Synthesis, characterization, fluorescence labeling and cellular internalization of novel amine-functionalized poly(ethylene glycol)-block-poly(ε-caprolactone) amphiphilic block copolymers
Jinliang
Yan
ab,
Zhaoyang
Ye
*a,
Houyong
Luo
a,
Min
Chen
a,
Yan
Zhou
a,
Wensong
Tan
a,
Yan
Xiao
b,
Yan
Zhang
b and
Meidong
Lang
*ab
aState Key Laboratory of Bioreactor Engineering, School of Bioengineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: zhaoyangye@ecust.edu.cn
bKey Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: mdlang@ecust.edu.cn
First published on 12th April 2011
Abstract
We report in this paper a facile way to prepare novel amine-functionalized monomethoxy-poly(ethylene glycol)-b-poly(ε-caprolactone) (mPEG-b-PCL) amphiphilic block copolymers, which are subsequently fluorescently labeled. In our synthetic route, monomethoxy-poly(ethylene glycol)-b-poly[ε-caprolactone-co-γ-(carbamic acid benzyl ester)-ε-caprolactone] [mPEG-b-P(CL-co-CABCL)] copolymers were synthesized viaring-opening polymerization (ROP) of ε-caprolactone (CL) and a newly developed monomer, γ-(carbamic acid benzyl ester)-ε-caprolactone (CABCL) at varied ratios using mPEG as macroinitiator and Sn(Oct)2 as catalyst. Subsequent deprotection upon removal of carbobenzoxy (Cbz) group yielded monomethoxy-poly(ethylene glycol)-b-poly(ε-caprolactone-co-γ-amino-ε-caprolactone) [mPEG-b-P(CL-co-ACL)] copolymers bearing primary amine functional groups on the PCL block. The structures of polymers were characterized with NMR, FT-IR and GPC techniques. These amphiphilic block copolymers self-assembled into micelles in aqueous solution and the critical micelle concentration (CMC) was dependent on the compositions of the copolymers. In addition, the particle size and morphology of micelles were studied with DLS and TEM, respectively. In vitro study demonstrated that the micelles were nontoxic to human fibroblasts based on MTT and live/dead assays. Furthermore, a proof-of-concept usage of amino groups for bioconjugation was illustrated by tagging the copolymer with a fluorophore, fluorescein isothiocyanate (FITC). Internalization of FITC-labeled micelles by fibroblast cells was observed under fluorescence microscopy. Through facile conjugation of chemical moieties such as drugs, peptides, proteins or fluorophores, micelles prepared with these amine-functionalized mPEG-b-PCL copolymers hold great promise in biomedical applications.
Introduction
In recent years, synthetic amphiphilic block copolymers composed of poly(ethylene glycol) (PEG) and poly(ε-caprolactone) (PCL) blocks have received great attention in biomedical applications.1–3 PEG itself possesses many unique properties such as great hydrophilicity, miscibility with diverse solvents, biocompatibility, low protein adsorption and cell non-adhesive properties.2,4 On the other hand, PCL is hydrophobic and also attractive in biomedical research due to biocompatibility, degradability and good mechanical properties.5–7 Upon integrating PEG and PCL, PEG-b-PCL amphiphilic copolymer-based micelles have demonstrated many advantageous properties, such as nanoscale size, core–shell structure, thermodynamic stability, biodegradability and high biocompatibility.2,3Currently emerging needs such as targeted drug delivery and in situ real-time tracking in biomedical fields call for micelles bearing functionalities which allow for sophisticated tasks in addition to drug incorporation.8 To date, polymeric micelles exhibiting environmental sensitivity (i.e. pH sensitivity, temperature sensitivity, magnetic sensitivity, electric sensitivity, etc.),9,10 targeting specificity,11,12 electrical charging13,14 and fluorescent labeling15,16 have been developed.
While bioconjugation represents an efficient means for functionalizing micelles,9,13,17 functionalization of PEG-b-PCL copolymers remains challenging due to the absence of reactive groups since only end hydroxyl groups are available for both PEG and PCL blocks.18 Most of studies have concentrated on modifying the end hydroxyl group of PCL block in PEG-b-PCL copolymers. For example, a targeting ligand, folic acid, was conjugated with PCL block following the conversion of end hydroxyl group of PCL into amino group. Such a modification improved the tumor targeting efficacy of the paclitaxel-loaded micelles.19 In another study, anticancer drug docetaxel (DTX) was coupled to the end of hydrophobic block of PEG-b-PCL copolymers, which resulted in higher DTX loading than that of PEG-b-PCL.20 On the other hand, fluorescent technology has greatly facilitated studying of biological processes.21,22 Through bioconjugation of fluorescent probes onto micelles prepared from PEG-b-PCL copolymers, solid evidence demonstrating endocytic internalization of micelles by cells has been documented.16,23 However, functionalization through end hydroxyl group is generally not favorable due to both limited number and restricted chemistries.24 Introduction of pendant functional groups to the PCL block in PEG-b-PCL copolymers represents an attractive strategy in conferring both sufficient and diverse reactive groups for bioconjugation.25,26 To the best of our knowledge, pendant functionalization of PCL block in PEG-b-PCL copolymers has been reported in few studies. Lavasanifar et al. synthesized mPEG-b-PCL copolymers bearing carboxyl groups on PCL chain.18 This carboxyl-functionalized copolymer permitted direct conjugation of a large amount of doxorubicin (DOX) molecules onto the core (i.e.PCL block) of PEG-b-PCL micelles, eventually leading to improved DOX loading capacity and release profile.27 In another study from the same group, amine side chains were attached onto the PCL block via these carboxylic groups, which enabled the complex formation with siRNA for gene delivery.13 In fact, many other different moieties have been also successfully conjugated onto PCL block through this carboxylic modification, conferring micelles with diverse properties.28–31 These studies prove that the pendant derivation of PCL block is advantageous in advancing the applications of PEG-b-PCL-based micelles.
Our lab has made great efforts in functionalizing PCL polymers for biomedical uses.32–36 Recently, we successfully synthesized a novel monomer, γ-(carbamic acid benzyl ester)-ε-caprolactone (CABCL).33 As a continued study to develop functional amphiphilic copolymers for preparing micelles, in the present paper, we described the synthesis of a series of mPEG-b-P(CL-co-CABCL) copolymers. Followed by removing the protective carbobenzoxy (Cbz) groups, pendant amine groups were generated, conferring amine-bearing PEG-b-PCL copolymers. The chemical structures and self-assembling properties of these block copolymers were characterized. Derivatization through the amine groups was demonstrated by covalently coupling with a model fluorophore, fluorescein isothiocyanate (FITC). In addition, the cytotoxicity and cellular uptake of the prepared micelles were studied on human dermal fibroblast cells. Our study illustrated an efficient methodology to introduce functional amine groups onto the core structure of PEG-b-PCL micelles, which will definitely broaden the utility of PEG-b-PCL micelles.
Experimental section
Materials
ε-Caprolactone (CL, Acros Organics, 99%) was dried over calcium hydride at room temperature for 48 h and distilled under reduced pressure just before use. Monomethoxy poly(ethylene glycol) (mPEG, Mn = 2000 Da) was purchased from Aldrich, further dried viaazeotropic distillation with anhydrous toluene and stored in a desiccator prior to use. The monomer CABCL was synthesized and purified according to the literature.33 Dulbecco's Modified Eagle's Medium (DMEM) was purchased from Gibco. FITC and 33 wt% HBr solution in glacial acetic acid were obtained from Acros Organics. Stannous octoate [Sn(Oct)2], 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Calcein AM and propidium iodide (PI) were purchased from Sigma Chemical Co. All other solvents and reagents were used as received.Measurements
NMR spectra were recorded on a Bruker AV 400M spectrometer at 25 °C. Deuterated chloroform and DMSO-d6 were used as the solvent for NMR measurements. The molecular weights of the obtained polymers were determined by gel permeation chromatography (GPC) on Waters 1515 GPC system with THF as eluent (flow rate: 1 mL min−1; 35 °C). Monodispersed polystyrene standards were used to generate the calibration curve. FT-IR spectra were recorded on a Nicolet 5700 instrument at frequencies ranging from 500 to 4000 cm−1. The copolymer samples were dissolved in chloroform and a film was cast on a KBr pellet by evaporation of the solvent.Synthesis of mPEG-b-P(CL-co-CABCL) block copolymers
mPEG-b-P(CL-co-CABCL) block copolymers were prepared by ring-opening polymerization (ROP) of CL and CABCL using mPEG as macroinitiator and Sn(Oct)2 as catalyst. In a typical reaction, mPEG (0.2 g, 0.1 mmol), CABCL (0.2 g, 0.76 mmol), and CL (0.8 g, 7 mmol) were added into a polymerization flask, which was initially fresh flamed and argon-purged for several times. The flask was placed in an oil bath at 50 °C with vigorous stirring until the mixture became homogeneous and clear. A catalytic amount of Sn(Oct)2 in dry toluene was added via a microlitre syringe. The flask was then hooked to a Schlenk line and an exhausting-refilling process was repeated several times to remove the trace toluene. Finally, the flask was sealed in a vacuum by flame-heating and immersed in a constant temperature oil bath at 130 °C for 24 h. The polymerization was quenched by immersing the flask in a cold water bath. The product was dissolved in a small amount of chloroform, precipitated in cold diethyl ether and isolated by filtration. The copolymer was dried under vacuum at room temperature until a constant weight was reached. Similarly, a series of copolymers were prepared by varying the feed ratios of CL to CABCL.Synthesis of mPEG-b-P(CL-co-ACL)
In a typical reaction, mPEG-b-P(CL-co-CABCL) (0.5 g) was dissolved in a 10 mL mixture of TFA/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, v/v), and a 4-fold molar excess of a 33 wt% solution of HBr in glacial acetic acid was added. The solution was stirred vigorously under argon for 1 h at 0 °C. Then, the yellow solution was concentrated under reduced pressure to remove the solvent. Finally, the residue was dissolved in a small amount of CHCl3 and precipitated with an excess of cold diethyl ether. The obtained yellow copolymer was dissolved in 5 mL THF and treated with 3 mL triethylamine (TEA) overnight at room temperature. The reaction mixture was filtered and the filtrate was dropped into a large volume of diethyl ether to precipitate. The white deprotected copolymer mPEG-b-P(CL-co-ACL), with free pendant amine groups was dried under vacuum at room temperature for 24 h.
:9, v/v), and a 4-fold molar excess of a 33 wt% solution of HBr in glacial acetic acid was added. The solution was stirred vigorously under argon for 1 h at 0 °C. Then, the yellow solution was concentrated under reduced pressure to remove the solvent. Finally, the residue was dissolved in a small amount of CHCl3 and precipitated with an excess of cold diethyl ether. The obtained yellow copolymer was dissolved in 5 mL THF and treated with 3 mL triethylamine (TEA) overnight at room temperature. The reaction mixture was filtered and the filtrate was dropped into a large volume of diethyl ether to precipitate. The white deprotected copolymer mPEG-b-P(CL-co-ACL), with free pendant amine groups was dried under vacuum at room temperature for 24 h.
Synthesis of FITC-labeled mPEG-b-P(CL-co-ACL)
mPEG-b-P(CL-co-ACL) was fluorescently labeled with FITC. Briefly, mPEG-b-P(CL-co-ACL) (50 mg), FITC (35 mg), and TEA (500 μL) were dissolved in 10 mL of dry DMF. The reaction mixture was then stirred at room temperature in dark for 24 h. The solution was concentrated, precipitated into an excess of cold ethanol and filtrated. The precipitate was dissolved in DMF and dialyzed (MWCO = 3500 Da) against distilled water to remove residual unreacted FITC (distilled water was refreshed every 12 h). The content of FITC in dialysate was monitored with a UV-vis spectrophotometer (UV-vis 8500 Techcomp, China) at an emission wavelength of 490 nm. The FITC-labeled copolymer was recovered by freeze-drying. The color of the copolymer changed from white to orange because of the conjugation of FITC.Micelle preparation and characterization
Micelles were prepared using the solvent evaporation method. Briefly, 20 mg of copolymer was dissolved in 2 mL of THF in a glass beaker. Under gentle stirring, 20 mL of double-distilled water was added drop-wise to the solution using a syringe. The mixture was left stirring overnight, allowing slow evaporation of THF. The residual THF was completely removed by dialysis against water for 24 h. To obtain micelle powders, the micelle solutions were frozen and lyophilized.CMCs of the copolymers were measured by a fluorescence method using pyrene as a probe. A predetermined amount of pyrene in acetone was added to volumetric flasks and the acetone was evaporated completely. The micelle solutions of copolymers with different concentrations were then added into these volumetric flasks, while the concentration of pyrene in each flask was maintained at 6.0 × 10−7 mol L−1. Then, the flasks were incubated overnight at 25 °C to equilibrate the pyrene in the micelles. The excitation spectra were recorded on a Fluorolog fluorescene spectrophotometer (Horiba Jobin Yvon, Inc.) with λem at 390 nm and slit width of 3 nm. The CMC value was calculated from the intersection of two tangent plots of intensity ratio I338/I333 as a function of the logarithm of polymer concentration.
Particle size and size distribution of micelles in aqueous solution were measured by dynamic light scattering (DLS) on a Malvern Zetasizer Nano (Malvern Instruments, Ltd., U.K.). The micellar solution was filtered through a 0.45 μm Millipore filter prior to analysis. The measurement was performed at a scattering angle of 90° at 25 °C. The sample concentration was maintained at 1.0 mg mL−1. All the measurements were repeated three times, and the values reported were the mean diameter ± standard deviation.
The zeta potential of mPEG-b-P(CL-co-ACL) micelles (Table 2, M4) (1 mg mL−1) was measured on a Zeta Potential/Particle Sizer Nicomp 380 ZLS (Santa Barbara, USA) at different pHs (2.0, 5.0, 7.0, 9.0 and 12.0). The pH was adjusted with NaOH (0.1 M) and HCl (0.1 M). The results were the mean of at least 3 measurements.
The morphology of micelles was observed on a TEM instrument (JEOL/JEM-2000EXII) operated at an accelerating voltage of 60 kV. A drop of micelle solution (1 mg mL−1) was deposited on a copper grid coated with carbon film and dried at ambient temperature.
Cell experiments
Results and discussion
Synthesis of mPEG-b-P(CL-co-CABCL)
With the objective to develop PEG-b-PCL containing functional reactive amine groups for preparing functional micelles, we utilized a newly developed monomer in our lab, namely CABCL.33 This monomer contained a protected amine group at the γ position of CL, providing the possibility of introducing pendant amine groups onto PCL backbone through copolymerization with CL. The procedure for the synthesis of CABCL monomer was shown in Scheme 1A. Firstly, benzyl chloroformate was used to protect the amino group of trans-4-aminocyclohexanol. Secondly, the hydroxyl group was oxidized into carbonyl group using Jones' reagent. Finally, the ketone was converted to the corresponding lactone through a Bayer-Villiger oxidation. The product in each step could be easily purified by recrystallization with good yield. The chemical structure of the obtained CABCL was confirmed by NMR (data not shown).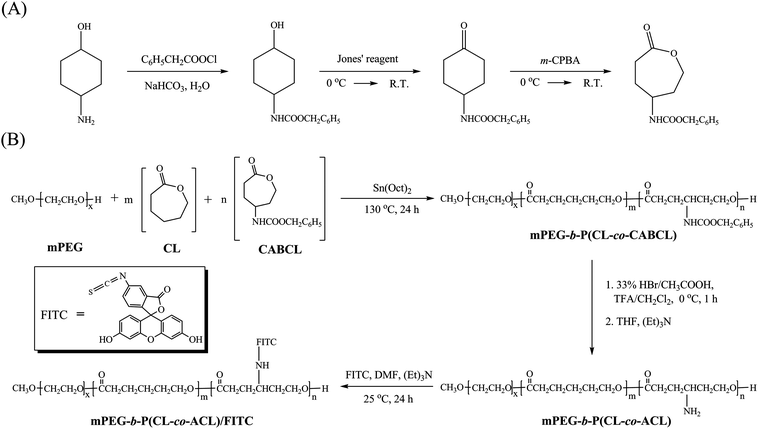 | ||
| Scheme 1 Synthetic routes for monomer γ-(carbamic acid benzyl ester)-ε-caprolactone (CABCL, γ-carbon-substituted ε-caprolactone monomer) (A) and block copolymers mPEG-b-P(CL-co-CABCL), mPEG-b-P(CL-co-ACL) as well as FITC-labeled mPEG-b-P(CL-co-ACL) (B). | ||
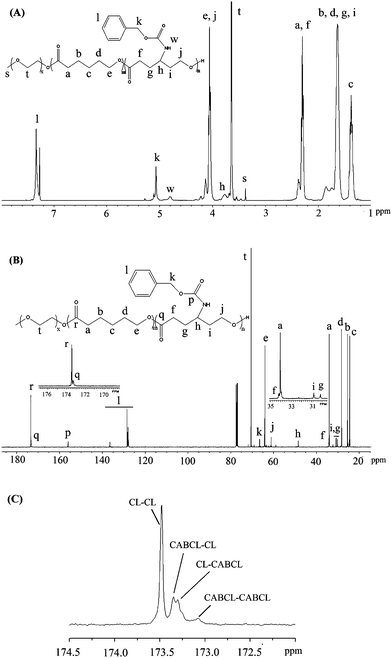
In our previous report, we demonstrated the successful copolymerization of CL with CABCL and yielded amine functionalized PCL.33 In the present work, we explored the feasibility of synthesizing novel amine-functionalized PEG-b-PCL-based amphiphilic block copolymers. As shown in Scheme 1B, mPEG-b-P(CL-co-CABCL) block copolymers, were synthesized through ROP of CL and CABCL using mPEG as macroinitiator and Sn(Oct)2 as catalyst in bulk at 130 °C. The structure of the copolymers was characterized by NMR. Fig. 1A illustrated a typical 1H NMR spectrum of a prepared copolymer (Table 1, entry 3). The hydrogen signals corresponding to EG, CL and CABCL repeating units were indicated, confirming the successful synthesis of mPEG-b-P(CL-co-CABCL). The 13C NMR spectrum of the copolymer (Table 1, entry 3) was also shown in Fig. 1B. In addition, an expanded spectrum corresponding to the ester carbonyl groups (172.5–174.5 ppm) (Fig. 1C) provided detailed information about the sequence distribution within the P(CL-co-CABCL) block of the copolymer. There were four resonance peaks corresponding to CL–CL, CABCL–CL, CL–CABCL, and CABCL–CABCL, respectively, which suggested that CL and CABCL were randomly distributed in the P(CL-co-CABCL) block.37 Moreover, Fig. 2 showed typical GPC traces of the starting macroinitiator (mPEG, Mn = 2000 Da) (I) and mPEG-b-P(CL-co-CABCL) (Table 1, entry 3) (II), which further confirmed the successful copolymerization.
| Entry | mol % of CABCL | mPEG-b-P(CL-co-CABCL) | mPEG-b-P(CL-co-ACL) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| In feedb | In polymerd | M n1 c | M n1 d | M n e | PDIe | Yield (%) | M n2 c | M n2 d | Yield (%) | |
| a Polymerization conditions: bulk, 130 °C, 24 h. The molar ratio of Sn(Oct)2 to monomer (CL + CABCL) was 1/1000. b f CABCL = (CABCL)/(CL + CABCL) (mol/mol). c Theoretical number-average molecular weight: Mn1 = 2000 × [1 + (WCL + WCABCL)/WmPEG]; Mn2 = Mn1c − n(MCbz − 1), where MCbz was the molecular weight of Cbz and n is the number of Cbz. d Determined by 1H NMR (CDCl3 as the solvent). e Determined by GPC (THF as the eluent). | ||||||||||
| 1 | 6 | 6 | 11 000 | 10 500 | 10 300 | 1.4 | 92 | 9 800 | 9 700 | 80 |
| 2 | 11 | 10.5 | 12 300 | 12 600 | 11 200 | 1.4 | 94 | 11 400 | 11 000 | 78 |
| 3 | 16 | 14.5 | 13 400 | 13 500 | 14 400 | 1.8 | 88 | 11 700 | 11 800 | 84 |
| 4 | 22 | 21.5 | 15 300 | 16 200 | 16 700 | 2.0 | 90 | 13 500 | 12 700 | 82 |
 | ||
| Fig. 1 1H NMR (A) and 13C NMR (B) spectra of mPEG-b-P(CL-co-CABCL) (Table 1, entry 3) in CDCl3 and an expanded 13C NMR spectrum (C) for the carbonyl carbon region (between 172.5–174.5 ppm) showing four resonance peaks corresponding to CL–CL, CABCL–CL, CL–CABCL, and CABCL–CABCL. | ||
 | ||
| Fig. 2 GPC profiles of mPEG with molecular weight of 2000 Da (profile I), and a prepared block copolymer mPEG-b-P(CL-co-CABCL) (profile II) (Table 1, entry 3). | ||
In the present study, for all copolymerization reactions, the molecular weight of mPEG was kept as 2000 Da and the mass ratio of CL/mPEG was kept constant. By varying the feed ratios of CL to CABCL, we prepared four copolymers. Table 1 summarized both the copolymerization conditions and characteristics of the copolymers with the feeding content of CABCL ranging from 6 to 22 mol% [fCABCL = (CABCL)/(CL + CABCL)]. Based on 1H NMR spectrum as shown in Fig. 1A, the copolymer composition was determined by calculating the relative peak areas of the methylene protons (–C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 2–C2–O–) at 3.60–3.68 ppm of the EG repeat units, the phenyl ring protons (–C65) at 7.31–7.40 ppm of the CABCL repeat units, and the methylene protons (–C2–O–) at 1.30–1.43 ppm of the CL units. According to 1H NMR analysis, the molar fractions of the CABCL unit in the copolymers2, 3 and 4 were all slightly lower than the initial feeding ratio (Table 1), which could be ascribed to the lower reactivity of CABCL than that of CL.33 As listed in Table 1, it was also shown that the experimental molecular weights calculated based on 1H NMR spectrum agreed well with the theoretical molecular weights. However, the molecular weights measured by GPC calibrated with poly(styrene) slightly deviated from theoretical values, which was possibly due to the difference in the hydrodynamic volumes of copolymers and poly(styrene) in THF.38 Nonetheless, the broader molecular weight distribution (high PDI values in Table 1) with higher CABCL content was possibly due to greater probability of the inter- or intramolecular transesterification side reactions for the substituted CL (i.e.CABCL).1,39–41
2–C2–O–) at 3.60–3.68 ppm of the EG repeat units, the phenyl ring protons (–C65) at 7.31–7.40 ppm of the CABCL repeat units, and the methylene protons (–C2–O–) at 1.30–1.43 ppm of the CL units. According to 1H NMR analysis, the molar fractions of the CABCL unit in the copolymers2, 3 and 4 were all slightly lower than the initial feeding ratio (Table 1), which could be ascribed to the lower reactivity of CABCL than that of CL.33 As listed in Table 1, it was also shown that the experimental molecular weights calculated based on 1H NMR spectrum agreed well with the theoretical molecular weights. However, the molecular weights measured by GPC calibrated with poly(styrene) slightly deviated from theoretical values, which was possibly due to the difference in the hydrodynamic volumes of copolymers and poly(styrene) in THF.38 Nonetheless, the broader molecular weight distribution (high PDI values in Table 1) with higher CABCL content was possibly due to greater probability of the inter- or intramolecular transesterification side reactions for the substituted CL (i.e.CABCL).1,39–41
Removal of protective groups
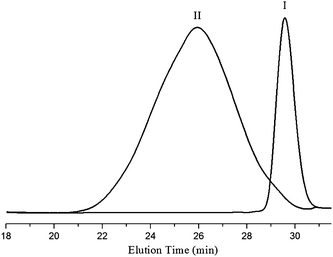
To generate amine groups, the Cbz protecting groups in mPEG-b-P(CL-co-CABCL) copolymers were removed by acidolysis with a 33 wt% solution of HBr solution in glacial acetic acid as described in the literature.42 In comparison with the 1H NMR spectrum of mPEG-b-P(CL-co-CABCL) (Fig. 1A), in the 1H NMR spectrum of the deprotected copolymer mPEG-b-P(CL-co-ACL) (Table 1, entry 3) (Fig. 8B), proton signals at 5.09 ppm (C6H5C2) and 7.29–7.40 ppm (C65CH2) disappeared whereas other signals were little changed. In addition, in the FT-IR spectra as shown in Fig. 3 (copolymers as listed in Table 1, entry 3), the disappearance of νCH vibration at 3064.6 cm−1 and 3033.2 cm−1 and γCH vibration at 742.3 cm−1 and 697.2 cm−1 of the phenyl rings in deprotected copolymer mPEG-b-P(CL-co-ACL) (Fig. 3, II) further confirmed the successful removal of Cbz groups from mPEG-b-P(CL-co-CABCL) (Fig. 3, I). Moreover, the appearance of both a shoulder band in the region of 3300–3000 cm−1 and a new peak of N–H wagging (737.5 cm−1) suggested the presence of amine groups. It is well known that for polymers having cationic groups, GPC is not always accurate for measuring molecular weight due to binding of cationic moieties with column packing materials and the use of polystyrene as calibration standards.14,43,44 Therefore, the molecular weights of the deprotected polymers were determined with NMR (Table 1).45 Notably, the molecular weights of the copolymers after deprotection agreed well with the theoretical values, indicating that the polymeric chain changed minimally under this deprotection condition. 42,46,47
 | ||
| Fig. 3 FT–IR spectra of mPEG-b-P(CL-co-CABCL) (I) and mPEG-b-P(CL-co-ACL) (II) (see Table 1, Entry 3 for more details of these copolymers). A shoulder band and a new peak at 737.5 cm−1 corresponding to N–H wagging in profile II indicated the presence of amine groups, and νCH vibration at 3064.6 cm−1 and 3033.2 cm−1 and γCH vibration at 742.3 cm−1 and 697.2 cm−1 of the phenyl rings in profile I were absent in profile II. | ||
Preparation and characterization of micelles
Amphiphilic block copolymers can self-assemble in aqueous solution to form micellar structures once the concentration is above the CMC.48 CMC is an effective parameter to evaluate the thermodynamic stability of micelles in aqueous solution. In micelles prepared from mPEG-b-PCL amphiphilic copolymers, the PCL segments constitute the hydrophobic core and the PEG segments serve as the hydrophilic shell stabilizing the core.3 CMC is normally determined by the fluorescence inclusion method using pyrene as a fluorescent probe.48 Following the formation of micelles, pyrene molecules preferentially partition into the hydrophobic core of micelles, which induces a red shift in the excitation spectrum of pyrene. According to Wilhelm et al.,49 the abrupt change in intensity ratio of peak at 338 nm to that at 333 nm (I338/I333) from the excitation spectra of pyrene indicates the onset of micellization and provides a quantitative method for determining the CMC.In the present study, all copolymers tested were capable of forming micelles demonstrating reasonable low CMC values (Table 2). Specifically, the CMC values of mPEG-b-P(CL-co-CABCL) copolymers were found to decrease with increasing CABCL content. The CMC decreased from 11.5 × 10−4 mg mL−1 (11.0 × 10−8 mol L−1) to 4.6 × 10−4 mg mL−1 (2.8 × 10−8 mol L−1) when the CABCL content increased from 6 to 22 mol% while maintaining the contents of both PEG and PCL same. Obviously, introduction of CABCL content increased the hydrophobicity of the PCL segment and thus showed a higher tendency for self-association.29 The lower CMC value indicates higher thermodynamic stability of micelles.48 Therefore, adjusting the fraction of CABCL groups in mPEG-b-P(CL-co-CABCL) copolymers could modulate the stability of resultant micelles. The CMCs of the deprotected copolymers were also measured and listed in Table 2. It was noted that the CMC of mPEG-b-P(CL-co-ACL) was much higher than that of respective parent copolymer. This suggested that the introduction of hydrophilic amine groups into hydrophobic PCL block resulted in a lower tendency of self-association of the copolymers, as observed by others, wherein a carboxylic group was introduced onto PCL block in a pendant manner.18Fig. 4A further illustrated typical plots of the intensity ratio I338/I333 from excitation spectraversus log concentrations of the copolymers (Table 1, entry 3) before and after deprotection, and the value of CMC increased from 5.6 × 10−4 mg mL−1 (4.2 × 10−8 mol L−1) to 21.4 × 10−4 mg mL−1 (18.1 × 10−8 mol L−1) after deprotection.
| Micellesa | mPEG-b-P(CL-co-CABCL) | mPEG-b-P(CL-co-ACL) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CMCb | Micellar size | PDId | CMCb | Micellar size (nm)d | PDId | ||||
| (mg mL−1) | (mol L−1)c | (nm)d | (mg mL−1) | (mol L−1)c | Peak I | Peak II | |||
| a Micelles were prepared from copolymers in Table 1 [M1 (entry 1), M2 (entry 2), M3 (entry 3), M4 (entry 4)]. b CMC was estimated by a fluorescence spectroscopic method using pyrene as the fluorescence probe. c M n from 1H NMR was used for calculation. d Average diameter (intensity mean) was determined by DLS measurement. | |||||||||
| M1 | 11.5 × 10−4 | 11.0 × 10−8 | 52.0 ± 0.2 | 0.12 | 18.6 × 10−4 | 19.2 × 10−8 | 101.5 ± 0.5 | — | 0.26 |
| M2 | 8.5 × 10−4 | 6.7 × 10−8 | 74.2 ± 0.5 | 0.27 | 13.8 × 10−4 | 12.5 × 10−8 | 106.0 ± 1.5 | — | 0.28 |
| M3 | 5.6 × 10−4 | 4.2 × 10−8 | 107.5 ± 1.5 | 0.18 | 21.4 × 10−4 | 18.1 × 10−8 | 48.7 ± 1.3 (5.6%) | 216.5 ± 1.5 (94.4%) | 0.33 |
| M4 | 4.6 × 10−4 | 2.8 × 10−8 | 132.5 ± 1.5 | 0.24 | 39.8 × 10−4 | 31.3 × 10−8 | 48.0 ± 2.6 (20.4%) | 241.0 ± 7.0 (79.6%) | 0.42 |
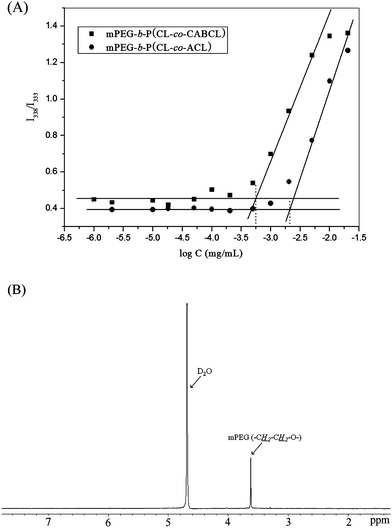 | ||
| Fig. 4 (A) Plots of the intensity ratio (I338/I333) as a function of log concentrations of mPEG-b-P(CL-co-CABCL) and mPEG-b-P(CL-co-ACL) were shown and CMC was obtained by drawing two tangent lines with the x-coordinate of the intersect equal to CMC value of specific copolymer as indicated with a dashed line. (B) 1H NMR spectrum of mPEG-b-P(CL-co-CABCL) copolymer micelles in D2O. (see Table 2, entry M3, before deprotection). | ||
Formation of micelles was also studied by 1H NMR. Fig. 4B showed a typical 1H NMR spectrum of micelles (M3 in Table 2, before deprotection) in D2O, which was prepared by lyophilization of micelles in aqueous solution followed by re-dispersion in D2O as described in a previous report.50 It was clear that signals corresponding to protons of hydrophobic block disappeared, while signals at 3.61 ppm representing protons of mPEG block remained unchanged. This result suggested the formation of core-shell structure of micelles where the hydrophobic block was constrained in the core due to its hydrophobic nature in aqueous environment and the core was surrounded by the solvated mPEG moieties because of their hydrophilicity.
The size and morphology of micelles in aqueous solution were investigated by DLS and TEM, respectively. As summarized in Table 2, for mPEG-b-P(CL-co-CABCL) copolymers, when the CABCL content increased from 6 to 22 mol %, an increase in micellar diameter from 52.0 nm to 132.5 nm was noticed (entry M1 to M4). Generally, amphiphilic copolymers with longer hydrophobic blocks form larger micelle particles.51mPEG-b-P(CL-co-CABCL) with higher CABCL content had longer hydrophobic chain length since the contents of mPEG and CL were kept constant in the present study. However, a bimodal distribution in particle size was observed for the deprotected copolymer micelles (Table 2, entry M3 and M4), which probably was a result of a secondary association of micelles.18 Typical particle size distribution was also shown in Fig. 5A and 5B for the protected and deprotected copolymer micelles (Table 2, entry M4), respectively. It was assumed that the amine groups in the deprotected copolymers would mostly be embedded in the hydrophobic core during the self-assembly process due to the tight association on the backbone of PCL block. As shown in Fig. 6, there was a dependence on pH conditions (between 2.0–12.0) for both Zeta potential and particle size of the micelles (Table 2, entry M4, after deprotection). At low pH (2.0), the Zeta potential of the micelles was slightly positive and an elevation in pH led to a decrease in Zeta potential and a slight increase in particle size from 162.8 nm to 196.0 nm. These results might suggest that some amine groups were possibly located in the core/shell interface or even exposed to the aqueous solution randomly, thus enabling the deprotonation of amine groups upon pH increase. The deprotonation of amine groups resulted in a slight decrease in Zeta potential and also favored hydrogen bonding interaction between amine groups, which possibly caused the secondary association. Lavasanifar et al. also suggested that the presence of carboxyl groups on the core-forming block of PEO-b-PCL induced the secondary association due to the hydrogen bonding interactions between carboxyl groups.18 From TEM images, it could be seen that the micelles (Table 2, entry M4, before deprotection) had spherical morphology with a diameter of around 100 nm for the protected micelles in dry state (Fig. 5C), which was smaller than that obtained by DLS measurement. For the deprotected micelles (Table 2, entry M4, after deprotection), micellar aggregates could be observed (Fig. 5D), which was consistent with the bimodal distribution of micellar size based on DLS measurements.
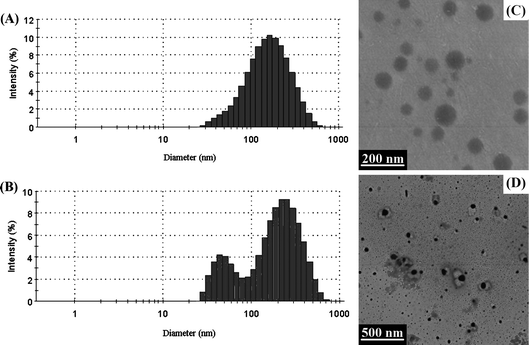 | ||
| Fig. 5 The size distribution determined by DLS (A, B) and TEM images (C, D) of micelles (Table 2, entry M4) prepared with protected copolymer (A, C) and corresponding deprotected copolymer (B, D), respectively. | ||
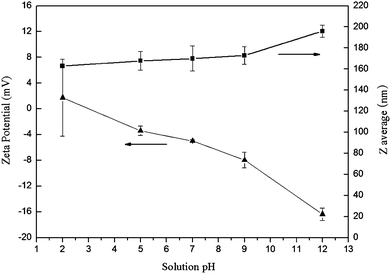 | ||
| Fig. 6 The effects of pH on Zeta potential (mV) and average diameter (Zaverage, nm) of mPEG-b-P(CL-co-ACL) (Table 2, entry M4, 1 mg mL−1). | ||
In vitro cytotoxicity of micelles
Biocompatibility is one of the most important factors for biomedical applications of synthetic polymers.52 In the present study, taking the micelles in Table 2, entry 3 (including micelles prepared with both protected and deprotected polymers) for example, the cytotoxicity against human dermal fibroblast cells was determined by MTT assay. As shown in Fig. 7A, the viability of cells in the presence of the two types of micelles was all around 100% after 24 h treatment with all tested concentrations (1/512–1.0 mg mL−1). Although positive charges on amine groups under physiological conditions can be toxic to cells,53 the non-toxicity of the micelles was possibly due to the fact that most of the amine groups were possibly buried in the micellar core structure or at least at the interface between the hydrophilic corona and hydrophobic core. A live/dead assay was also performed to demonstrate the biocompatibility of micelles prepared with these block copolymers. In the live/dead assay, live cells have intracellular esterases that convert non-fluorescent, cell-permeable calcein AM to an intensely green fluorescent calcein. Cleaved calcein is retained within cells. However, PI can only enter cells with damaged membranes and produce red fluorescence when bound to nucleic acids. As shown in Fig. 7B–E, after both “live” staining with 2 μM calcein-AM and “dead” staining with 2 μM PI, it was observed that almost all fibroblast cells incubated with copolymer micelles (1 mg mL−1) for 24 h remained viable and there were almost no dead cells, which was consistent with the results of MTT assay.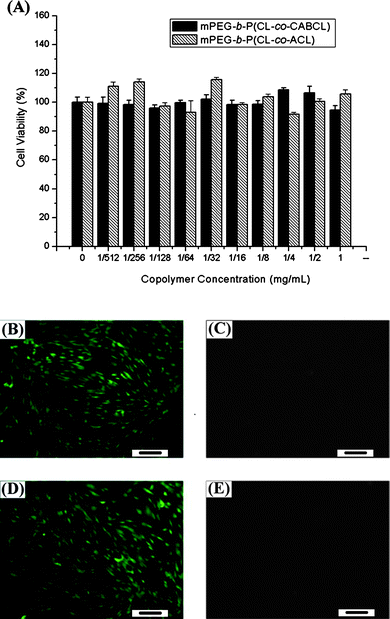 | ||
| Fig. 7 The cytotoxicity of the micelles (Table 2, entry M3) on human fibroblast cells after 24 h of treatment was evaluated with an MTT assay (A). In addition, a live/dead assay was performed to study the viability of cells treated with either protected (B, C) or deprotected (D, E) copolymers (Table 2, entry M3). Cells were observed under fluorescence microscopy with live cells stained green (B, D) and dead cells red (C, E). Scale bar, 100 μm. | ||
Bioconjugation of FITC with mPEG-b-P(CL-co-ACL)
The free pendant amino groups on the copolymer provide a platform for further functionalization via bioconjugation. FITC is a widely used fluorophore in molecular imaging. In the present study, FITC-labeled mPEG-b-PCL was prepared viathiourea bond formation between the reactive amino groups of mPEG-b-P(CL-co-ACL) (Table 1, entry 3) and the isothiocyanate group of FITC as schematically shown in Scheme 1. In order to make sure that FITC was covalently attached to the copolymer rather than physically entrapped, ethanol (a good solvent for FITC) was chosen as the precipitant to remove unconjugated FITC. The residual FITC was further removed by dialysis, and the dialysate was monitored by measuring the absorbance at 490 nm until no absorbance could be observed in dialysate. Furthermore, the characteristic peaks of FITC and mPEG-b-P(CL-co-ACL) could be observed in the 1H NMR spectrum of the mPEG-b-P(CL-co-ACL)/FITC copolymer (Fig. 8A–C), indicating that FITC had been successfully covalently conjugated with the copolymer. In addition, the molar ratio of CL repeating unit to FITC moiety in the copolymer was 7.9 based on the analysis of the 1H NMR spectrum, indicating a nearly 80% substitution of amine groups. Moreover, the UV-vis absorption spectra for FITC and the FITC-labeled copolymer in aqueous solution showed the maximum absorption at 487 nm and 480 nm, respectively, while there was no absorption for mPEG-b-P(CL-co-ACL) (data not shown). The small blue shift (∼7 nm) for the FITC-labeled copolymer could be attributed to both the covalent binding of the FITC molecules on the copolymer and the change of microenvironment after confinement within the micellar structure, thus confirming the successful attachment of FITC onto the copolymer.54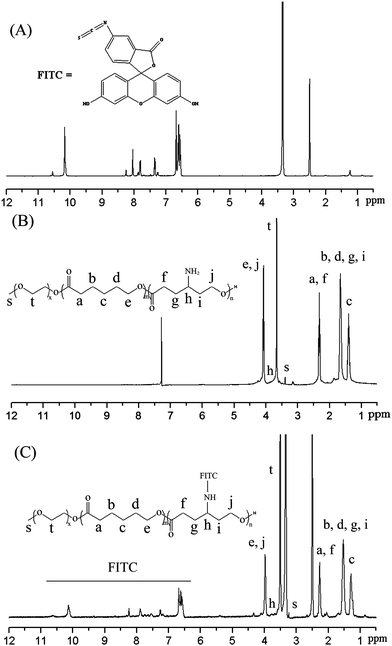 | ||
| Fig. 8 1H NMR spectra of FITC in DMSO-d6 (A), mPEG-b-P(CL-co-ACL) in CDCl3 (B), and mPEG-b-P(CL-co-ACL)/FITC in DMSO-d6 (C). (Table 1, entry 3). | ||
Cellular uptake of FITC-labeled micelles
It is well known that the internalization of synthetic polymer micelles in cells takes place mainly through endocytosis.16,19 We took advantage of FITC labeling (green color) to study the cellular uptake of these micelles by human fibroblasts. Fig. 9 illustrated the fluorescence images of fibroblast cells incubated with the FITC-labeled micelles (prepared from mPEG-b-P(CL-co-ACL) in Table 1, entry 3, Zaverage = 67.5 nm, PDI = 0.061, 0.5 mg mL−1) for 2 h. It clearly demonstrated that these micelles could be efficiently taken up by the cells (Fig. 9A). In addition, the subcellular location of internalized micelles was mainly within the cytoplasm and diffusion into nuclei (counter-stained with PI) was also noticeable (Fig. 9B), resulting from the escape of the micelles from endosome/lysosome structures. These results suggested that these amine-bearing polymeric micelles hold great potential as drug delivery vehicles. Furthermore, with such fluorescence labeling, a real-time surveillance of the intracellular trafficking of drug-loaded micelles can be easily implemented.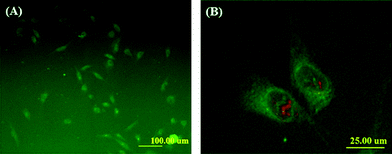 | ||
| Fig. 9 Cellular uptake of FITC-labeled micelles (FITC-labeled copolymers were derived from mPEG-b-P(CL-co-ACL) as in Table 1, entry 3). Human fibroblast cells were incubated for 2 h with the micelles at a concentration of 0.5 mg mL−1 and then were observed under fluorescence microscopy with 100 × magnification (A). In addition, an expanded view was also displayed (B), where the green color is FITC-labeled copolymers/micelles and the red color is cell nuclei. | ||
Conclusions
In summary, a family of novel amphiphilic (mPEG-b-PCL)-based block copolymers bearing functional carbobenzoxy and amine groups on PCL block have been successfully synthesized through ROP. The copolymer structures were investigated in detail using NMR, GPC and FT-IR measurements. The self-assembling of these block copolymers into micellar structures was also confirmed. The amine-functionalized copolymers were amenable to bioconjugation chemistry, e.g., FITC labeling. In vitro studies revealed that these micelles were biocompatible and could be internalized by fibroblast cells. Collectively, these amphiphilic copolymers were believed to be of great potential in biomedical and pharmaceutical applications.Acknowledgements
This research was supported by the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of China (20804015, 31000424), Specialized Research Fund for the Doctoral Program of Higher Education (200802511021, 20100074120009), Shanghai Pujiang Program (10PJ1402200), “Shu Guang” Project of Shanghai Municipal Education Commission, the Natural Science Foundation of Shanghai (08ZR1406000) and Program for Changjiang Scholars and Innovative Research Team in University (IRT0825).References
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- X. Wei, C. Gong, M. Gou, S. Fu, Q. Guo, S. Shi, F. Luo, G. Guo, L. Qiu and Z. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS.
- M. A. Petersen, L. G. Yin, E. Kokkoli and M. A. Hillmyer, Polym. Chem., 2010, 1, 1281–1290 RSC.
- S. Theiler, M. Teske, H. Keul, K. Sternberg and M. Moller, Polym. Chem., 2010, 1, 1215–1225 RSC.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- P. F. Gou, W. P. Zhu and Z. Q. Shen, Polym. Chem., 2010, 1, 1205–1214 RSC.
- C. K. Huang, C. L. Lo, H. H. Chen and G. H. Hsiue, Adv. Funct. Mater., 2007, 17, 2291–2297 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- F. Suriano, R. Pratt, J. P. K. Tan, N. Wiradharma, A. Nelson, Y. Y. Yang, P. Dubois and J. L. Hedrick, Biomaterials, 2010, 31, 2637–2645 CrossRef CAS.
- D. Q. Wu, B. Lu, C. Chang, C. S. Chen, T. Wang, Y. Y. Zhang, S. X. Cheng, X. J. Jiang, X. Z. Zhang and R. X. Zhuo, Biomaterials, 2009, 30, 1363–1371 CrossRef CAS.
- X. B. Xiong, H. Uludag and A. Lavasanifar, Biomaterials, 2009, 30, 242–253 CrossRef CAS.
- W. L. Zhang, J. L. He, Z. Liu, P. H. Ni and X. L. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1079–1091 CrossRef CAS.
- S. Parimi, T. J. Barnes, D. F. Callen and C. A. Prestidge, Biomacromolecules, 2010, 11, 382–389 CrossRef CAS.
- L. B. Luo, J. Tam, D. Maysinger and A. Eisenberg, Bioconjugate Chem., 2002, 13, 1259–1265 CrossRef CAS.
- X. L. Hu, S. Liu, Y. B. Huang, X. S. Chen and X. B. Jing, Biomacromolecules, 2010, 11, 2094–2102 CrossRef CAS.
- A. Mahmud, X. B. Xiong and A. Lavasanifar, Macromolecules, 2006, 39, 9419–9428 CrossRef CAS.
- E. K. Park, S. Y. Kim, S. B. Lee and Y. M. Lee, J. Controlled Release, 2005, 109, 158–168 CrossRef CAS.
- A. S. Mikhail and C. Allen, Biomacromolecules, 2010, 11, 1273–1280 CrossRef CAS.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS.
- P. Watson, A. T. Jones and D. J. Stephens, Adv. Drug Delivery Rev., 2005, 57, 43–61 CrossRef CAS.
- R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS.
- Z. G. Xie, H. L. Guan, C. H. Lu, X. S. Chen and X. B. Jing, Acta Biomater., 2005, 1, 635–641 CrossRef.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- V. Darcos, S. El Habnouni, B. Nottelet, A. El Ghzaoui and J. Coudane, Polym. Chem., 2010, 1, 280–282 RSC.
- A. Mahmud, X. B. Xiong and A. Lavasanifar, Eur. J. Pharm. Biopharm., 2008, 69, 923–934 CrossRef CAS.
- M. Shahin and A. Lavasanifar, Int. J. Pharm., 2010, 389, 213–222 CrossRef CAS.
- A. Falamarzian and A. Lavasanifar, Macromol. Biosci., 2010, 10, 648–656 CrossRef CAS.
- A. Falamarzian and A. Lavasanifar, Colloids Surf., B, 2010, 81, 313–320 CrossRef CAS.
- A. Mahmud, S. Patel, O. Molavi, P. Choi, J. Samuel and A. Lavasanifar, Biomacromolecules, 2009, 10, 471–478 CrossRef CAS.
- Y. Y. He, Y. Zhang, C. H. Gu, W. F. Dai and M. D. Lang, J. Mater. Sci.: Mater. Med., 2010, 21, 567–574 CrossRef CAS.
- J. L. Yan, Y. Zhang, Y. Xiao, Y. Zhang and M. D. Lang, React. Funct. Polym., 2010, 70, 400–407 CrossRef CAS.
- W. F. Dai, J. Y. Zhu, A. Y. Shangguan and M. D. Lang, Eur. Polym. J., 2009, 45, 1659–1667 CrossRef CAS.
- J. S. Li, Y. Zhang, J. Chen, C. H. Wang and M. D. Lang, J. Macromol. Sci., Part A: Pure Appl. Chem., 2009, 46, 1103–1113 Search PubMed.
- W. F. Dai, H. Huang, Z. Z. Du and M. D. Lang, Polym. Degrad. Stab., 2008, 93, 2089–2095 CrossRef CAS.
- D. Mecerreyes, B. Atthoff, K. A. Boduch, M. Trollsas and J. L. Hedrick, Macromolecules, 1999, 32, 5175–5182 CrossRef.
- S. El Habnouni, V. Darcos and J. Coudane, Macromol. Rapid Commun., 2009, 30, 165–169 CrossRef CAS.
- Y. C. Wang, S. Y. Shen, Q. P. Wu, D. P. Chen, J. Wang, G. Steinhoff and N. Ma, Macromolecules, 2006, 39, 8992–8998 CrossRef CAS.
- A. Mahapatro, A. Kumar and R. A. Gross, Biomacromolecules, 2004, 5, 62–68 CrossRef CAS.
- K. M. Stridsberg, M. Ryner and A. C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CAS.
- S. Blanquer, J. Tailhades, V. Darcos, M. Amblard, J. Martinez, B. Nottelet and J. Coudane, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5891–5898 CrossRef CAS.
- A. Postma, T. P. Davis, A. R. Donovan, G. X. Li, G. Moad, R. Mulder and M. S. O'Shea, Polymer, 2006, 47, 1899–1911 CrossRef CAS.
- B. C. Anderson, S. M. Cox, P. D. Bloom, V. V. Sheares and S. K. Mallapragada, Macromolecules, 2003, 36, 1670–1676 CrossRef CAS.
- M. V. D. Banez, K. L. Robinson, M. Vamvakaki, S. F. Lascelles and S. P. Armes, Polymer, 2000, 41, 8501–8511 CrossRef.
- Z. G. Xie, C. H. Lu, X. S. Chen, L. Chen, X. L. Hu, Q. Shi and X. B. Jing, Eur. Polym. J., 2007, 43, 2080–2087 CrossRef CAS.
- B. Nottelet, A. El Ghzaoui, J. Coudane and M. Vert, Biomacromolecules, 2007, 8, 2594–2601 CrossRef CAS.
- J. Z. Du, L. Y. Tang, W. J. Song, Y. Shi and J. Wang, Biomacromolecules, 2009, 10, 2169–2174 CrossRef CAS.
- M. Wilhelm, C. Zhao, Y. Wang, R. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- Y. C. Wang, L. Y. Tang, T. M. Sun, C. H. Li, M. H. Xiong and J. Wang, Biomacromolecules, 2008, 9, 388–395 CrossRef CAS.
- X. T. Shuai, H. Ai, N. Nasongkla, S. Kim and J. M. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS.
- H. Chen, L. Yuan, W. Song, Z. K. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS.
- D. Putnam, C. A. Gentry, D. W. Pack and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1200–1205 CrossRef CAS.
- H. Wang, D. Wingett, M. H. Engelhard, K. Feris, K. M. Reddy, P. Turner, J. Layne, C. Hanley, J. Bell, D. Tenne, C. Wang and A. Punnoose, J. Mater. Sci.: Mater. Med., 2009, 20, 11–22 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |