No matter the order of monomer addition for the synthesis of well-defined block copolymers by sequential group transfer polymerization using N-heterocyclic carbenes as catalysts†
Jean
Raynaud
ab,
Na
Liu
ab,
Maréva
Fèvre
ab,
Yves
Gnanou
ab and
Daniel
Taton
*ab
aCentre National de la Recherche Scientifique, Laboratoire de Chimie des Polymères Organiques, 16 avenue Pey-Berland, F-33607, Pessac cedex, France. E-mail: taton@enscbp.fr
bUniversité de Bordeaux, Laboratoire de Chimie des Polymères Organiques, IPB-ENSCBP, F-33607, Pessac cedex, France
First published on 18th May 2011
Abstract
Unsaturated N-heterocyclic carbenes (NHCs) such as 1,3-bis(di-isopropyl)imidazol-2-ylidene (1) and 1,3-bis(di-tert-butyl)imidazol-2-ylidene (2) are shown to catalyze the sequential group transfer polymerization (GTP) of (meth)acrylic monomers. A variety of block copolymers including not only alkyl methacrylate but also alkyl acrylate monomer units as well as blocks deriving from N,N-dimethylacrylamide and methacrylonitrile were thus obtained at room temperature, using 1-methoxy-2-methyl-1-trimethylsiloxypropene (MTS) as initiator in THF as solvent. Block copolymerizations could be achieved, starting indifferently from the GTP of the acrylic monomer to that of the methacrylic one or vice versa, that is, regardless of the order of addition of the two monomers, in contrast to most examples of block copolymer synthesis by “controlled/living” sequential polymerization. It is postulated that these NHC-catalyzed GTPs of (meth)acrylics proceed via a single step concerted-like associative mechanism, involving the formation of thermodynamically unstable intermediates or transition states, likely hypervalent siliconates, with no detectable anionic enolates formed.
Introduction and scope
Block copolymers have widespread applications as the result of their self-assembling properties either in bulk or in thin films or in a selective solvent for one block, providing a great variety of ordered morphologies or nanosized objects.1–4 From a synthetic viewpoint, linear block copolymers are mainly prepared by so-called “controlled/living” sequential polymerization also referred to as block copolymerization involving consecutive addition of the monomers.1–6 Alternate synthetic methods include the coupling of preformed polymer segments possessing antagonist end-groups,1–7 the switch from one to another polymerization mechanism,8 and the use of dual (“double-headed”) initiators.9–11Sequential polymerization most often requires adding monomers in a certain order to achieve well-defined materials, especially when monomers exhibiting a distinct reactivity have to be polymerized.1–6,12–15 A key rule is that the rate of cross-addition must be higher than the rate of propagation of the second monomer. For instance in living anionic polymerization (LAP), monomers must be added in the order of their increasing electro-affinity which means that the sequential polymerization must be carried out in the following order: styrene ≈ butadiene > methacrylates > acrylates ≈ vinyl pyridine.2,6,15 Similar rules apply to block copolymer synthesis by controlled radical polymerization (CRP). In atom transfer radical polymerization (ATRP), for instance, the order is as follows: acrylonitrile > methacrylates > styrene ≈ acrylates > acrylamides.12–14 As for the reversible addition fragmentation chain transfer (RAFT) polymerization, a general rule is that the block giving the best leaving polymer radical must be synthesized first for efficient cross-over.13
Solutions to reverse the order of addition of monomers have seldom been reported. The basic idea is that the reactivity of growing species arising from the first block must be “boosted” to favor re-initiation over propagation. Teyssie et al.16 thus synthesized poly(ethylene oxide)-b-polystyrene by LAP, after modification of the living potassium poly(ethylene oxide) chains with a disilacyclobutane, which allowed them to polymerize styrene as the second monomer by LAP. Matyjaszewski et al. used the “halogen exchange” technique17 for the sequential ATRP of methyl acrylate and methyl methacrylate, in this order: a poly(methyl methacrylate) block was first grown from a bromine-terminated (instead of chlorine-terminated) poly(methyl acrylate) precursor in the presence of CuCl (instead of CuBr).
Recently, Yamago et al. have reported organotellurium-mediated CRP (acronym = TERP) as being insensitive toward the order of addition of monomers than any other CRP methods.18,19 Both TERP and RAFT proceed by a degenerative transfer.18–20 It has been suggested that the origin of the difference between RAFT and TERP regarding block copolymer synthesis is due to a difference in the mechanism operating in both cases.18,19TERP is claimed to involve a concerted-like mechanism, forming a hypervalent tellurium intermediate. In contrast, RAFT polymerization implies a three-step mechanism generating a radical intermediate, whose stability strongly depends on its polymer moieties.20
In this contribution, we show that the sequential group transfer polymerization (GTP) of (meth)acrylic monomers shows similarities with TERP, regarding the order of addition of monomers for block copolymer synthesis, when catalyzed by specific N-heterocyclic carbenes (NHCs).
GTP has been proposed in the mid 1980s as a convenient method to control the polymerization of (meth)acrylic monomers at ambient temperature and above.21–25 It is based on the repetition of Mukaiyama–Michael reactions26 involving the addition of silyl ketene acetal (SKA) onto an incoming (meth)acrylic monomer. GTP is generally catalyzed by a metal-free nucleophile (Lewis base) or a metal-based Lewis acid, for methacrylic and acrylic monomers, respectively.21,22,24,25,27,28 The absence of a unique catalytic system that could be generalized to both families of monomers did not permit, however, the synthesis of block copolymers featuring both monomer units by sequential GTP.21,22,24,25,27–29
In recent years, new developments in GTP catalysis have been disclosed.30–37 In particular, Hedrick, Waymouth et al.31 and our group30,36,38 have reported that GTP can be catalyzed by NHCs, also potent catalysts in ring-opening polymerizations.39–45 These reports have also reopened the debate on the mechanism that occurs in GTP.21,30,31,36,38,46–48 While Hedrick et al. have postulated a dissociative mechanism forming truly enolate-type species using NHC 3 as catalyst,31 we have proposed that GTP occurs by an associative mechanism when catalyzed by NHC 1 or 2 (Scheme 1).30,36,38 Both associative and dissociative mechanisms not only provide a distinct signature in terms of polymerization kinetics but also dramatically influence the order of addition of monomers for the purpose of block copolymer synthesis.21,22,24,25,28,30,31,36,38,46,47 Here we demonstrate that NHCs 1 and 2 can trigger the sequential GTP regardless of the order of addition of the monomers (GTP of methacrylics first, followed by GTP of acrylics or vice versa). These results may again be explained by the formation of unstable hexavalent siliconate intermediates in a transition state, via a single step associative GTP mechanism utilizing NHC 1 or 2.
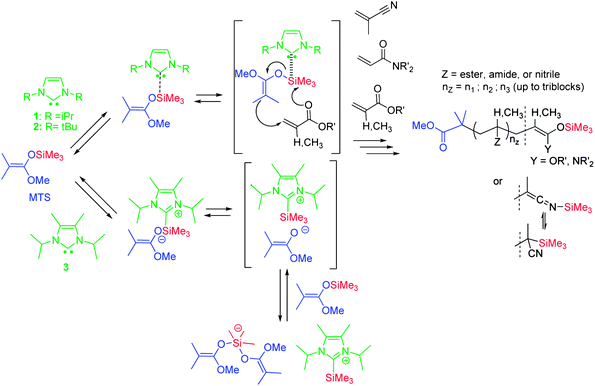 | ||
| Scheme 1 Associative (top) versus dissociative (bottom) mechanism of group transfer polymerization catalyzed by N-heterocyclic carbenes. | ||
Experimental
Materials
Methyl methacrylate (MMA), N,N-dimethylaminoethyl methacrylate (DMAEMA), n-butylacrylate (nBA), tert-butylacrylate (tBA), N,N-dimethylaminoethylacrylate (DMAEA), N,N-dimethylacrylamide (DMA), and methacrylonitrile (MAN) were all purchased in Aldrich or Fluka (purity: 97–99%) and were distilled over CaH2 into burettes. All other reagents were purchased from Aldrich. Methyl trimethylsilyl-ketene acetal (MTS; 95%) was distilled over CaH2 and stored into a Schlenk tube kept at low temperature in a glovebox. Tetrahydrofuran (THF) (technical grade) was distilled over Na/benzophenone and toluene over polystyryl lithium prior to use. Dimethylformamide (DMF; technical grade) was cryo-distilled over CaH2 and stored over activated molecular sieves, and freshly cryo-distilled prior to use. NHCs 1 and 2 were prepared by slightly modifying already reported procedures:49 the di-isopropyl-imidazolium salt was deprotonated with NaH and a catalytic amount of tBuOK and the di-tert-butyl-imidazolium salt with nBu-Li. Then the NHC 1 was purified by distillation under vacuum, the NHC 2 by sublimation under vacuum. Solutions of these catalysts were kept in a glove-box under argon atmosphere.Instrumentation
1H NMR (400 MHz) spectra were recorded on a Bruker AC-400 spectrometer in appropriate deuterated solvents. Molar masses of PMMAs, PtBAs and PnBAs were determined by size exclusion chromatography (SEC) using a 3-column set of TSK gel TOSOH (G4000, G3000, G2000 with pore sizes of 20, 75 and 200 Å respectively, connected in series) calibrated with PMMA standards with THF as eluent (1 mL min−1) and trichlorobenzene as a flow marker at 25 °C, using both refractometric and UV detectors (Varian).SEC in DMF was used for the characterization of PDMAEMAs, PDMAEAs, PDMAs and PMANs, using a 3-column set of TSK gel TOSOH (G4000, G3000, G2000 with pore sizes of 20, 75 and 200 Å respectively, connected in series) calibrated with polystyrene (PS) standards with DMF as eluent (0.8 mL min−1) and toluene as a flow marker at 60 °C, in the presence of LiBr (1 g L−1) using both refractometric and UV detectors (Varian).
Group transfer polymerization of methyl methacrylate
Polymerization of MMA was carried out under a dry and inert atmosphere using Schlenk equipments. In a typical polymerization, 70 μL of a 1 M solution of NHC 1 (7 × 10−5 mol; 2 mM) and 70 μL of MTS (3.4 × 10−4 mol; 10 mM) were introduced via a syringe in a vacuumed flame-dried Schlenk special apparatus equipped with a withdrawal digit on the side of the main flask (ESI†), kept in a glove-box under an argon atmosphere. After removal of the Schlenk from the glove-box, 30 mL of dry THF were added under vacuum. After homogenization, 4 mL (3.7 × 10−2 mol, final concentration 1 M) of MMA were introduced at 25 °C. The addition proceeded discontinuously over 5 minutes. After addition of first droplets, the color of the solution turned pink and then red-orange. At precise time intervals, aliquots were withdrawn thanks to the vacuum flame-dried digit. A droplet of degassed MeOH was then introduced, and the reaction mixture in the digit became colorless. The aliquot was removed from the withdrawal digit attached to the flask. Its volume was given by transfer to a small tarred container with a precise syringe. Conversions were determined by 1H NMR (ESI†).Group transfer polymerization of n-butyl acrylate and tert-butyl acrylate
The polymerization procedures are similar to the one used for the NHC-catalyzed GTP of MMA. In a typical polymerization, 100 μL of a 0.1 M solution of NHC 2 (10−5 mol; 0.3 mM) and 20 μL of MTS (10−4 mol; 2.5 mM) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removal of the Schlenk from the glove-box, 30 mL of dry THF were added under vacuum. After homogenization, 3.5 mL (2.4 × 10−2 mol, giving a final concentration of 0.7 M) of nBA were introduced at 25 °C. A droplet of degassed MeOH was introduced after completion of the reaction.Group transfer polymerization of N,N-dimethylaminoethyl methacrylate and N,N-dimethylaminoethyl acrylate
The polymerization procedures are similar to that described for the GTP-derived PMMAs. In a typical polymerization, 180 μL of a 0.1 M solution of NHC 1 (1.8 × 10−5 mol; 0.5 mM) and 150 μL of MTS (7.5 × 10−4 mol; 20 mM) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removal of the Schlenk from the glove-box, 30 mL of dry THF were added under vacuum. After homogenization, 6 mL (3.6 × 10−2 mol, giving a final concentration of 1 M) of DMAEMA were introduced at 25 °C. A droplet of degassed MeOH was introduced after completion of the reaction.Group transfer polymerization of N,N-dimethylacrylamide
In a typical polymerization, 100 μL of a 0.1 M solution of NHC 1 (10−5 mol; 0.3 mM) and 20 μL of MTS (10−4 mol; 2.5 mM) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removal of the Schlenk from the glove-box, 30 mL of dry THF were added under vacuum. After homogenization, 2.5 mL (2.4 × 10−2 mol, giving a final concentration of 0.7 M) of DMA were introduced at 25 °C. The reaction was quenched with degassed MeOH.Group transfer polymerization of methacrylonitrile
The polymerization procedures are similar to the one used for PMMA but using DMF as solvent to insure homogeneous conditions. For instance, in a typical polymerization, 180 μL of a 0.1 M solution of NHC 1 (1.8 × 10−5 mol; 0.5 mM) and 150 μL of MTS (7.5 × 10−4 mol; 20 mM) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removal of the Schlenk from the glove-box, 30 mL of dry DMF were added under vacuum. After homogenization, 3 mL (3.5 × 10−2 mol; 1 M) of MAN were introduced at 25 °C. A droplet of degassed MeOH was introduced after completion of the reaction.Synthesis of PnBA-b-PMMA by sequential GTP
In a typical polymerization experiment (entry 3, Table S1†), 50 μL of a 10−1 M solution of NHC 2 (5 × 10−6 mol or ∼1 mg of pure NHC 2, that is, a final concentration of 1.5 × 10−4 M) and 50 μL of MTS (2.5 10−4 mol corresponding to a concentration of 7 × 10−3 M) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removing the Schlenk from the glove-box prior to the polymerization, 30 mL of dry THF were added under vacuum, then 1.5 mL (1.0 × 10−2 mol) of nBA were introduced at room temperature drop wise. An aliquot was withdrawn after three hours of reaction, deactivated by a droplet of degassed MeOH and analyzed to check the completion of the reaction. Onto the living PnBA, 1.3 mL (1.2 × 10−2 mol) of MMA were slowly added. An aliquot was withdrawn after two days of reaction for characterization purpose.Synthesis of PMMA-b-PDMAEMA by sequential GTP
In a typical polymerization experiment (entry 9, Table S1†), 125 μL of a 10−1 M solution of NHC 1 (1.25 × 10−5 mol or ∼2 mg of pure NHC 1, that is, a final concentration of 3.7 × 10−4 M) and 50 μL of MTS (2.5 × 10−4 mol corresponding to a concentration of 7 × 10−3 M) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removing the Schlenk from the glove-box prior to the polymerization, 30 mL of dry THF were added under vacuum, then 3.8 mL (3.5 × 10−2 mol) of MMA were introduced at room temperature drop wise. An aliquot was withdrawn after three hours of reaction, deactivated by a droplet of degassed MeOH and analyzed to check the completion of the reaction. Onto the living PMMA, 3 mL (1.8 × 10−2 mol) of DMAEMA were slowly added. An aliquot was withdrawn after two days of reaction for characterization purpose.Synthesis of PMMA-b-PMAN by sequential GTP
In a typical polymerization experiment (entry 12, Table S1†), 125 μL of a 10−1 M solution of NHC 1 (1.25 × 10−5 mol or ∼2 mg of pure NHC 1, that is, a final concentration of 2.8 × 10−4 M) and 50 μL of MTS (2.5 10−4 mol corresponding to a concentration of 5.5 × 10−3 M) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removing the Schlenk from the glove-box prior to the polymerization, 30 mL of dry THF were added under vacuum, then 15 mL (10−1 mol) of MMA were introduced at room temperature drop wise. An aliquot was withdrawn after one day of reaction, deactivated by a droplet of degassed MeOH and analyzed to check the completion of the reaction. Onto the living PMMA, 9 mL (10−1 mol) of MAN were slowly added. An aliquot was withdrawn after two extra days of reaction for characterization purpose.Synthesis of a PMMA-b-PtBA-b-PDMA triblock copolymer by sequential GTP
In a typical polymerization experiment (entry 13, Table S1†), 17.5 μL of a 6.6 × 10−1 M solution of NHC 1 (1.16 × 10−5 mol or ∼1.75 mg of pure NHC 1, that is, a final concentration of 3.7 × 10−4 M) and 47 μL of MTS (2.3 × 10−4 mol corresponding to a concentration of 7.7 × 10−3 M) were introduced via a syringe in a vacuumed flame-dried Schlenk kept in a glove-box under an argon atmosphere. After removing the Schlenk from the glove-box prior to the polymerization, 30 mL of dry THF were added under vacuum, then 0.8 mL (7.5 mmol) of MMA were introduced at room temperature drop wise. An aliquot was withdrawn after one day of reaction, deactivated by a droplet of degassed MeOH and analyzed to check the completion of the reaction. Onto the living PMMA, 0.8 mL (5.5 mmol) of tBA were slowly added. An aliquot was withdrawn after three extra days of reaction for characterization purpose. Onto the living PMMA-b-PtBA, 0.5 mL (4.8 mmol) of DMA were slowly added. An aliquot was withdrawn after seven extra days of reaction for characterization purpose.Results and discussion
Webster et al. originally suggested to name it “group transfer polymerization” a mechanism involving the repetitive transfer of an initiator moiety upon addition of each monomer unit, after they observed that the trimethylsilyl group remained with the chain it started with during polymerization.21,25,29 In other words, an associative (concerted) mechanism of GTP involving a transfer of the trialkylsilyl moiety from the polymer chain-end to the just inserted monomer was originally put forward (Scheme 1). However, this pathway was subsequently questioned, in particular by Quirk and Ren who proposed that GTP occurred by a dissociative mechanism forming minute amounts of propagating enolates.47 These anionic species would be the real active centers and be temporarily trapped by silyl ketene acetals, forming dormant bis(enolato)siliconates (Scheme 1).In fact, the mechanism of GTP dramatically depends on the overall polymerization conditions and, in particular, on the nature of the catalyst.21,24,25,30–38,47,48,50 NHCs have themselves been reported to induce either the dissociative or the associative mechanism, with NHCs 3 and 1–2, respectively (see Scheme 1), due to a difference in nucleophilicity/silicophilicity between these different carbene catalysts. In these independent studies, Hedrick, Waymouth et al. and our group have also reported that NHCs can efficiently catalyze GTP of both acrylic and methacrylic monomers, in sharp contrast to the best catalysts used so far.28,30,31,36,38,51–54
Here NHCs 1 and 2 are shown to trigger the sequential GTP of acrylics and methacrylics using a unique catalytic system. These sequential GTPs utilizing NHC catalysts 1 and 2 were performed at room temperature in THF as solvent, in the presence of 1-methoxy-2-methyl-1-trimethylsiloxypropene (MTS) as initiator, providing an easy access to colorless and metal-free all acrylic-based block copolymers. Results of these block copolymerizations are summarized in Fig. 1. Fig. 2 also shows the size exclusion chromatography (SEC) traces of block copolymers, including those of the precursors (first blocks). Complementary information can be found in Table S1 (ESI†). A wide range of block copolymers could thus be derived, with an excellent control over molar masses and dispersities. The first block-forming GTPs were conducted to complete monomer conversion, as checked by 1H NMR (ESI†), before adding the second monomer.
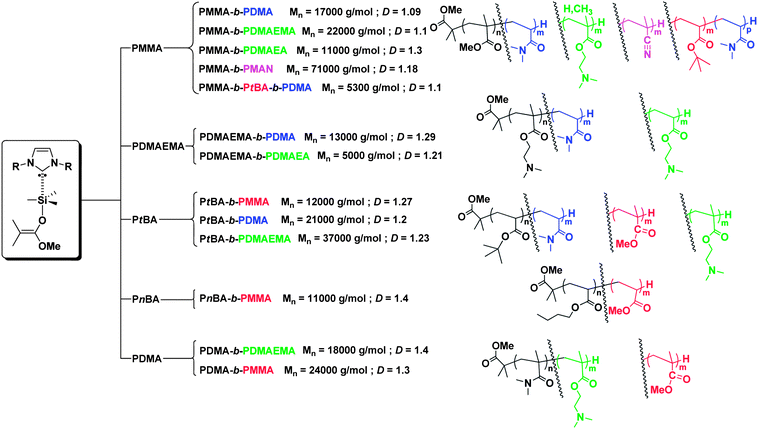 | ||
| Fig. 1 Block copolymers obtained via sequential group transfer polymerization in THF using NHCs as catalysts and MTS as an initiator. | ||
 | ||
| Fig. 2 SEC traces (RI detection) of block copolymers obtained by sequential GTP catalyzed by NHC 1 and initiated by MTS. The first block is shown in red and block copolymer in blue (see also Table S1, ESI†). | ||
PtBA-b-PDMA, PtBA-b-PDMAEMA and PMMA-b-PDMA block copolymers were analyzed using THF as eluent of SEC (calibration with PMMA standards), because the first blocks (PtBA and PMMA) were long enough to avoid possible interactions with the SEC columns. In contrast, DMF was used as eluent in the presence of LiBr at 60 °C to characterize other diblock copolymers. Calibration of the latter SEC device with polystyrene standards was obviously not fully adapted. The apparent values thus obtained for the first block were nonetheless used to calculate the block copolymer composition by 1H NMR spectroscopy (see below). A clear shift towards the higher molar mass domain after GTP of the second monomer was systematically observed, while dispersity remains quite low, attesting to an effective crossover (Fig. 2).
The slight broadening of molar mass distributions observed when growing polyacrylate as the second block may be explained by a faster rate of propagation of the acrylic monomer compared to the rate of (re)activation of the silyl ketene acetal chain ends.
Analysis by 1H NMR of all of the copolymers confirmed the presence of both blocks. A typical 1H NMR spectrum of a PDMA-b-PDMAEMA block copolymer derived by sequential GTP with all peaks' assignment is given in Fig. 3. The overall composition could be calculated from the relative integrations of the characteristic peaks of both blocks, assuming that SEC provides a good estimate of the molar mass of the first block.
 | ||
| Fig. 3 1H NMR spectrum of a PDMAEMA-b-PDMA block copolymer (entry 6, Table S1†) and calculation of its overall composition. | ||
Of particular interest, block copolymers featuring both acrylate and methacrylate monomer units could be prepared by sequential GTP, regardless of the order of addition of the two monomers. In other words, well-defined materials were obtained starting from the GTP of the acrylic monomer to that of the methacrylic one and vice versa. For instance, well-defined PnBA-b-PMMA, PtBA-b-PDMAEMA, PnBA-b-PMMA, and PDMA-b-PDMAEMA block copolymers could be obtained using NHCs 1 and 2, respectively (Fig. 2; see also entries 2 and 3, Table S1, ESI†). Synthesis of such block copolymers in this order would not have been possible with regular GTP catalysts.22,24,25,31 Access to block copolymers featuring blocks with pendant functional groups, such as poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA), poly[2-(dimethylamino)ethylacrylate] (PDMAEA), poly(N,N-dimethylacrylamide) (PDMA), and even poly(methacrylonitrile) (PMAN) was also possible. We have recently demonstrated that the NHC-catalyzed GTP of the corresponding monomers (DMAEMA, DMAEA, DMA and MAN) can be successfully achieved.38 In spite of the poor solubility of PMAN in many organic solvents, the PMMA-b-PMAN block copolymer could be synthesized in THF by sequential GTP, the length of the PMMA block being long enough to entail solubilization and analysis of the diblock copolymer in this solvent.
The synthesis by sequential GTP of ABA-type triblock copolymers, namely, PMMA-b-PnBA-b-PMMA and PMMA-b-PtBA-b-PMMA was already described in our previous reports.30,36 Here, the NHC 1-catalyzed sequential GTP was applied for the first time to the synthesis of an ABC-type, namely, PMMA-b-PtBA-b-PDMA triblock copolymer. Engineering of ABC-type triblock copolymers is well-documented; a variety of sub-micron size range morphologies can be contemplated as a result of their self-assembly in bulk or in solution.55,56Fig. 4 shows the SEC traces of materials obtained after crossing-over from the first to the second and to the third block, attesting to the well-defined character of the final PMMA-b-PtBA-b-PDMA sample (see the ESI† for its NMR characterization).
 | ||
| Fig. 4 SEC traces (RI detection) of the PMMA-b-PtBA-b-PDMA triblock copolymer obtained by sequential GTP catalyzed by NHC 1 and initiated by MTS. The first block is shown in red, diblock in blue and triblock in green (see also Table S1, entry 13, ESI†). | ||
As emphasized above, block copolymer synthesis by “controlled/living” sequential polymerization most often requires adding the monomers in a certain order for monomers that exhibit different reactivities.1–6,12–15 In the context of GTP, a dissociative mechanism forming true enolates as active species would not have permitted the synthesis of well-defined polyacrylate-b-polymethacrylate in this order. Indeed, polyacrylate living ends stand lower in the ladder of reactivity compared to polymethacrylate ones, the re-initiation could not have been quantitative.6 By making use of NHC 3 as GTP catalyst, which was reported to induce such a dissociative mechanism (Scheme 1), Hedrick, Waymouth et al. did note an incomplete crossover from the PtBA block to the PMMA one and eventually obtained ill-defined PtBA-b-PMMA block copolymers.31 In contrast, we show here that NHCs 1 and 2-catalyzed GTPs are indifferent toward the order of addition of the acrylic and methacrylic monomers. In our previous reports, we provided arguments showing that both NHCs 1 and 2 favor the associative mechanism, on the basis of kinetic investigations.30,36,38 Although the exact structure of intermediates formed by such an associative mechanism has never been clearly established,21,24,25,30,31,34,36,38,47,48 the existence of unstable hypervalent silicon intermediates has been postulated. Such hypervalent siliconates could explain the direct synthesis of (meth)acrylic block copolymers by sequential GTP, irrespective of the order of addition of the monomers, via a concerted-like (associative) mechanism. Background literature in molecular chemistry supports the existence of such hypervalent siliconates.39,57 Hence, re-initiation of a methacrylate polymerization by acrylic-type SKA polymer chains is made possible, yielding relatively well-defined polyacrylate-b-polymethacrylate materials.
Although more investigations are needed to elucidate the nature of these intermediates in particular when crossing-over from one monomer to another by sequential GTP, the formation of enolate-type species via a dissociative mechanism can be ruled out with NHCs 1 and 2 used as GTP catalysts for reasons mentioned above.
Conclusions
Miscellaneous (meth)acrylic-based block copolymers could be readily synthesized by sequential group transfer polymerization using unsaturated NHCs as catalysts. Monomers that can be block copolymerized include both alkyl acrylates and methacrylates, as well as N,N-dimethylacrylamide and methacrylonitrile. These NHC-organocatalyzed GTPs offer several advantages, such as the production of colorless and metal-free (co)polymer materials that can be synthesized at room temperature from a single NHC catalyst, an effective crossover during the block copolymer synthesis regardless of the order of monomer addition. That quite unique feature can be rationalized by the fact that the catalysis by NHCs 1 and 2 induces a concerted-like associative mechanism, likely forming hypervalent unstable siliconate intermediates. Although such intermediates have not been directly identified, the very possibility of reversing the order of monomer addition and growing a methacrylic block from an acrylic monomer is another piece of indirect evidence of the presence of such unstable siliconate intermediates. Should true enolates be formed in the presence of NHCs, a complete cross-over from active species generated by monomers of higher electro-affinity to those produced by monomers of lesser electro-affinity could not have been achieved. Another potential advantage of the associative mechanism over the dissociative one in GTP may be a control of the stereoselectivity; this will be the topic of a forthcoming contribution. These new developments in GTP catalysis are expected to rejuvenate this “controlled/living” polymerization method for a use in macromolecular synthesis.Notes and references
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- I. W. Hamley, Prog. Polym. Sci., 2009, 34, 1161–1210 CrossRef CAS.
- K. Matyjaszewski and A. H. E. Müller, Prog. Polym. Sci., 2006, 31, 1039–1040 CrossRef CAS.
- K. Matyjaszewski and A. H. E. Müller, Controlled and Living Polymerizations: From Mechanisms to Applications, Wiley-VCH, 2009 Search PubMed.
- D. Taton and Y. Gnanou, in Block Copolymers in Nanoscience, ed. M. Lazzari, G. Liu and S. Lecommandoux, Wiley-VCH Verlag GmbH & Co, Darmstadt, 2006, p. 9 Search PubMed.
- Y. Yagci and M. Atilla Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- C. J. Hawker, J. L. Hedrick, E. E. Malmstrom, M. Trollsas, D. Mecerreyes, G. Moineau, P. Dubois and R. Jerome, Macromolecules, 1998, 31, 213–219 CrossRef CAS.
- K. V. Bernaerts and F. E. Du Prez, Prog. Polym. Sci., 2006, 31, 671–722 CrossRef CAS.
- N. Chagneux, T. Trimaille, M. Rollet, E. Beaudoin, P. Gerard, D. Bertin and D. Gigmes, Macromolecules, 2009, 42, 9435–9442 Search PubMed.
- K. A. Davis and K. Matyjaszewski, Statistical, gradient and segmented copolymers by controlled/living radical polymerizations, Adv. Polym. Sci., 2002, 159, 1.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- H. L. Hsieh and R. P. Quirk, Anionic Polymerization: Principles and Practical Applications, Marcel Dekker Inc., New York, 1996 Search PubMed.
- T. Zundel, J. Baran, M. Mazurek, J.-S. Wang, R. Jerome and P. Teyssie, Macromolecules, 1998, 31, 2724–2730 Search PubMed.
- D. A. Shipp, J.-L. Wang and K. Matyjaszewski, Macromolecules, 1998, 31, 8005–8008 CrossRef CAS.
- S. Yamago, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1–12 Search PubMed.
- S. Yamago, Chem. Rev., 2009, 109, 5051–5068 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, 2008 Search PubMed.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706–5708 CrossRef CAS.
- D. Y. Sogah, W. R. Hertler, O. W. Webster and G. M. Cohen, Macromolecules, 1987, 20, 1473–1488 CrossRef CAS.
- O. W. Webster, Science, 1991, 251, 887–893 CrossRef CAS.
- W. J. Brittain, Rubber Chem. Technol., 1992, 65, 580–600 Search PubMed.
- O. W. Webster, in Advances in Polymer Science (New Synthetic Methods), 2004, pp. 1–34 Search PubMed.
- T. Mukaiyama, Angew. Chem., Int. Ed., 2004, 43, 5590–5614 CrossRef CAS.
- W. R. Hertler, D. Y. Sogah, O. W. Webster and B. M. Trost, Macromolecules, 1984, 17, 1415–1417 CrossRef CAS.
- I. B. Dicker, G. M. Cohen, W. B. Farnham, W. R. Hertler, E. D. Laganis and D. Y. Sogah, Macromolecules, 1990, 23, 4034–4041 CrossRef CAS.
- O. W. Webster, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2855–2860 CrossRef CAS.
- J. Raynaud, A. Ciolino, A. Baceiredo, M. Destarac, F. Bonnette, T. Kato, Y. Gnanou and D. Taton, Angew. Chem., Int. Ed., 2008, 47, 5390–5393 CrossRef CAS.
- M. D. Scholten, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2008, 41, 7399–7404 CrossRef CAS.
- Y. Zhang and E. Y. X. Chen, Macromolecules, 2008, 41, 36–42 CrossRef CAS.
- Y. Zhang and E. Y. X. Chen, Macromolecules, 2008, 41, 6353–6360 CrossRef CAS.
- E. Y. X. Chen, Chem. Rev., 2009, 109, 5157–5214 CrossRef CAS.
- R. Kakuchi, K. Chiba, K. Fuchise, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 8747–8750 Search PubMed.
- J. Raynaud, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5996–6005 CrossRef CAS.
- K. Fuchise, R. Sakai, T. Satoh, S.-i. Sato, A. Narumi, S. Kawaguchi and T. Kakuchi, Macromolecules, 2010, 43, 5589–5594 Search PubMed.
- J. Raynaud, N. Liu, Y. Gnanou and D. Taton, Macromolecules, 2010, 43, 8853–8861 Search PubMed.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, J. Am. Chem. Soc., 2009, 131, 3201–3209 CrossRef CAS.
- J. Raynaud, C. Absalon, Y. Gnanou and D. Taton, Macromolecules, 2010, 43, 2814–2823 Search PubMed.
- J. Raynaud, W. N. Ottou, Y. Gnanou and D. Taton, Chem. Commun., 2010, 46, 3203–3205 RSC.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018–4025 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, H. Li, E. C. Hagberg, R. M. Waymouth and J. L. Hedrick, in N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley VCH, 2006, p. 275 Search PubMed.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- R. P. Quirk and G. P. Bidinger, Polym. Bull., 1989, 22, 63–70 Search PubMed.
- R. P. Quirk and J. Ren, Macromolecules, 1992, 25, 6612–6620 Search PubMed.
- A. H. E. Müller, Macromolecules, 1994, 27, 1685–1690 Search PubMed.
- A. J. Arduengo, III, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef CAS.
- D. M. Haddleton, M. C. Crossman, K. H. Hunt, C. Topping, C. Waterson and K. G. Suddaby, Macromolecules, 1997, 30, 3992–3998 CrossRef CAS.
- D. Y. Sogah and O. W. Webster, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 927–931 Search PubMed.
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 2364 Search PubMed.
- A. I. Triftaridou, D. Kafouris, M. Vamvakaki, T. K. Georgiou, T. C. Krasia, E. Themistou, N. Hadjiantoniou and C. S. Patrickios, Polym. Bull., 2007, 58, 185–190 Search PubMed , and references herein.
- N. A. Hadjiantoniou, T. Krasia-Christoforou, E. Loizou, L. Porcar and C. S. Patrickios, Macromolecules, 2010, 43, 2713–2720 CrossRef CAS.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymer Morphology, John Wiley & Sons, Inc., 2003 Search PubMed.
- C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371–1448 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00077b |
| This journal is © The Royal Society of Chemistry 2011 |