Optimization of N-carboxyanhydride (NCA) polymerization by variation of reaction temperature and pressure†
Gijs J. M.
Habraken
a,
Karel H. R. M.
Wilsens
a,
Cor E.
Koning
a and
Andreas
Heise
*ab
aEindhoven University of Technology, Laboratory of Polymer Chemistry, Den Dolech 2, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands
bDublin City University, School of Chemical Sciences, Glasnevin, Dublin 9, Ireland. E-mail: andreas.heise@dcu.ie
First published on 18th April 2011
Abstract
Several new methods for the controlled ring-opening polymerization of N-carboxyanhydrides (NCA ROP) have been established in the recent past, all of which based on the normal amine mechanism. In this paper the optimal NAM polymerization conditions were investigated combining the high-vacuum and the low-temperature for the NCAs of γ-benzyl-L-glutamate (BLG), Nε-benzyloxycarbonyl-L-lysine (ZLL), L-alanine (Ala), β-benzyl-L-aspartate (BLA), O-benzyl-L-serine (BLS), and O-benzyl-L-threonine (BLT). The polymerizations were followed by FTIR, size exclusion chromatography (SEC) and MALDI-ToF-MS to provide information of the monomer conversion, polymer molecular weight and chain composition as a function of pressure and temperature. It was found that the studied NCAs could be divided into two groups: in the first group monomers of BLG, ZLL and Ala polymerized considerably faster when a lower pressure of 1 × 10−5 bar was applied. MALDI-ToF-MS analysis confirmed that the formation of side products for these monomers mostly started after full monomer conversion. The second group of monomers, i.e. BLA, BLS and BLT, polymerized considerably slower than the first group and no effect was observed from the lower pressure. On the other hand, the number of side reactions was significant at 20 °C, so that the polymerizations for the latter monomers should preferably be done at 0 °C. By combining both methods, multiblock polypeptides were synthesized including a tetrablock of PBLG-b-PAla-b-PZLL-b-PBLA with a polydispersity of 1.3.
Introduction
The interest in well-defined synthetic polypeptide architectures from the ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA) has increased significantly in the last ten years.1,2 This can be ascribed to the enormous potential arising from the combination of synthetic polypeptide segments with either synthetic or natural building blocks. For example, numerous reports have been published on the synthesis and self-organization of polypeptide block copolymers and polypeptide conjugates into vesicles, micelles and nanoparticles.3–15 The increased attention that these materials have received was clearly facilitated by the development of synthetic methods that allowed for the control of the ring-opening polymerization of NCAs. Generally, nucleophiles like primary amines can initiate the NCA polymerization and produce polypeptides with a molecular weight determined by the amine to NCA ratio (normal amine mechanism, NAM). However, prone to side-reactions with solvents, end-group termination and competing polymerization mechanisms, the reactions often lack the level of control necessary to synthesize more complex polymer architectures like block copolymers. Generally, two different approaches were taken to prevent chain termination in NCA polymerization. When compared to the NAM, the first approach builds on different reaction mechanisms by end-group protection and the formation of a dormant chain ends. For example, the metal mediated NCA ROP coordinates the NCA monomer to the chain end, where it then protects the end-group from side reactions.16 In the silazane method the end-group is protected by a trimethylsilyl carbamate and coordinates the monomer for insertion, while releasing CO2.17 The ammonium halide initiators proposed by Schlaad and Dimitrov propagate following the NAM, but have a protected end group in the dormant state.18While all three methods rely on the addition of a regulating agent, control of NCA polymerization can also be achieved by optimizing the NAM conditions themselves, for example by lowering the reaction temperature or applying a high vacuum.19–21 Mechanistically, the first step in the NAM is the attack of a primary (nucleophilic) amine on the C-5 position of the NCA, causing the ring to open. This is followed by the transformation of the carbamic ion and decomposition of the carbamic acid under liberation of CO2 (Scheme 1). The newly formed primary amine then propagates the polymerization. As stated in the literature the carbamic acid is only stable at lower temperatures, while the carbamic ion is a more stable form depending on the solvent.2,22 For the efficiency of the high-vacuum technique several reasons have been proposed. The first one is the acceleration of the reaction by efficient removal of CO2.2 Secondly, with the removal of CO2, a side-reaction involving DMF, resulting in formaldehyde and dimethylamine, is suppressed.23 In the key publication on the high-vacuum technique, the high control of the polymerization was shown for γ-benzyl-L-glutamate (BLG) NCA and was evident from the linear increase of the molecular weight and the low polydispersity.21 However, expansion to other NCAs or an investigation of the chain composition, such as the end groups, was not carried out. Recently, the synthesis of poly(O-benzyl-L-tyrosine) with diaminohexane under vacuum conditions was compared to glove-box conditions. The reaction products obtained after 24 hours were investigated by MALDI-ToF-MS, and the formation of formamide end-groups by reaction of the peptide with the solvent DMF was found even for the high-vacuum technique.24 For the reaction performed in the glove-box many more side reactions were identified, such as the formation of ureido acid-terminated products and polymers from the competing activated monomer mechanism.
 | ||
| Scheme 1 NCA ROP according to the normal amine mechanism (NAM), initiated by benzylamine, and structures of the NCAs investigated in this study. | ||
The effect of decreased reaction temperature on the reduction of side-reactions in NCA polymerization was also shown recently. Capillary electrophoresis (CE) was used to determine the chain compositions of poly(t-Boc-L-lysine) after 48 hours.19,20 At 0 °C the polypeptide end-groups had retained their primary amines compared to the reactions performed at room temperature, where formyl and ureido-acid end groups were found. We recently conducted a systematic study on the effect of temperature on the polymerization of several NCAs.25 Depending on the type of NCA (i.e.amino acid) MALDI-ToF-MS analysis showed side reactions such as intramolecular terminations, chain composition alterations and the formation of cyclics at polymerization temperatures of 20 °C. These reactions were almost completely absent at a polymerization temperature of 0 °C.
While the low-temperature NCA polymerization thus provides an advantage in terms of structural control, a drawback of the polymerization at 0 °C is the long reaction time. On the other hand, the application of vacuum at higher temperatures seems to increase the reaction kinetics but the literature suggests that some side reactions still occur.20 From the literature a direct comparison between both methods cannot be made easily because different analytical methods (SEC, CE, MALDI-ToF-MS) were used to prove the degree of control of the polymerizations. In addition, different NCA monomers were studied, which makes comparison more difficult due to different side reactions occurring for different monomers, different polymerization kinetics, and chain conformations in the reaction solutions.
The goal of this work was to optimize the NAM by performing NCA ROPs for several NCA monomers by combining the low-temperature NCA polymerization with the high-vacuum technique. Polymerizations were carried out at 0 °C and 20 °C under nitrogen at atmospheric pressure and a high vacuum of 1 × 10−5 bar. The results were used to come to a recommendation of the most ideal reaction conditions for individual NCAs, and were validated by the synthesis of well-defined tetra-block copolymers in the most optimal conditions.
Experimental
Materials
N ε-Benzyloxycarbonyl-L-lysine, L-alanine, benzylamine (99.5%; purified by redistillation), α-pinene (98%) and bis(trichloromethyl) carbonate (triphosgene) 99% were purchased from Aldrich. L-Glutamic acid, γ-benzyl ester, L-aspartic acid, β-benzyl ester, O-benzyl-L-threonine hydrogenchloride salt and O-benzyl-L-serine were supplied by Bachem. DMF (extra dry), ethyl acetate, n-heptane and diethyl ether were purchased from Biosolve. All chemicals were used without any purification unless otherwise noted. DMF and ethyl acetate were used directly from the bottle and stored under an inert, dry atmosphere. NCAs were synthesized as previously reported unless otherwise noted.25Methods
FTIR measurements were performed on a PerkinElmer SpectrumOne FTIR Spectrometer, using a universal ATR sampling accessory (resolution 1 cm−1). Samples for IR spectroscopy were taken from the reaction mixtures and analyzed without any further modification.NMR analyses of NCA monomers were performed on a Mercury 400 in deuterated chloroform. NMR analyses of the polypeptide products were performed in deuterated TFA.
1,1,1,3,3,3-Hexafluoroisopropanol size exclusion chromatography (HFIP SEC) was performed on a system equipped with a Waters 1515 Isocratic HPLC pump, a Waters 2414 refractive index detector (40 °C), a Waters 2707 autosampler and a PSS PFG guard column followed by 2 PFG-linear-XL (7 μm, 8 × 300 mm) columns in series at 40 °C. HFIP (Apollo Scientific Limited) with potassium trifluoroacetate (3 g L−1) was used as eluent at a flow rate of 0.8 ml min−1. The molecular weights were calculated against polymethyl methacrylate standards (Polymer Laboratories, Mp = 580 Da up to Mp = 7.1 × 106 Da).
N,N-Dimethylacetamide size exclusion chromatography (DMAc SEC) was measured on a Waters Alliance system equipped with a Waters 2695 separation module, a Waters 2414 refractive index detector (40 °C), a Waters 2996 photodiode array detector, a PSS GRAM guard column followed by 2 PSS GRAM columns in series of 100 Å (10 μm particles) and 3000 Å (10 μm particles) respectively at 60 °C. DMAc was used as an eluent at a flow rate of 1 ml min−1. The molecular weights were calculated against polystyrene standards (Polymer Laboratories, Mp = 580 Da up to Mp = 7.1 × 106 Da). Before SEC analysis was performed, the samples were filtered through a 0.2 μm PTFE filter (13 mm, PP housing, Alltech).
Matrix Assisted Laser Desorption/Ionization-Time of Flight-Mass Spectroscopy (MALDI-ToF-MS) analysis was carried out on a Voyager DE-STR from Applied Biosystems (laser frequency 20 Hz, 337 nm and a voltage of 25 kV). The matrix material used was trans-2-(3-(4-t-butyl-phenyl)-2-methyl-2-propenylidene)malononitrile (DCTB) (40 mg ml−1). Potassium trifluoroacetic acid (KTFA) was added as a cationic ionization agent (5 mg ml−1). The polymer sample was dissolved in HFIP (1 mg ml−1), to which the matrix material and the ionization agent were added (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5), and the mixture was placed on the target plate. Samples were precipitated from the reaction medium in diethyl ether, filtered and placed in a freezer before measuring.
:1:5), and the mixture was placed on the target plate. Samples were precipitated from the reaction medium in diethyl ether, filtered and placed in a freezer before measuring.
Results and discussion
NCA ROP conversions as a function of temperature and pressure
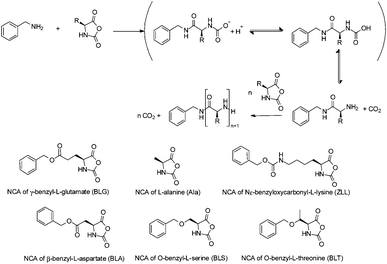
To determine optimal polymerization conditions with respect to temperature and pressure, a series of amino acid NCAs, as shown in Scheme 1, was investigated. All NCA monomers were prepared following the same procedure using triphosgene and α-pinene as HCl scavenger.26,27 For the synthesis of BLT NCA from the amino acid HCl salt, an extra addition of α-pinene was required. All monomers were recrystallized before storage in the refrigerator in a desiccator.In order to monitor the NCA conversion under different polymerization conditions, FTIR spectroscopy was used. Upon ring opening the NCA carbonyl peaks of the anhydride at 1786 cm−1 and 1857 cm−1 are reduced while amide peaks of the polypeptide products increase. A calibration curve was made by plotting the area under the NCA carbonyl peak at 1786 cm−1 against the NCA solution concentration for all used NCAs. In the reactions under high vacuum there is the possibility that the actual concentration is altered by the evaporation of solvent (DMF), which could lead to an error in the calculation of the NCA conversion. Therefore an additional calibration plot was made for the benzyl-group containing NCAs using the signal at 700 cm−1 to correct for any change in NCA monomer concentration. Since the quantity of benzyl groups does not change during the polymerization, the absorption of this group could be used as an internal standard for the polymerization of the well-soluble polypeptides PZLL, PBLG and PBLT. The resulting calibration plot is shown in Fig. 1, from which it can be observed that the peak areas measured as a function of concentration are almost identical for all NCAs.
 | ||
| Fig. 1 Calibration plot of the peak areas of the carbonyl peak at 1786 cm−1 (filled symbols) and benzyl peaks at 700 cm−1 (open symbols) versus the NCA monomer concentration (mol l−1) in DMF for the NCAs of BLG (■), Ala (▲), ZLL (●), BLA (▼), BLS (◆) and BLT (◀). For the NCA abbreviations, see Scheme 1. | ||
For the polymerization experiments, NCA monomer solutions were prepared and a solution of benzylamine initiator added. This stock solution was quickly divided over four Schlenk tubes, two of which were polymerized under nitrogen at a temperature of 0 °C and 20 °C, respectively. The other two tubes were polymerized at the same two temperatures but exposed to a pressure of 1 × 10−5 bar. Samples were taken from the reaction solution by a μl Finn pipet at time intervals and analyzed by SEC in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) for molecular weight analysis and by FTIR for monomer conversion analysis using the calibration plot. The high vacuum reaction flask was purged with nitrogen gas prior to sampling and subsequently evacuated again. Different degrees of polymerization (DP) were aimed at by altering the monomer to initiator ratio. As a standard a target DP of 40 was used for all polymers. In addition, for polymers well soluble in DMF, such as PBLG, reactions aiming at higher DPs of 100 and 400 were performed.
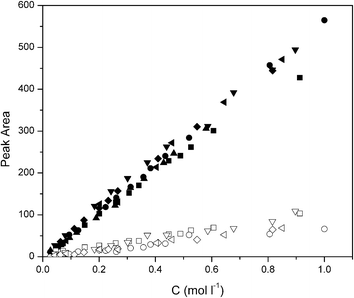
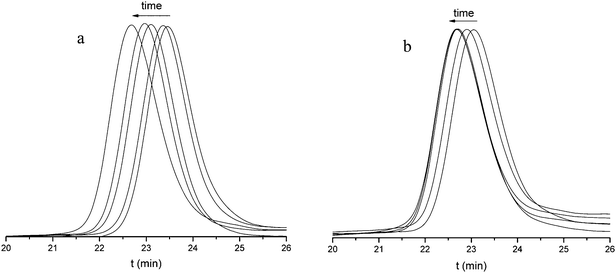
A typical result is shown in Fig. 2 and 3 for the polymerization of BLG with DP = 100. Inspection of Fig. 3 shows that at both temperatures of 0 °C and 20 °C the NCA conversion is higher, i.e. the reaction is faster, at the lower pressure (Fig. 3a and b). This effect is more pronounced at the higher temperature of 20 °C. At both temperatures and pressures the molecular weight increases linearly with the NCA conversion, and low polydispersity indices (PDI) < 1.2 suggest good control over the polymerization (Fig. 2, and Fig. 3c and d). In order to be able to compare the results, the points of full conversion were determined for the different reaction conditions and NCAs and listed in Table 1. For BLG (DP = 100), the time to reach full conversion was 70 h at 0 °C irrespective of the pressure, while it was much shorter for the higher temperature, namely 12 h under nitrogen at 1 bar and 5 hours and 20 minutes at 1 × 10−5 bar, respectively.
 | ||
| Fig. 2 HFIP SEC results of BLG polymerization (DP: 100): (a) at 20 °C under nitrogen and (b) at 20 °C under high vacuum. | ||
 | ||
| Fig. 3 Polymerization of BLG NCA at a monomer of BLG NCA to initiator ratio of 100: (a) monomer conversion at 0 °C, (b) monomer conversion at 20 °C, (c) Mnvs. conversion at 0 °C, and (d) Mnvs. conversion at 20 °C. (▲) Reaction under a high vacuum of 1 × 10−5 bar and (■) reaction under nitrogen at 1 bar. | ||
| NCA | DP | Concentration/M | Temp./°C | M n a/g mol−1 | PDI | Timea (N2) | Timea (HV) |
|---|---|---|---|---|---|---|---|
| a Time to reach full conversion determined by FTIR. In cases where full conversion was not reached, the maximum conversion is mentioned in brackets. | |||||||
| BLG | 40 | 0.38 | 0 | 8900 | 1.08 | 30 h | 23 h |
| BLG | 40 | 0.38 | 20 | 10900 |
1.17 | 3 h | 2 h 30 minutes |
| BLG | 100 | 0.76 | 0 | 21800 |
1.08 | 70 h | 70 h |
| BLG | 100 | 0.76 | 20 | 23500 |
1.16 | 12 h | 5 h 20 minutes |
| BLG | 400 | 0.76 | 0 | 78800 |
1.14 | 237 h (93%) | 237 h (99%) |
| BLG | 400 | 0.76 | 20 | 80000 |
1.08 | 31 h 15 minutes | 23 h 15 minutes |
| ZLL | 40 | 0.38 | 0 | 10700 |
1.12 | 47 h 10 minutes | 21 h 40 minutes |
| ZLL | 40 | 0.38 | 20 | 10300 |
1.17 | 2 h 36 minutes | 1 h 45 minutes |
| Ala | 40 | 0.38 | 0 | 5700 | 1.69 | 32 h | 22 h |
| Ala | 40 | 0.38 | 20 | 6700 | 1.80 | 5 h | 3 h 30 minutes |
| BLA | 40 | 0.38 | 0 | 10500 |
1.20 | 79.5 h (97%) | 79.5 h (93%) |
| BLA | 40 | 0.38 | 20 | 11900 |
1.09 | 30 h 30 minutes | 30 h 30 minutes |
| BLA | 100 | 0.76 | 0 | 22200 |
1.28 | 336 h (88.6%) | 336 h (91.7%) |
| BLA | 100 | 0.76 | 20 | 16600 |
1.17 | 54 h | 54 h |
| BLT | 40 | 0.76 | 0 | 10400 |
1.09 | 480 h (80%) | 480 h (80%) |
| BLT | 40 | 0.76 | 20 | 11000 |
1.11 | 192 h (93%) | 192 h (80%) |
| BLS | 40 | 0.38 | 0 | 8300 | 1.26 | 412 h (93%) | 412 h (90%) |
| BLS | 40 | 0.38 | 20 | 7500 | 1.23 | 76 h (90%) | 76 h (89%) |
Due to the good solubility of PBLG in DMF, a polymerization to high molecular weight was attempted. When the initiator to monomer ratio was increased to 400, the time to reach full monomer conversion was considerably longer. In fact, the reaction at 0 °C did not even reach full conversion after 237 hours, while at 20 °C full conversion was obtained after about 31 h under nitrogen and 23 h under high vacuum (Table 1). Noticeable was the high viscosity of the reaction medium at 0 °C, which probably led to a physical retardation of the polymerization. Especially for the high vacuum polymerizations at 20 °C it was found that the polymerization solution was concentrated by 23% due to solvent evaporation, while for the polymerization at 0 °C it was only 2–3%. However, as stated above, due to the use of an internal standard in the FTIR calculations, this had no influence on the determination of the monomer conversion. The obtained molecular weights were in the range of the expected molecular weights for this and all other polymerizations.
The results shown for the BLG NCA clearly confirm the positive effect of the high vacuum on the polymerization kinetics of this monomer. All other investigated NCAs can be divided into two groups, i.e. those where a similar effect was observed and those where the high vacuum did not lead to an acceleration of the polymerization. Examples of the latter group are the NCAs of BLA, BLS and BLT. Compared to the polymerization of BLG NCA, the monomer conversions of these NCAs were considerably slower under identical conditions and levelled off more quickly (Fig. 4). First order kinetic plots of the reactions did not show linear increases for Ala, BLS and ZLL and BLA NCA at 0 °C (see ESI†), suggesting a decrease in the available primary amine end groups for propagation. For BLA and BLS this can predominantly be ascribed to the low solubility of the formed polypeptides as manifested in a precipitation or gelation observed during the polymerization. For PBLS this resulted in a bimodal distribution (Fig. S8†). This is more evident at the lower polymerization temperature, where full conversion could not be reached for either of these NCAs. Furthermore, BLT is known for its slow polymerization kinetics and did require long reaction times to reach full monomer conversion.28 However, the polymer has an excellent solubility in many different solvents, including DMF, and the molecular weight increase was linear with monomer conversion. For these three monomers the difference between the high vacuum and nitrogen conditions was insignificant.
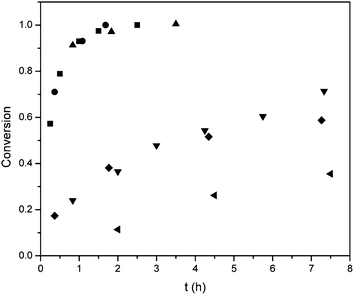 | ||
| Fig. 4 Conversion plots of BLG (■), Ala (▲), ZLL (●), BLA (▼), BLS (◆), BLT (◀) (0.76 M) for a monomer to initiator ratio of 40 at 20 °C and 1 × 10−5 bar. | ||
In contrast, a clear effect of the vacuum was seen for BLG NCA, as discussed above, and for ZLL and Ala. Interestingly all these monomers have their rapid polymerization in common (Fig. 4). A fast polymerization was observed for the Ala NCA, but due to precipitation during the polymerization the reaction resulted in a broad PDI of the polymer.
When seeking for an explanation of the different effects of the CO2 removal on the two groups of monomers, it is noticeable that the slow polymerizing monomers have a hydrogen bond accepting group next to the β-carbon of the NCA monomer (γ-carbon for β-benzyl-L-aspartate). It was already hypothesized for the slow propagation of L-threonine that the nucleophilicity of the corresponding amine group at the chain end could be lower, thereby lowering the propagation speed.16 Intrachain hydrogen bonds between the Asp, Thr and Ser side groups and the primary amine could decrease the nucleophilicity of the amine chain end.29 The specific side groups could also affect the carbonyl, making it less electrophilic. Another important influence on the polymerization kinetic might arise from the secondary structures of the polypeptides in the studied solvent. DMF is known to be an inefficient hydrogen bond breaking solvent for some of the polypeptide species (PBLG, PZLL).30 Preliminary FTIR measurements of the polypeptides in DMF (and chloroform for comparison) did indicate that different secondary structures were present in the reaction medium (Fig. S10 and S11†). Although the solvent peaks are partly overlapping the amide signals, α-helices in the amide I bond regime, and β-sheets as well as the α-helices in the amide II bond regime could be assigned. For PBLS and PZLL the presence of β-sheets was found, while for PBLG, PBLA and PZLL α-helices were identified. PBLT did not show to form a secondary structure. For PAla FTIR did not offer any conclusive result on the polypeptide structure in DMF due to precipitation, but it is expected to contain both β-sheets and α-helices.31 In general, the α-helical forming polypeptides appear to have higher polymerization reactivity, but that cannot explain the slow polymerization speed of PBLA.
At this point we cannot offer a conclusive explanation for the different polymerization speeds but it is reasonable to assume a combination of reasons as discussed above. Recently, density functional theory (DFT) calculations were used for the determination of the NCA ring opening mechanism.32 Possibly, DFT calculations could help to find an answer to the reactivity differences found here.
Polypeptide structure analysis as a function of temperature and pressure
In our previous work, the occurrence of side reactions in the polymerization of various NCAs was investigated by detailed MALDI-ToF-MS analysis after a standard reaction time of four days. It was found that there is an increase in these side and termination reactions when the polymerization temperature is increased to 20 °C, while at 0 °C almost no side reactions were observed.25 Here we expand this investigation to a structural analysis of samples taken during polymerization and after full conversion has been reached. This provides important information as to when a reaction has to be stopped at a given reaction temperature to avoid termination reactions or other unwanted structural changes on the polymer. A series of MALDI-ToF-MS spectra was recorded for samples taken from the NCA polymerization mixtures listed in Table 1 with a targeted DP of 40 at 20 °C. It should be noted that quantification of MALDI-ToF-MS spectra is not possible, and the obtained data were thus compared only qualitatively. The samples were taken directly after full monomer conversion was reached and subsequently after several hours and days. Fig. 5 depicts the example of the BLG NCA polymerization. Besides the expected peaks of the PBLG with amino end-groups (A), the peak series of PBLG with pyroglutamate (B) end-groups can be identified (Scheme 2). This reaction is caused by the intramolecular amidation of the amino end-group with the adjacent benzyl ester side chain of the PBLG, and consequently terminates the chain growth. While this reaction is totally absent at 0 °C even after four days, from Fig. 5 it can be seen that the formation of the pyroglutamate at 20 °C increased over time but only after all monomer was consumed. A first detectable signal for the pyroglutamate end-group was apparent three hours after full monomer conversion was reached (six hours reaction time). No difference was observed between the reactions under nitrogen and the high vacuum. This suggests that this termination reaction is absent as long as the monomer is present and that the polymerization of BLG NCA is well controlled at 20 °C. Lowering the reaction temperature only provides an advantage if the polymer is to be stored in the reaction medium for a longer period of time or if monomer conversion is not carefully monitored.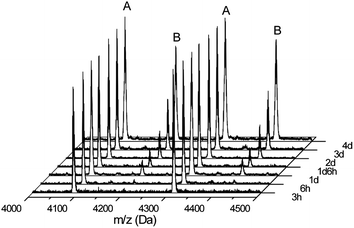 | ||
| Fig. 5 MALDI-ToF-MS spectra of samples taken from the polymerization reaction of BLG NCA at 20 °C under nitrogen at different times after complete monomer conversion. The spectra were normalized using peak A. | ||
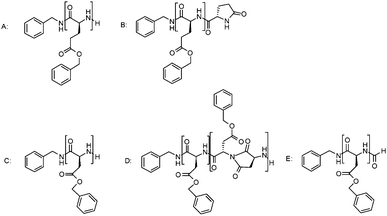 | ||
| Scheme 2 Structures of reaction products formed in various NCA polymerizations. Letters refer to structural assignments in MALDI-ToF-MS spectra, presented in Fig. 5 and 6. | ||
For PZLL no side reaction products were found, with the exception of a small amount of cyclic structures under both conditions at 20 °C, while these structures were absent at 0 °C. PAla showed no side reactions at all over time.33 Both polymerizations appear to remain controlled for a longer period of time. For the other investigated NCAs such as the BLA, BLS and BLT more side reactions were found directly after full conversion at 20 °C. As already demonstrated in our previous work, the formation of succinimides is a side reaction that occurs in the case of PBLA.25Fig. 6 shows that the formation of succinimides (peak D) begins directly after full conversion is obtained irrespective of the pressure applied during the polymerization. However, the formation of formamide-terminated amine groups due to the reaction with the solvent DMF seems to be slightly lower under high vacuum conditions due to the removal of dimethylamine, while under both conditions it increased over time. For BLS the formation of the cyclic structures was found. This was not influenced by the reaction conditions suggesting that it occurred during the polymerization. The side reactions identified in the BLT NCA polymerization include cyclics formation, water- and dimethylamine-initiated polymers as well as formamide terminated chains.34 Compared to the 0 °C reaction under nitrogen atmosphere, there were significantly more side products at 20 °C (see ESI†). For PBLA, PBLS and PBLT the results suggest that to overcome the multiple side reactions the polymerizations should preferably be performed at 0 °C.
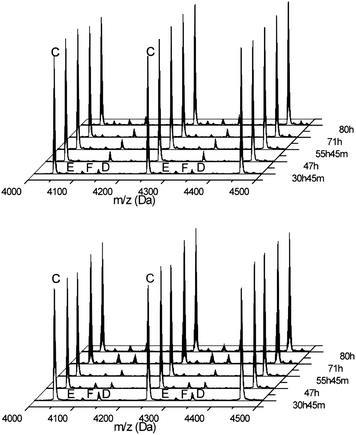 | ||
| Fig. 6 MALDI-ToF-MS spectra of samples taken from the polymerization reaction of BLA NCA at 20 °C and 1 × 10−5 bar (top) and under nitrogen (bottom) at different reaction times. The spectra were normalized using peak C. | ||
Optimized polymerization conditions
For the different NCAs examined in this work the polymerization characteristics in terms of reaction kinetics and structural control have been determined. When taking both factors into account recommendations on the preferred polymerization conditions can be given as summarized in Table 2. For example, BLG NCA can be best polymerized under high vacuum at 20 °C since those conditions allow a fast polymerization and a short reaction time. However, if further extension of the PBLG block is desired and monomer conversion is not monitored, polymerization at 0 °C is advantageous as it prevents end-group termination. ZLL and Ala NCA polymerization also work best at 20 °C under high vacuum. Generally, the reaction at 0 °C should be used if termination reactions occur more frequently, as in BLA, BLS and BLT. For these NCAs the vacuum did not increase the polymerization kinetics and can thus be omitted. It should be noted that reaction control is also drastically affected by the precipitation or formation of secondary structures during the polymerization.+ = high, + = good, − = moderate, −− = low; HV = high vacuum
| NCA | 0 °C, N2 | 0 °C, HV | 20 °C, N2 | 20 °C, HV | Remarks | ||||
|---|---|---|---|---|---|---|---|---|---|
| C | S | C | S | C | S | C | S | ||
| BLG | ++ |
−− |
++ |
− | + | + | + | ++ |
Limited time and DP for 20 °C. Viscous for higher DPs at both temperatures |
| ZLL | ++ |
−− |
++ |
− | + | + | + | ++ |
Minor formation of cyclics under all conditions |
| Ala | ++ |
−− |
++ |
− | + | + | + | ++ |
Precipitation at both temperatures |
| BLA | ++ |
− | ++ |
− | −− |
+ | −− |
+ | Precipitation at both temperatures |
| BLS | ++ |
− | ++ |
− | −− |
+ | −− |
+ | Precipitation at both temperatures |
| BLT | ++ |
−− |
++ |
−− |
−− |
− | −− |
− | Extremely slow |
The optimized polymerization conditions obtained from the homo polymerizations were validated by performing the synthesis of a tetrablock copolymer PBLG80-b-PAla25-b-PZLL80-b-PBLA40 (Table 3). The monomer conversion was followed by FTIR spectroscopy, and individual NCA monomers were added to the polymerization solution consecutively when full conversion was reached of the previous NCA monomer without intermediate work-up. The polymerization was started with the synthesis of the well-soluble PBLG block at 20 °C under high vacuum. After three hours, full monomer conversion was reached, and for a sample withdrawn from the reaction solution the Mn of the PBLG block was determined to be 15700 g mol−1 with a PDI of 1.06 (entry 1A, Table 3). Ala NCA was then directly added to the polymerization solution and the reaction continued for 90 min at 20 °C under high vacuum. For the PAla block the number of repeating units was kept low (25) to maintain the solubility of the block copolymer for the subsequent monomer addition of the ZLL NCA. Consequently, only a small increase of the molecular weight to 18400 g mol−1 (PDI: 1.07) was obtained (entry 1B, Table 3). From Fig. 7 it can be seen that a full shift of the SEC trace to higher molecular weight was achieved from the PBLG macroinitiator to the diblock copolymer. Without further work-up ZLL NCA was added to the reaction and polymerized at 20 °C under high vacuum for 3.5 hours. A larger block of 80 monomer units was aimed for in order to obtain a significant increase in the molecular weight. Indeed, the SEC trace of the triblock copolymer shows a correspondingly significant shift to higher molecular weight as compared to the diblock copolymer macroinitiator. The trace remains narrow (PDI 1.14) and a total molecular weight of 34700 g mol−1 was determined (entry 1C, Table 3). However, the trace also reveals the presence of a small amount of remaining macroinitiator, suggesting a non-quantitative initiation. FTIR of the amide II bonds shows the presence of high α-helix content, without the presence of β-sheets (Fig. S23†). The incomplete macroinitiation is therefore attributed to the poor solubility of the PAla block. Finally, a PBLA block was polymerized at 0 °C over a much longer time scale of 168 h due to progressively lowered concentration of the initiating amine end group with every monomer addition and lower reaction temperature. The reaction for the last block did not reach full conversion. The entire procedure yielded the final tetrablock copolymer with a total Mn of 36400 g mol−1 and a PDI of 1.30 (entry 1D, Table 3). Other tri- and tetrablock copolymers with similar combinations were attempted using the same conditions for the blocks of 1A to 1D with similar results. Although this approach pushes the limits of block copolymer synthesis by NCA polymerization, the applied reaction conditions resulted in a fast and very controlled synthesis.
| Entry | Polymer | Conditions | RT/h | M n a/g mol−1 | M w a/g mol−1 | PDIa |
|---|---|---|---|---|---|---|
| a DMAc SEC calibrated with polystyrene standards. b Reactions conditions for entries 2 to 4 were similar as described for the blocks 1A to 1D. | ||||||
| 1A | PBLG80 | 20 °C, HV | 3 | 15700 |
16600 |
1.06 |
| 1B | PBLG80-b-PAla25 | 20 °C, HV | 1.5 | 18400 |
19700 |
1.07 |
| 1C | PBLG80-b-PAla25-b-PZLL80 | 20 °C, HV | 3.5 | 34700 |
41900 |
1.14 |
| 1D | PBLG80-b-PAla25-b-PZLL80-b-PBLA40 | 0 °C, N2 | 168 | 36400 |
47300 |
1.30 |
| 2b | PBLG100-b-PZLL100-b-PAla20 | 20 °C, HV | 72 | 47100 |
54400 |
1.15 |
| 3 | PBLG50-b-PAla15-b-PZLL50-b-PBLA40 | 20 °C, HV/0 °C, N2 | 96 | 30700 |
36400 |
1.19 |
| 4 | PBLG100-b-PAla15-b-PZLL100-b-PBLA40 | 20 °C, HV/0 °C, N2 | 168 | 47500 |
51100 |
1.31 |
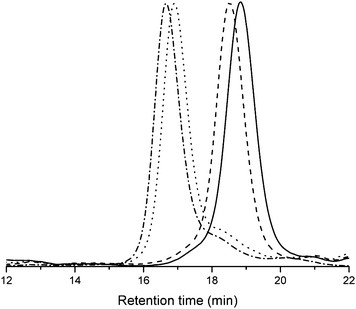 | ||
| Fig. 7 DMAc-SEC results of PBLG80-b-PAla25-b-PZLL80-b-PBLA40 tetrablock copolymerization. Traces represent the sequentially extended block copolymers from right to left (entries 1A to 1D in Table 3). | ||
Conclusions
The polymerization of several NCA monomers was investigated to determine the optimal reaction conditions with respect to the temperature and the pressure in DMF. The studied NCAs can be divided into two groups: in the first group monomers of BLG, ZLL and Ala polymerized considerably faster when a lower pressure of 1 × 10−5 bar was applied. MALDI-ToF-MS analysis confirmed that the formation of side products for these monomers mostly started after full monomer conversion. This was particularly prominent for the pyroglutamate formation in PBLG. The second group of monomers, i.e. BLA, BLS and BLT, polymerized considerably slower than the first group and no effect was observed from the lower pressure. On the other hand, the number of side reactions was significant at 20 °C, so that the polymerizations for the latter monomers should preferably be done at 0 °C. The temperature and pressure can thus be used to balance polymerization kinetics and structural control in NCA polymerizations. However, monomer conversion is an important parameter in the reaction control. The information obtained in this study was then applied for the synthesis of tetrablock copolymer with low polydispersity and high structural control comprising PBLG, PAla, PZLL and PBLA blocks by combining different reaction conditions. The combination of the parameters of temperature and pressure proved to be useful for a more efficient controlled ring-opening polymerization of NCAs.Acknowledgements
This work forms part of the research program of the Dutch Polymer Institute (DPI, project number 610). AH is a SFI Stokes Senior Lecturer (07/SK/B1241).References
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS
.
- N. Hadjichristidis, H. Iatrou, M. Pitskalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528–5578 CrossRef CAS
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS
- H. Iatrou, H. Frielinghaus, S. Hanski, N. Ferderigos, J. Ruokolainen, O. Ikkala, D. Richter, J. Mays and N. Hadjichristidis, Biomacromolecules, 2007, 8, 2173–2181 CrossRef CAS
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS
- M. I. Gibson and N. Cameron, Angew. Chem., Int. Ed., 2008, 47, 5160–5162 CrossRef CAS
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS
- H.-A. Klok and S. Lecommandoux, Adv. Polym. Sci., 2006, 202, 75–111 CAS
- R. J. I. Knoop, G. J. M. Habraken, N. Gogibus, S. Steig, H. Menzel, C. E. Koning and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3068–3077 CrossRef CAS
- H. Schlaad, Adv. Polym. Sci., 2006, 202, 53–73 CAS
- S. Steig, F. Cornelius, P. Witte, C. E. Koning, A. Heise and H. Menzel, Chem. Commun., 2005, 5420–5422 RSC
- F. Audouin, R. J. I. Knoop, J. Huang and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 6402–6410
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS
- H. Lu and J. J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221–1224 CrossRef CAS
- W. Vayaboury, O. Giani, H. Cottet, S. Bonaric and F. Schué, Macromol. Chem. Phys., 2008, 209, 1628–1637 CrossRef CAS
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS
-
H. R. Kricheldorf, α-Aminoacid-N-Carboxyanhydrides and Related Heterocycles, Springer-Verlag, Berlin, 1987 Search PubMed
- J. Muzart, Tetrahedron, 2009, 65, 8313–8323 CrossRef CAS
- D. L. Pickel, N. Politakos, A. Avgeropoulos and J. M. Messman, Macromolecules, 2009, 42, 7781–7788 CrossRef CAS
- G. J. M. Habraken, M. Peeters, C. H. J. T. Dietz, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 514–524 RSC
- N. M. B. Smeets, P. L. J. van der Weide, J. Meuldijk, J. A. J. M. Vekemans and L. A. Hulshof, Org. Process Res. Dev., 2005, 9, 757–763 Search PubMed
- F. Corneille, J.-L. Copier, J.-P. Senet, Y. Robin, EP 1201659, 2002.
- M. I. Gibson and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2882–2891 CrossRef CAS
- B. Brulc, E. Žagar, M. Gadzinowski, S. Słomkowski and M. Žigon, Macromol. Chem. Phys., 2011, 212, 550–562 CrossRef CAS
- A. Teramoto and H. Fujita, Adv. Polym. Sci., 1975, 18, 68–147
- H. R. Kricheldorf, M. Mutter, F. Maser, D. Müller and H. Förster, Biopolymers, 1983, 22, 1357–1372 CrossRef CAS
- J. Ling and Y. Huang, Macromol. Chem. Phys., 2010, 211, 1708–1711 CrossRef CAS
- H. R. Kricheldorf, C. V. Lossow and G. Schwarz, Macromol. Chem. Phys., 2004, 205, 918–924 CrossRef CAS
- H. R. Kricheldorf, C. v. Lossow and G. Schwarz, Macromol. Chem. Phys., 2005, 206, 282–290 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Conversion, molecular weight and kinetic plots of all monomers, MALDI-ToF, FTIR spectra of homopolymers and SEC plots of block copolymers. See DOI: 10.1039/c1py00079a |
| This journal is © The Royal Society of Chemistry 2011 |