a residual oil. The resulting acid chloride was dissolved in 30 mL of anhydrous ether and added dropwise over a period of one hour to a 25 mL solution of pyridine and hydroxyethyl methacrylate in anhydrous ether at room temperature and let the reaction proceeds for 5 hours. The ethereal solution was filtered to remove the pyridine salt and extracted with 1 M aqueous sodium bicarbonate. The combined organic phase was dried over magnesium sulfate, rotoevaporated and dried in a vacuum oven at room temperature to obtain methacryloylethyl phenylglyoxylate (MEPG) as a yellow viscous liquid (41% yield). During the entire reaction process, precautions were taken to minimize exposure to light. 1H NMRδ 1.90 (s, 3H), 4.48 (m, 2H), 4.68 (m, 2H), 5.61 (m, 1H), 66.17 (m, 1H), 7.47–7.55 (m, 2H), 7.64–7.68 (m, 1H), 7.99–8.04 (m, 2H).RAFT polymerization of MEPG
1.57 g (0.006 mol) MEPG, 0.016 g (7.5 × 10−5 mol) RAFT agent, and 0.0018 g (1.125 × 10−5 mol) AIBN were added to a Schlenk flask under nitrogen atmosphere. The mixture was stirred for 24 hours at 60 °C. The resulting polymer was dissolved in THF, and then precipitated with methanol and dried under vacuum. GPC analysis (in THF, polystyrene standards): Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 160 and Mw/Mn = 1.23.
160 and Mw/Mn = 1.23.
Photopolymerizations
All photoreactions were conducted using a Rayonet Photochemical Reactor (Southern N.E. Ultraviolet Company in Middletown, Connecticut). It is comprised of a circular chamber with 16 bulbs evenly distributed around the circumference with 8 bulbs of each 254 nm and 354 nm UV lamps. All samples were placed on a platform in the middle of the chamber and exposed to light for the desired time. 4′′ Pyrex test tubes were used to allow visualization of gas bubbles.Carbon monoxide release tests
In order to quantify the amount of carbon monoxide released from the decarbonylation reactions, a known amount of sample was introduced in a 10 mL flask equipped with a stopcock. The flask was closed and photocured for 20 minutes. One end of a Draeger detector tube (SKC Inc.) was connected to the stopcock, while the other end connected to a 100 cm3 Draeger pump. The stopcock was opened and the gas was extracted through the detector tube which gives a direct reading of the carbon monoxide released by a color change from white to green.Polymerization kinetics
The degree of conversion was determined from FT-IR data collected using a Galaxy series 2020 spectrometer. A small drop of each sample containing the monomer mixture was placed between two transparent tape strips, which were pressed between two NaCl crystals to produce a thin layer. The samples were irradiated for a desired time period. Absorbance spectra were recorded after the end of irradiation with 32 scans at a resolution of 1 cm−1. The amount of vinyl double bonds remaining in the sample after irradiation corresponds to the intensity of the peak at 1637 cm−1. The degree of conversion was calculated based on the decrease of 1637 cm−1 absorption as shown in eqn (1):5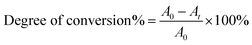 | (1) |
Polymerization shrinkage measurements
Density measurements using a 10 mL glass pycnometer were used to determine the polymerization shrinkage of the resin. First, the total volume of the pycnometer was determined using deionized water. Second, a known amount of uncured monomer mixture (higher density than water) was added to the glass pycnometer and the exact weight was recorded. The pycnometer was then filled with deionized water and the total weight (water and sample) was recorded. By determining the volume of the water displaced by the monomer mixture, the density of the latter was determined (dmonomer). After photocuring the samples using pyrex tubing (1/4′′ inner diameter), the tubing was cut and the polymer density (dpolymer) was determined in a similar manner to the method described above. The polymerization shrinkage was calculated using eqn (2).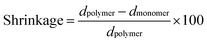 | (2) |
Results and discussion
Synthesis and polymerization
The MEPG was successfully synthesized with high purity according to literature procedures. Furthermore, the MEPG was polymerized by reversible addition–fragmentation chain transfer (RAFT) polymerization6,7 (Scheme 1) to obtain poly(MEPG) with low polydispersity. While a low polydispersity is not a requirement in terms of generating CO from the polymer, it was expected that by limiting the molecular weight and having a well-defined distribution would minimize the effect of increased viscosity during the curing of the BisGMA/TEGDMA systems, thus allows a more direct comparison between the addition of MEPG and poly(MEPG). 2-Cyanoisopropyl dithiobenzoate was used as the RAFT agent and azobisisobutyronitrile (AIBN) as an initiator. The molecular weight analysis of the polymer by gel permeation chromatography (GPC) gave a number-average molecular weight (Mn) of 11160 g mol−1, and a polydispersity (Mw/Mn) of 1.23 (where Mw is the weight-average molecular weight).
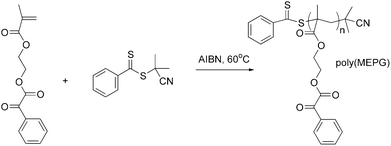 | ||
| Scheme 1 RAFT polymerization of MEPG. | ||
Crosslinking kinetics and photochemistry of MEPG
In order to visually identify the decarbonylation property of MEPG in the presence of a crosslinking monomer mixture (often used in dental resins), 5 wt% of MEPG was added to a 1:1 wt/wt BisGMA/TEGDMA mixture.8 Without the addition of any other initiator, the mixture was photolyzed using a Rayonet reactor equipped with 254 nm and 354 nm UV lamps for 5 minutes in an air atmosphere (alkylphenyl phenylglyoxylates typically absorb in the 350 nm region9). The resulting mixture polymerized, transitioning to a viscous gel and then into a glassy solid after the first minute of the photocuring process. After 5 minutes, the material appeared to harden completely with a significant amount of entrapped gas bubbles (carbon monoxide). In a control experiment, where no MEPG was added to the BisGMA/TEGDMA, no polymerization occurred. It is well known10,11 that α-keto esters, including alkyl phenylglyoxylates,9,12 when exposed to light undergo both intermolecular and intramolecular hydrogen abstraction that yield several radical species. The intramolecular hydrogen abstraction produces a biradical, while the intermolecular hydrogen abstraction produces several radicals such as hydroxyphenylcarboxy radicals and benzoyl formate radicals. The latter lose carbon monoxide and form benzoyl radicals. The polymerization reaction due to the presence of MEPG in the dimethacrylate monomer mixture was initiated by one or more of the above mentioned radicals.
Hu and Neckers9 studied the photochemistry of ethyl phenylglyoxylate and its use as a radical photoinitiator for diacrylate monomers for negative photoimaging applications. When a benzene solution of 0.5% mass of ethyl phenylglyoxylateversusdi(ethylene glycol) dimethacrylate was irradiated with UV light in the absence of oxygen, the monomer solution was converted into a “non-mobile” polymer gel with an extent of polymerization exceeding 95% after 300 seconds. The major product was found to be diethyl 2,3-dihydroxy-2,3-diphenyl succinate without carbon monoxide release. The study also revealed that the polymerization reaction is initiated by both benzoyl and phenyl radicals. This suggests that MEPG initiated the polymerization of the BisGMA/TEGDMAvia both radicals as well.
It is also possible that a polymer bearing a phenylglyoxylate functional group would initiate radical polymerization since poly(MEPG) would give yield to carbon monoxide and radical formation as well. Hu and Neckers13 have made polymers based on (meth)acryloylethyl phenylglyoxylates using conventional radical polymerization and studied photo-induced intra-molecular crosslinking. In our study, 5 wt% of poly(MEPG) in the BisGMA/TEGDMA mixture (1:1 weight ratio) was irradiated for 5 min with UV light in an air atmosphere. The resin mixture started hardening after one minute of the curing process, with a significant amount of carbon monoxide formation after 5 min of irradiation as shown in Fig. 1. These images are representative of gas bubble formation in resins with larger amounts of MEPG and poly(MEPG).
 | ||
| Fig. 1 BisGMA and TEGDMA (1:1) photolyzed for 5 min in the presence of (a) 5% MEPG and (b) 5% poly(MEPG). Images are representative of gas bubble formation in resins with higher content of MEPG and poly(MEPG). | ||
With the successful initiation of the polymerization using MEPG or poly(MEPG) demonstrated, the degree of conversion as a function of time was studied in order to provide a better understanding of the polymerization. Fig. 2 shows the conversion vs. time plots for several experiments. When 5% MEPG was irradiated in the presence of BisGMA/TEGDMA mixture (1:1 wt ratio), the degree of polymerization increases with time and reaches a plateau after 5 minutes of curing attaining 80% conversion, while 65% conversion was achieved after only one minute of curing time. This degree of conversion is significantly higher than the degree of conversion of commercial BisGMA/TEGDMA-based resins which normally ranges between 50 and 65%.14 The extent of polymerization for polymeric materials plays an important role not only in determining the mechanical properties of the final polymer, but also a high degree of conversion minimizes the presence of the residual monomer present in the composite which might cause undesirable effects.
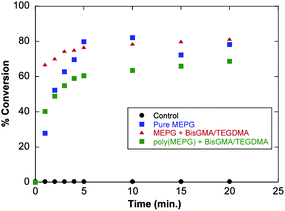 | ||
| Fig. 2
Polymerization kinetics of MEPG and poly(MEPG) in BisGMA:TEGDMA mixtures. | ||
The limiting conversions are primarily due to the decrease in mobility of the species involved in the polymerization (both monomeric and polymeric). Anseth et al.15 studied the kinetics of several multi(meth)acrylate monomers such as diethylene glycol dimethacrylate and showed that the termination reaction mechanism (reaction of two macroradicals) becomes diffusion controlled starting at 5% double bond conversion and completely diffusion controlled at 10% conversion, which explains the rapid increases in the double bond conversion observed for the studied systems. However, as the viscosity of the mixture increases, the propagation reaction also becomes diffusion controlled which leads to a limiting final degree of conversion, as seen in Fig. 2.
Furthermore, the presence of BisGMA in the monomer system plays an important role in determining the kinetic behavior of the double bond conversion. Sideridou et al.5 studied the degree of conversion of several multifunctional monomers in the presence of camphorquinone and showed that the degree of conversion of BisGMA exhibits much higher initial polymerization reactivity than TEGDMA. After only 10 seconds, 22.9% conversion was observed for the BisGMA versus 13.7% for TEGDMA. However, as the polymerization proceeds, TEGDMA polymerizes at a much faster rate than BisGMA. This is a clear indication that the mobility of the reactants and the radical species in the reacting medium play a major role in determining the kinetic behavior of the polymeric materials, thus shaping the final mechanical properties.
When pure MEPG was cured in the present study, with no dimethacrylate added (i.e. no BisGMA or TEGDMA), the degree of conversion showed a much slower rate, reaching only 25% after the first minute, and 60% after 4 minutes. The conversion attained a plateau after 5 minutes, and reached 80% after 20 minutes of curing. This clearly indicates the ability of MEPG to self-polymerize in the absence of any other monomer of initiator. Although the double bond degree of conversion was lower than in the presence of dimethacrylate monomer mixture, a maximum degree of conversion similar to the system that contains 5% MEPG in BisGMA/TEGDMA was achieved.
When 5 wt% of poly(MEPG) in the 1:1 w/w BisGMA/TEGDMA mixture was irradiated, a much lower rate of conversion was observed, reaching only 60% conversion after 5 minutes and less than 70% after 20 minutes of curing. This degree of conversion is lower than that obtained in the pure MEPG and 5% MEPG in BisGMA/TEGDMA experiments. In the case of poly(MEPG), the chromophore functional groups (α-keto ester) are in close proximity to each other, with a restricted mobility due to the polymer backbone, whereas in the MEPG, the generated radicals have more degrees of freedom. Hu et al.16 showed that the photolysis of poly(MEPG) in benzene yields a crosslinked polymer, which results from the intermolecular hydrogen abstraction between pendent phenylglyoxylate functional groups (Scheme 2). Thus, it is possible that the low degree of polymerization we observed for the system containing 5 wt% poly(MEPG) is due to (intramolecular) radical coupling within polymer chains.
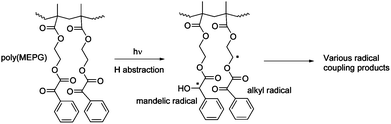 | ||
| Scheme 2 Photolysis of polyMEPG and the resulting radical coupling. | ||
Polymerization shrinkage
Since the main objective of this study was to examine the possible reduction in polymerization shrinkage, we performed experiments to quantify shrinkage upon curing. The gas bubble formation as shown in Fig. 1 is a clear indication that using MEPG or poly(MEPG) in the crosslinking monomer system would most likely increase the volume of the materials, thus reducing the polymerization shrinkage. However, since the poly(MEPG)-initiated system showed a lower monomer conversion compared to the MEPG-initiated system, the shrinkage measurements were limited to the latter system.As determined by density measurements, the polymerization shrinkage of the BisGMA/TEGDMA decreases with increasing MEPG concentration (Fig. 3). When the BisGMA/TEGDMA mixture contains only camphorquinone (1 wt%) and dimethylaminoethyl methacrylate (1 wt%), our results gave a polymerization shrinkage of 8.6 ± 0.4%. This value is in agreement with the published polymerization shrinkage.17 Our work further reveals that when 1 wt% of MEPG was added to the BisGMA/TEGDMA mixture without camphorquinone and dimethylaminoethyl methacrylate, no significant change in the polymerization shrinkage was observed, which is possibly due to the small amount of carbon monoxide that is released at this concentration. Similar observation was made when 5 wt% MEPG was added. However, when 10 wt% MEPG was added to the BisGMA/TEGDMA mixture, the polymerization shrinkage decreased significantly to 5.8 ± 0.4%. With 20 wt% MEPG, the polymerization shrinkage was further reduced to 4.3 ± 0.1%. However, when a higher concentration of MEPG was added (45 wt%), the polymerization shrinkage was reduced to 3.2 ± 1.1%, which corresponds to 63.1% reduction of the shrinkage.
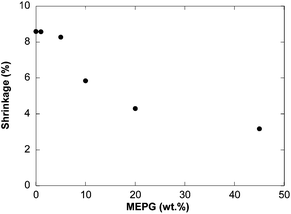 | ||
| Fig. 3 Polymerization shrinkage as a function of MEPG wt% in BisGMA/TEGDMA mixtures. | ||
The results clearly show that the polymerization shrinkage decreases with increasing MEPG concentration. The use of alkyl phenylglyoxylates, specifically ethyl phenylglyoxylate, as a photoinitiator has been reported in the literature9 for the development of a negative photoimage system; however to the best of our knowledge, this is the first time that a photochemical reaction has been used for both initiating a chemical reaction and controlling the volume change of the resulting polymeric material. As shown in Fig. 3, the volume shrinkage in the polymeric material can be easily controlled by the amount of photolabile monomer (MEPG) used, which determines the amount of carbon monoxide released.
Using Draeger carbon monoxide detector tubes, carbon monoxide release tests were conducted using both MEPG and ethylbenzoyl formate (EBF), which is a commercially available alkyl phenylphenylglyoxylate. EBF should undergo decarbonylation reaction in a similar manner to MEPG (Scheme 3). It was found that after curing 1.50 mmol of MEPG (no solvent or comonomers) for 20 minutes an estimated 3 ppm of carbon monoxide was released. Similarly, when 3.14 mmol EBF were irradiated for 20 minutes (also with no solvent added), the amount of carbon monoxide released was approximately 2 ppm. Using greater quantities of EBF yielded more CO, as expected (e.g. 9.4 mmol gave 5 ppm CO, 19.0 mmol gave 7 ppm of CO). The amounts of CO released by MEPG and EBF did not correlate with each other presumably because as MEPG decarbonylates, it also polymerizes, thus causing CO to become trapped within the polymer matrix. Regardless, these results clearly support the polymerization shrinkage measurements showing the volume increase of the resin when the concentration of MEPG was increased.
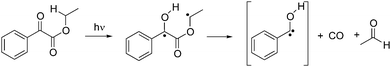 | ||
| Scheme 3 Photolysis of EBF. | ||
Several more experiments showed that the polymerization shrinkage is reduced when inorganic filler is added to the resin mixture. For instance, when we added 20% silica to the sample that contains 10% MEPG in BisGMA/TEGDMA, the polymerization shrinkage was reduced from 5.8% to 4%. Adding even more silica particles to the resin mixture might be desirable since commercial resins may contain more than 80% weight inorganic filler.18 Thus, it is expected that polymerization shrinkage will be further reduced with a higher concentration of inorganic filler in our MEPG/BisGMA/TEGDMA mixture.
Conclusions
The goal of this work was to reduce the polymerization shrinkage that results from the change of the intermolecular distance between the monomer units when covalently bonded in polymer chain upon photocuring. To the best of our knowledge, we show for the first time that an α-keto ester can be used to reduce the polymerization volume shrinkage in resins. MEPG showed a dramatic effect on the volume shrinkage of the resin upon photocuring with UV light. In addition, MEPG can initiate free radical polymerization upon exposure to UV light, and yielded a higher degree of conversion of the classical resin monomers. Further work is needed to determine the effect of the gas formation on the mechanical properties of the resin, and ideally devise a process in which the CO gas is evenly distributed within the resin.Acknowledgements
We thank the Department of Chemistry and Biomolecular Science at Clarkson University for support for KO and JJF, and the Center for Advanced Materials Processing (CAMP), a New York State Center for Advanced Technology, for support of DAS and REP.References
- C. J. Kleverlaan and A. J. Feilzer, Dent. Mater., 2006, 21, 1150–1157.
- J. R. Condon and J. L. Ferracane, Biomaterials, 2002, 23, 3807–3815 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- M. Nakajima and J. P. Anselme, J. Org. Chem., 1983, 48, 1444–1448 CrossRef CAS.
- I. Sideridou, V. Tserki and G. Papanastasiou, Biomaterials, 2002, 23, 1819–1829 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- D. A. Shipp, J. Macromol. Sci., Polym. Rev., 2005, 45, 171–194.
- A. J. Feilzer and B. S. Dauvillier, J. Dent. Res., 2003, 82, 824–828 CrossRef CAS.
- S. Hu and D. C. Neckers, J. Mater. Chem., 1997, 7, 1737 RSC.
- G. L. Closs and R. J. Miller, J. Am. Chem. Soc., 1978, 100, 3483–3494 CrossRef CAS.
- P. A. Leermakers and G. F. Vesley, J. Am. Chem. Soc., 1963, 85, 3776–3779 CrossRef CAS.
- S. Hu, X. Wu and D. C. Neckers, Macromolecules, 2000, 33, 4030–4033 CrossRef CAS.
- S. Hu and D. C. Neckers, Macromolecules, 1998, 31, 322–327 CrossRef CAS.
- A. Peutzfeldt, Eur. J. Oral Sci., 1997, 105, 97–116 CrossRef CAS.
- K. S. Anseth, C. M. Wang and C. N. Bowman, Macromolecules, 1994, 27, 650–655 CrossRef CAS.
- S. Hu, A. Mejiritski and D. C. Neckers, Chem. Mater., 1997, 9, 3171–3175 CrossRef CAS.
- M. Atai, D. C. Watts and Z. Atai, Biomaterials, 2005, 26, 5015–5020 CrossRef CAS.
- H. Y. Chen, J. Manhart, R. Hickel and K. H. Kunzelmann, Dent. Mater., 2001, 17, 253–259 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |