Reversible click chemistry at the service of macromolecular materials†
Alessandro
Gandini
*,
Armando J. D.
Silvestre
and
Dora
Coelho
CICECO and Department of Chemistry, University of Aveiro, Campus de Santiago, 3810-193, Aveiro, Portugal. E-mail: agandini@ua.pt; Fax: +351 234 370 084; Tel: +351 234 370 693
First published on 18th May 2011
Abstract
The application of the reversible Diels–Alder (DA) reaction to the realm of furan polymers has recently bloomed, because of its potential for the preparation of a wide variety of novel macromolecular materials based on renewable resources, possessing, among other properties, thermoreversible, mendable and recycling features. In this study, the synthesis and characterisation of novel furan–maleimide monomers, viz. AB-type molecules, and their polycondensation by means of the DA reaction, are presented. These systems represent an interesting alternative to the traditional linear DA polycondensations, because they ensure the ideal initial stoichiometry. The behaviour of two A–B monomers was investigated with the maleimide group protected in the form of a furan–DA adduct in order to obtain a stable monomer and thus avoid premature polymerization. Their polycondensation was then followed after the in situ deprotection at high temperature, followed by the cooling to the appropriate temperature for the DA polymerization to occur.
Introduction
The last few decades have witnessed an impressive surge of research related to the use of furan derivatives as alternative chemical synthons and commodities, mostly in the shape of monomers, not only because of the ubiquitous availability and continuous natural replenishing of their precursors, viz. the vegetable biomass, but also because they can provide original macromolecular structures not readily available via petrochemistry.1–6One of the peculiar chemical features of the furan heterocycle is its pronounced dienic character, which makes it particularly suited to intervene in the [4 + 2] Diels–Alder (DA) cycloaddition reaction as the dienic partner. This reversible click reaction has therefore been applied to the realm of furan polymers with growing frequency, because of its potential in the preparation of a wide variety of novel macromolecular materials from renewable resources, possessing, among others, the common and noteworthy feature of thermal reversibility within a readily accessible domain of temperatures, leading to promising applications like self-mendability and network recyclability. In addition to reviews on the topic,4–7 important recent contributions to the specific furan–maleimide coupling system include thermoresponsive hydrogels,8 hydrogels for tissue engineering,9 phase-change materials,10 healing of fiber-reinforced composites,11 the control of surface energy and wettability,12 and a careful study of the stereoisomeric effects on thermo-remendable networks.13
The relevant feature of the DA reaction in the present context is its reversible character (Scheme 1), where the temperature is a key factor in determining the position of the equilibrium, which can be drastically shifted from predominant adduct formation (DA reaction), up to ca. 60 °C, to the predominant reversion to its precursors (retro-DA reaction), above ca. 100 °C.4–6
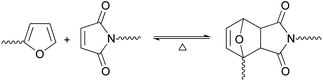 | ||
| Scheme 1 The DA equilibrium between growing species bearing, respectively, furan and maleimide end groups. | ||
Different polymer syntheses using di- and poly-functional monomers bearing the two complementary moieties, viz. the furan heterocycle (A) as the diene and the maleimide function (B) as the dienophile,4–6 have been tackled in our laboratory.14–16 In a first investigation, the kinetic and mechanistic features of the DA/retro-DA cyclic reaction were studied on a model system involving monofunctional compounds (–A + –B) and then applied to an A–A + B–B linear polymerization,14 as well as to non-linear An + Bm (n or m ≥ 2) systems.15 The present investigation deals in detail with two linear AB-type polycondensations, as a follow-up to a preliminary communication, which set the stage on this approach.16
The use of single monomers bearing both the furan and the maleimide moieties in their structures, viz. A–B systems, represents an interesting alternative to more traditional two-monomer linear polycondensations, because it provides the ideal initial stoichiometry.2,4,6 The first attempts to prepare this type of AB-type monomer were reported in the early nineties by Diakoumakos and Mikroyannidis,17 but their results were puzzling in that the spectroscopic characterization of the monomers did not provide convincing evidence about their expected structure, and no NMR spectra of the final materials could be taken, because they were not soluble in any tested solvent, despite the fact that they should have had a linear macromolecular structure, given the difunctional character of their precursors. A few years later, this same issue was briefly investigated by Goussé and Gandini,18 who prepared, characterized and polymerized the simplest A–B monomer of this series, viz. N-2-furfurylmaleimide, but the single methylene bridge joining the two complementary DA moieties made the molecule difficult to handle and hence the study was not pursued. Gaina and Gaina19 also tackled such A–B systems, although their attention was focussed on the aromatization of the ensuing DA adducts leading to thermally stable polymers,4 and again little attention was devoted to provide clear-cut evidence of the monomer and polymer structures involved.
More viable AB structures (Fig. 1) have now been synthesized in our laboratory,16 using an amino acid, viz.β-alanine, as the source of the maleimide moiety, so that both precursors to the monomers would be derived from renewable resources. Given the intrinsically reactive nature of these AB structures, one of the functional moieties had to be protected to avoid premature polymerization and hence the maleimide group was masked in the form of its furan–DA adduct, in order to obtain storage-stable latent monomers ready to be activated.
 | ||
| Fig. 1 Structure of the protected A–B monomers. | ||
The stereochemical aspects related to the relative abundance of exo and endo forms of the adducts are not a relevant issue in this context, since both play the same role as chain links between monomer units. Hence, the results presented and discussed here refer to the formation or the decoupling of a given adduct taken as the sum of its two stereoisomers.
Experimental
3-Aminopropanoic acid (β-alanine), exo-3,6-epoxy-1,2,3,6-tetrahydrophthalic anhydride (furan–maleic anhydride DA adduct) and anhydrous sodium carbonate were purchased from Acros Organics, whereas N,N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), furfuryl alcohol, furfurylamine, 1,1,2,2-tetrachloroethane (TCE) and its deuterated counterpart (TCE-d2) were purchased from Sigma-Aldrich. All were used without further purification.3-(exo-3,6-Epoxy-1,2,3,6-tetrahydrophthalimido) propanoic acid (3) and monomer (2-furfuryl)-3-(exo-3,6-epoxy-1,2,3,6-tetrahydrophthalimido) propanoate (5) [mp 67.4 °C (DSC)] were synthesized as described previously.16
IR spectra were taken with a Mattson 7000 spectrophotometer working in the ATR mode; electronic spectra with a temperature-controlled Jasco V-560 spectrophotometer using 1 cm Hellma suprasil cells equipped with a 9.9 mm quartz spacer and a quartz-to-pyrex graded seal; and 1H and 13C NMR spectra with a Bruker Avance spectrometer working, respectively, at 300.13 and 75.47 MHz. The coupling constants (J) are given in Hz. DSC thermograms were run in a Perkin Elmer Diamond DSC Power Compensation Dynamic Scanning Calorimeter, using heating and cooling rates of 20 °C min−1 in the temperature range of −90 to 200 °C under a nitrogen atmosphere. The equipment was calibrated with In and Pb standards. The software used for processing the acquired information was Pyris v.7.0.0.0110 2004. The determination of polymer molecular weights and their distributions by size-exclusion chromatography (SEC) called upon the use of a Varian PL-GPC 110 equipped with an IR-PD 2020 light scattering detector. DMA was used as the solvent with a sample concentration of about 6 mg mL−1. A calibration curve was obtained with five near-monodisperse polystyrene standards (1 mg mL−1) of molecular weights comprised between 1700 and 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
Synthesis of N-(2-furfuryl)-3-(exo-3,6-epoxy-1,2,3,6-tetra-hydrophthalimido) propanamide (7)
A procedure similar to that reported for 5 was followed.9 A solution of 3 (4.74 g, 20 mmol), furfurylamine (6) (3.85 g, 40 mmol) and DMAP (1.95 g, 16 mmol) in dry dichloromethane (20 mL) was prepared and after 5 min of stirring, DCC (4.54 g, 22 mmol) was added to it and the reaction mixture left for 4 hours at room temperature under N2. The precipitated dicyclohexylurea (DCU) formed during the reaction was removed by filtration and the filtrate washed with 0.5 M aqueous HCl (2 × 5 mL) and a saturated sodium hydrogen carbonate aqueous solution (2 × 5 mL). The organic phase was dried over anhydrous sodium sulfate before vacuum removal of the solvent. Additional DCU was precipitated by adding ethyl acetate to the residue and removed by filtration. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The expected product was obtained as a white solid (6.12 g, 97.0%). Compound 7 was stable and could not undergo DA self-condensation since the maleimide moiety was protected as its furan adduct. Mp 138.3 °C (DSC). FTIR-ATR: νmax/cm−1: 3337 (NH), 3117 and 3006 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2933 (asCH2), 2857 (sCH2), 1770, 1694 and 1644 (3 × CO), 1429 and 1376 (CC), 1340, 1340, 1169 and 851 (C–N), 1221 and 1016 (C–O). 1H NMR (300 MHz; CDCl3, Me4Si): δ/ppm = 7.34 (1H, d, 3J 2.0, furan 5′-H), 6.50 (2H, s, CHCHCH), 6.32 (1H, dd, 3J 3.2, 1.8, furan 4′-H), 6.23 (d, 1H, 3J 3.6, furan 3′-H), 6.01 (1H, br s; NH), 5.22 (2H, s, CHCHCH), 4.39 (2H, d, J 5.5, NHCH2-2-furan), 3.80 (2H, 3J 7.1, NCH2CH2), 2.83 (2H, s, CHCHCH), 2.49 (2H, t, 3J 7.1, NCH2CH2). 13C NMR (75 MHz; CDCl3, Me4Si): δ/ppm = 175.93 (COamide), 169.27 (COmaleimide), 151.02 (furan C-2′), 142.15 (furan C-5′), 136.46 (CHCHCH), 110.46, 107.59 (furan C-3′, C-4′), 80.94 (CHCHCH), 47.32 (CHCHCH), 36.47 (NCH2CH2), 35.21 (NHCH2-2-furan), 34.04 (NCH2CH2).
CH), 2933 (asCH2), 2857 (sCH2), 1770, 1694 and 1644 (3 × CO), 1429 and 1376 (CC), 1340, 1340, 1169 and 851 (C–N), 1221 and 1016 (C–O). 1H NMR (300 MHz; CDCl3, Me4Si): δ/ppm = 7.34 (1H, d, 3J 2.0, furan 5′-H), 6.50 (2H, s, CHCHCH), 6.32 (1H, dd, 3J 3.2, 1.8, furan 4′-H), 6.23 (d, 1H, 3J 3.6, furan 3′-H), 6.01 (1H, br s; NH), 5.22 (2H, s, CHCHCH), 4.39 (2H, d, J 5.5, NHCH2-2-furan), 3.80 (2H, 3J 7.1, NCH2CH2), 2.83 (2H, s, CHCHCH), 2.49 (2H, t, 3J 7.1, NCH2CH2). 13C NMR (75 MHz; CDCl3, Me4Si): δ/ppm = 175.93 (COamide), 169.27 (COmaleimide), 151.02 (furan C-2′), 142.15 (furan C-5′), 136.46 (CHCHCH), 110.46, 107.59 (furan C-3′, C-4′), 80.94 (CHCHCH), 47.32 (CHCHCH), 36.47 (NCH2CH2), 35.21 (NHCH2-2-furan), 34.04 (NCH2CH2).
Polycondensation of the A–B monomers (deprotected 5 and 7)
A typical procedure applied to the use of UV spectroscopy follows: a 0.2 M solution of 5 (31.7 mg, 0.1 mmol) or 7 (31.6 mg, 0.1 mmol) was prepared using TCE (0.5 mL) as solvent and left at 110 °C for 24 hours under a nitrogen atmosphere. This step promoted the retro-DA elimination of furan from the stabilized monomer and its ensuing maleimide function was hence set free to polymerize with the furan counterpart. The solution was then transferred to a 0.1 mm Suprasil cell, which was left thereafter in the temperature-controlled spectrophotometer at 65 °C. UV spectra were taken every 30 min during the first 3 hours, then every hour for 24 hours and finally after 3 days.A typical procedure applied to the use of NMR spectroscopy follows: a 0.2 M solution of 5 (63.5 mg, 0.2 mmol) or 7 (63.3 mg, 0.2 mmol) in TCE-d2 (1.0 mL) was prepared and left at 110 °C for 24 hours under a nitrogen atmosphere, in order to promote the elimination of furan from the adduct moiety and set the ensuing maleimide function free to polymerise with its furan counterpart. The solution was transferred to an NMR tube and the corresponding 1H NMR spectrum taken immediately at room temperature. The tube was then kept in an oil bath at 65 °C for 96 hours, and spectra taken at regular intervals. Thereafter, the system was allowed to return to room temperature and left for seven more days, when its 1H NMR spectrum suggested near-complete reaction.
The ensuing polymers 8 and 9 were precipitated in an excess of petroleum ether (b.p. 40–60 °C), filtered and dissolved in dichloromethane. The solvent was removed, the products vacuum dried and characterized by DSC and SEC.
Results and discussion
Synthesis of the A–B monomers
The steps leading to the protected A–B monomers 5 and 7 are depicted in Scheme 2. Similar procedures were used to prepare these structures and both were then characterized by FTIR and NMR spectroscopy. | ||
| Scheme 2 Synthesis of the protected AB monomers 5 and 7. | ||
Their FTIR spectra were consistent with the expected structures, through the presence of the relevant peaks (Fig. 2), viz. the maleimide CO bands at 1775 and 1693 cm−1 and the furan heterocycle bands (3120, 1015, 915, 734, 593 cm−1), accompanied by the CO ester band at 1736 cm−1 for 5, and the characteristic amide bands at 3337, 1644, 1539, 693 cm−1 for 7. The absence of bands arising from COOH, OH and NH2 moieties confirmed that any residual reagent had indeed been removed by the adopted synthetic protocol.

These results were further corroborated by the 1H and 13C NMR spectra of 5 and 7. The typical signals of the DA adduct moiety at δ 6.5, 5.1 and 2.9 ppm respectively, assigned to protons 7-H (double bond), 6-H (bridgehead) and 5-H (fused rings), were displayed in both their 1H NMR spectra, thus confirming the success of the protection of the maleimide moiety.
In addition to these peaks, the most relevant resonances included those of the furan protons at δ 7.3 (5′-H), 6.5 (4′-H) and 6.2 (3′-H) ppm, as well as those assigned to the methylene protons of the furfuryl moiety at δ 5.0 ppm as a singlet (OCH2-2-furan) for 5 (Fig. 3a), and at δ 4.4 ppm as a doublet (NHCH2-2-furan) for 7 (Fig. 4a).
 | ||
| Fig. 3 1H NMR spectra of 5 (0.2 M in *TCE-d2), (a) before and (b) after the in situ deprotection of the maleimide moiety by the retro-DA reaction (**traces of DCU). | ||
 | ||
| Fig. 4 1H NMR spectra of 7 (0.2 M in *TCE-d2), (a) before and (b) after the in situ deprotection of the maleimide moiety by the retro-DA reaction (**traces of DCU). | ||
In their 13C NMR spectra, the corresponding adduct carbons resonated at δ 137 (C-7), 81 (C-6), and 48 ppm (C-5), together with those of the two carbonyl peaks at δ 176 and 171 ppm. Additionally, resonances were recorded for the CO groups, at δ 175 and 169 ppm, as well as those of the furan carbons at δ 151, 142, 110 and 107 ppm, and of the methylene carbon of the furfuryl moiety at δ 58 ppm for 5, and at 32 ppm for 7.
DA polycondensation of the A–B monomers
As shown in Scheme 3, the DA polycondensation of the A–B monomers 5 and 7 was initiated after the in situ deprotection of the maleimide moiety at about 110 °C (retro-DA reaction), followed by cooling to 65 °C. Fig. 3 and 4 show, respectively, the 1H NMR spectra of 5 and 7 taken before and after their in situ deprotection. Apart from traces of residual DCU (δ 1.0–2.0 ppm) and evidence of the incipient polymerization, the spectra of both unprotected AB structures clearly confirmed their complete deprotection through the presence of the maleimide CHCHprotons, at δ 6.6 ppm, as well as the disappearance of the adduct peaks at δ 6.5, 5.1 and 2.9 ppm.
 | ||
| Scheme 3 Deprotection of the AB monomers and their DA polymerization. | ||
These A–B polycondensations were then followed by both UV and 1H NMR spectroscopy. The former technique provided quantitative information on the yield-vs.-time evolution of the DA reactions by monitoring the progressive decrease in the intensity of the maleimide peak at ∼300 nm, associated with the corresponding loss of conjugation between the two carbonyl groups and the CC unsaturation separating them, which accompanied the formation of the DA adduct. As expected, the initial solutions did not show any absorption at that wavelength, because the maleimide moiety was still protected. Then, after the in situ deprotection, the systems displayed the characteristic maleimide peak, which decreased progressively as the DA proceeded (Fig. 5). The presence of an isosbestic point at 260 nm (not clearly observed here) indicated that these DA polycondensations were not accompanied by any side reaction. However, in the case of polymer 9, the formation of a precipitate was observed during the reaction, which affected the corresponding spectra.
 | ||
| Fig. 5 Progressive decrease in the maleimide peak absorbance following the evolution of the DA polymerization of the unprotected AB monomer 8 in TCE at 65 °C, with an optical path of 0.01 cm (the blue line corresponds to the initial protected monomer solution). | ||
The evolution of these A–B systems was also followed by 1H NMR spectroscopy to assess the structural evolutions in a more detailed way. As an example, the progress of the DA polycondensation of deprotected 5 is shown in Fig. 6. The spectrum taken at t = 0 corresponds to the monomer solution after the in situ deprotection of its maleimide moiety by the retro-DA reaction. The expected pattern was observed in both systems, namely the progressive decrease in the peak intensity related to the monomer's relevant protons, viz. at δ 7.4 (5′-H), 6.7 (5-H), and 6.4–6.3 (3′-H, 4′-H) ppm and the corresponding increase in the signals of the corresponding adduct at δ 5.3 (e-H), 6.5–6.2 (c-H, d-H), and 3.6–2.9 (f-H, g-H) ppm. Concurrently, as the reaction proceeded, the viscosity of the medium increased appreciably.
 | ||
| Fig. 6 Time evolution of the 1H NMR spectra for the A–B polymerization of unprotected 5 (0.2 M) at 65 °C in *TCE-d2 (**traces of DCU). The initial spectrum (t = 0), taken at room temperature, corresponds to the deprotected monomer. The top spectrum was taken after 3 days at 65 °C, followed by 4 days at room temperature. | ||
After the system had reached high conversions, the retro-DA reaction was followed by 1H NMR spectroscopy at 110 °C for 24 hours, and within 6 hours the solution already displayed the resonances of the starting monomer (Fig. 7), which showed that the equilibrium was being indeed shifted backward, although not completely, since after 24 hours 34% of adducts were still present. Thereafter, the system was brought back to 65 °C in order to promote a second polymerization, and left for three more days. The 1H NMR of the ensuing solution was similar to that taken at the end of the first polymerization, thus confirming the reversible nature of the system (Fig. 7).
 | ||
| Fig. 7 1H NMR spectra taken during the A–B retro-DA depolymerization of polymer 8 (24 h at 110 °C). The spectrum run after 6 h shown here was essentially identical to that taken after 24 h. The top spectrum represents the second DA polycondensation and was taken after 3 days at 65 °C (*TCE-d2; **traces of DCU). | ||
The same study was carried out with 7, and the results displayed similar features, except for the occurrence of polymer 9precipitation during the reaction which led to a general “disappearance” of the polymer peaks, since this was no longer in solution.
Polymers 8 and 9 (Scheme 3), isolated by precipitation in an excess of petroleum ether, filtration and drying under vacuum, were characterized by DSC and SEC. Table 1 gives the only detected Tg, together with the molecular weight distribution and polydispersity (PD) values for polymer 8. Given the occurrence of the retro-DA reaction above 80 °C, the DSC analyses were stopped at that temperature.
Whereas polymer 8 gave a Tg of about 45 °C (Fig. 8), no such transition was detected for polymer 9 (nor indeed any melting peak) below 80 °C. This considerable increase in the Tg of 9 can be explained by the presence of amide (–CONH–) groups in its backbone, which strongly favoured intermolecular hydrogen bonding, as in all polyamides, compared with the corresponding polyesters. The lack of melting features below 80 °C was complemented by an X-ray analysis of 9, which showed the typical features associated with a semi-crystalline material, as illustrated in Fig. 9, in tune with the relative ease with which polyamides crystallize compared with polyesters, for the same reason, related to ordered interchain packing through hydrogen bonding. The melting of these ordered microphases was not observed because its occurrence was preceded by their depolymerisation through the retro-DA reaction, i.e. kinetic decomposition features impeded the occurrence of the thermodynamic phase transition (as with, e.g. cellulose or aromatic polyamides). This behaviour was further corroborated by the difference in solubility between 8 and 9, since only the latter precipitated out of solution during its formation because of its higher interchain cohesive energy.
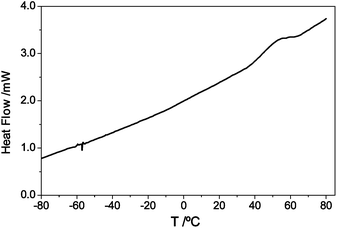 | ||
| Fig. 8 DSC thermogram of polymer 8. | ||
 | ||
| Fig. 9 X-Ray diffractogram of polymer 9. | ||
The SEC of polymer 8 gave a low molecular weight, but the PD value around 2 was consistent with a linear polycondensation. Its distribution curve (Fig. 10) displayed several narrow peaks, each with a PD value around 1, i.e., corresponding to specific compounds, instead of a distribution of homologues. These peaks were therefore assigned to cyclic oligomeric structures formed during the DA polycondensation, since a growth associated exclusively with linear molecules of different sizes would not have yielded such features in the corresponding SEC tracings.

The use of relatively low concentrations (<1 M) obviously favoured the formation of these cycles, which influenced negatively the final polymer molecular weights, as well as lowering their Tg by acting as internal “plasticizers”.
Since polymer 9 was not completely soluble in the solvent used for the SEC analysis, it was impossible to determine its molecular weight and the possible presence of similar cyclic oligomers, although the sharp peaks observed in its X-ray diffractogram could also be attributed to these cyclic structures, and not only to the semi-crystalline features of the linear macromolecules. Further study is however required to confirm this hypothesis. The loss of solubility of the growing chains of 9 clearly gave rise to modest molecular weights, since the precipitated products could not pursue their polycondensation.
Conclusions
These DA systems are excellent examples of reversible click chemistry applied to macromolecular syntheses by DA polycondensation. The two furan–maleimide A–B monomers described here were viable structures for these polymerizations, provided the protection of one of their reactive ends was applied in order to avoid their premature polymerization during their synthesis, purification and storage. The results obtained from both UV and 1H NMR spectroscopy corroborated the thermal reversibility and reproducibility, as well as the absence of significant side reactions in both forward and backward pathways, even after several cycles. Work is in progress to extend the scope of this investigation to other A–B monomers, as well as to AB2 structures capable of generating thermally reversible hyperbranched macromolecules.Acknowledgements
Dora Coelho thanks FCT for her Doctorate Grant (SFRH/BD/28271/2006) and for the “Apoio do Programa Nacional de Re-equipamento Científico” (REEQ/515/CTM/2005 POCI 2010, fonte FEDER).References
- Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008 Search PubMed.
- A. Gandini, Macromolecules, 2008, 41, 9491 CrossRef CAS; A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- (a) A. Gandini, in Encyclopedia of Polymer Science and Engineering, John Wiley & Sons, New York, 1986, vol. 7, p. 454 Search PubMed; (b) A. Gandini, ACS Symp. Ser., 1990, 433, 195 CAS; (c) M. N. Belgacem and A. Gandini, Prog. Polym. Sci., 1997, 22, 1203 CrossRef CAS; (d) C. Moreau, A. Gandini and M. N. Belgacem, Top. Catal., 2004, 27, 9; (e) A. Gandini and N. M. Belgacem, in ref. 1, ch. 6, pp.115–152.
- M. N. Belgacem and A. Gandini, ACS Symp. Ser., 2007, 954, 280.
- A. Gandini, in Green Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction, ed. R. T. Mathers and M. A. Meier, Wiley-VCH, Weinheim, 2011, pp. 29–58 Search PubMed; A. Gandini, in Films and Coatings from Renewable Resources, ed. D. Plackett, Wiley-VCH, 2011, pp. 180–209 Search PubMed.
- A. Gandini, Polym. Chem., 2010, 1, 245 RSC.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41 RSC; J. A. Syrett, C. Remzi Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978 RSC; C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643 CrossRef CAS; A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417 Search PubMed.
- H.-L. Wei, J. Yang, H.-J. Chu, Z. Yang, C.-C. Ma and K. Yao, J. Appl. Polym. Sci., 2011, 120, 974 Search PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Biomacromolecules, 2011, 12, 824 Search PubMed.
- J. P. Swanson, S. Rozvadovsky, J. E. Seppala, M. E. Mackay, R. E. Jensen and P. J. Costanzo, Macromolecules, 2010, 43, 6135 Search PubMed.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, ACS Appl. Mater. Interfaces, 2010, 2, 1141 Search PubMed; A. M. Peterson, R. E. Jensen and G. R. Palmese, Compos. Sci. Technol., 2011, 71, 586 Search PubMed.
- P. T. Dirlam, G. A. Strange, J. A. Orlicki, E. D. Wetzel and P. J. Costanzo, Langmuir, 2010, 26, 3942 CrossRef CAS.
- J. Canadell, H. Fischer, G. de With and R. A. M. van Benthem, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3456 Search PubMed.
- A. Gandini, D. Coelho and A. J. D. Silvestre, Eur. Polym. J., 2008, 44, 4029 CrossRef CAS.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. J. D. Silvestre, J. Mater. Chem., 2009, 19, 8656 RSC.
- A. Gandini, D. Coelho and A. J. D. Silvestre, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2053 CrossRef CAS.
- (a) C. D. Diakoumakos and J. A. Mikroyannidis, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2559 CrossRef CAS; (b) J. A. Mikroyannidis, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 125 CrossRef CAS and 2017; (c) C. D. Diakoumakos and J. A. Mikroyannidis, Eur. Polym. J., 1994, 30, 465 CrossRef CAS.
- C. Goussé and A. Gandini, Polym. Bull., 1998, 40, 389 CrossRef CAS.
- V. Gaina and C. Gaina, Rev. Roum. Chim., 2003, 48, 881–890 Search PubMed.
Footnote |
| † Part 3. Diels–Alder polycondensation of furan/maleimide AB monomers. |
| This journal is © The Royal Society of Chemistry 2011 |