DOI:
10.1039/C1PY00073J
(Paper)
Polym. Chem., 2011,
2, 1107-1113
Recent advances in sunlight induced polymerization: role of new photoinitiating systems based on the silyl radical chemistry
Received
11th February 2011
, Accepted 14th February 2011
First published on 24th March 2011
Abstract
In the present paper, the recent development of new silyl chemistry based photoinitiating systems for sunlight induced polymerization is presented. The inherent issues of the effects of a low light intensity and oxygen inhibition are discussed. Some selected examples leading to tack free coatings in free radical photopolymerization (FRP) and cationic photopolymerization (CP) are provided. The use of the silyl radical chemistry to overcome the O2 inhibition is presented. The sunlight induced polymerization might be a promising way for practical low cost and environmentally friendly curing processes. The use of renewable epoxy monomers (such as epoxidized soybean oil (ESO), epoxidized linseed oil (ELO) or limonene dioxide (LDO)) in combination with a sunlight induced process also appears particularly worthwhile for green chemistry applications.
Introduction
Light induced polymerization reactions are very well known1,2 from many years in industrial sectors such as radiation curing, laser imaging, microelectronics, medicine or optics and used for various applications e.g. in coatings, varnishes, paints, adhesives, graphic arts, printing plates, stereolithography, photoresists, laser direct imaging, computer-to-plate technology, holographic optical elements, tooth repair… They involve a polymerizable matrix and a more or less complex photoinitiating system (PIS) that contains at least a photoinitiator (PI) or a photosensitizer (PS): PI (or PS) has to absorb the light. Upon excitation, PI becomes excited (PI*) and directly generates (1) a reactive species RS△ (e.g. a radical R˙, a cation, an anion, an acid, a base as a function of the starting molecule). When PS is excited, the excitation has to move from PS* to PI by energy (2a) or electron transfer(2b): the same RS△ is formed or new ion radicals are created.| | | PI + hν → PI* → R˙ → → → polymer | (1) |
| | | PS + hν → PS* → PI* → R˙ → → → polymer | (2a) |
| | | PS + hν → PS* → PS˙+ + PI˙− → → → polymer | (2b) |
In the radiation curing area, free radical photopolymerization (FRP) is the most popular compared to cationic photopolymerization (CP). A photopolymerization either corresponds to a photoinduced polymerization process (a chain reaction occurs after the absorption of one photon) or a photocrosslinking reaction (the formation of each crosslink between two macromolecular chains requires one photon). When using multifunctional monomers or oligomers, the photoinduced polymerization reaction also leads to crosslinking reactions, thereby creating a polymer network.
The light sources were originally based (and still are) on the mercury lamps (high pressure, medium pressure and low pressure) that are still largely used together with doped Hg lamps or microwave powered lamps. The high flux of photons (typically >1.2 W cm−2 in the 280–445 nm range for a Hg lamp that satisfactorily matches the absorption of PIS) are delivered in the UV/near visible range. The visible light emission of Hg lamps is generally lost or weakly recovered. Xe and Hg–Xe lamps can be interesting for visible light irradiations (400 nm < λ < 800 nm). The possible presence of the usually available PS allows to extend the photosensitivity of the matrix towards longer wavelengths. The past development of various continuous or pulsed laser sources, the more recent design of many laser diodes with output energy >10 mW cm−2 operating at selected wavelengths (405, 457, 473, 532, 635 nm…) and the proposal of the new LED technology also allow today large possibilities of excitation from the UV to the near infra-red. The sun is a cheap but a low intensity source of light (typically <2 mW cm−2 but strongly affected by the weather and the location) that might be of interest for particular outdoor applications: using sunlight is a dream that could come true.
In FRP, a drawback concerns the well known oxygen inhibition.1 This is less the case, however, for thiol–ene reactions and their related radical polymerization.2c According to their lifetimes, excited states are more or less quenched by O2. Both the initiating R˙ and propagating radicals R–Mn˙ are scavenged by O2. This leads to highly stable peroxyl radicals ROO˙ (R–MnOO˙) which cannot participate in any further polymerization initiation reactions but can yield hydroperoxidesROOH (R–MnOOH) through hydrogen abstraction with a polymer chain or a hydrogen donor. The radical/O2 interaction is a nearly diffusion controlled reaction. The polymerization only starts in the film when oxygen is consumed. In highly viscous (e.g. ∼14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cP for an epoxy acrylate) or thick samples, the re-oxygenation process is quite slow leading to an efficient polymerization after an inhibition period. On the opposite, in very low viscosity (e.g. ∼70–100 cP for trimethylolpropane triacrylate (TMPTA)) or thin samples, the re-oxygenation remains efficient leading to reduced monomer conversions. In addition, when the light intensity is reduced, the initial O2 consumption decreases and the amount of initiating radicals becomes lower. Much more initiating radicals are thus scavenged by O2 and as a consequence, the oxygen inhibition has a larger dramatic effect on the polymerization profile. The same holds true in free radical promoted cationic photopolymerization (FRPCP).3 It consists in producing a radical R˙ (e.g. from a usual radical photoinitiator) which in turn should be oxidized by an onium salt Ph2I+, the resulting cation R+ being the polymerization initiating structure (3). The radical source can be easily selected to match the irradiation wavelength. Due to its versatility, FRPCP is certainly one of the most interesting and promising ways for exposure at λ > 400 nm (e.g. the search and design of suitable compounds as energy or electron donors in photosensitized CP can be a rather complex task).
500 cP for an epoxy acrylate) or thick samples, the re-oxygenation process is quite slow leading to an efficient polymerization after an inhibition period. On the opposite, in very low viscosity (e.g. ∼70–100 cP for trimethylolpropane triacrylate (TMPTA)) or thin samples, the re-oxygenation remains efficient leading to reduced monomer conversions. In addition, when the light intensity is reduced, the initial O2 consumption decreases and the amount of initiating radicals becomes lower. Much more initiating radicals are thus scavenged by O2 and as a consequence, the oxygen inhibition has a larger dramatic effect on the polymerization profile. The same holds true in free radical promoted cationic photopolymerization (FRPCP).3 It consists in producing a radical R˙ (e.g. from a usual radical photoinitiator) which in turn should be oxidized by an onium salt Ph2I+, the resulting cation R+ being the polymerization initiating structure (3). The radical source can be easily selected to match the irradiation wavelength. Due to its versatility, FRPCP is certainly one of the most interesting and promising ways for exposure at λ > 400 nm (e.g. the search and design of suitable compounds as energy or electron donors in photosensitized CP can be a rather complex task).
| | | R˙ + Ph2I+ → R+ + Ph˙ + Ph–I | (3) |
Nevertheless, using high light intensity sources as in the UV Curing area (Hg lamps, doped Hg lamps, Hg bulb lamps) and high PI concentrations allow to satisfactorily cure high (and to a lesser extent), low viscosity monomer/oligomer films under air for conventional coating applications on an industrial ground. The situation is very different if one considers applications where low intensities are available (e.g. with sunlight) or quite low viscosity monomers (particular acrylates or cationic monomers) or thin films have to be employed. In fact, a very strong oxygen inhibition occurs and polymerization under air with PIS usually considered as efficient cannot even be observed.4 This is the reason why the use of sunlight in the radiation curing field, despite efforts during the last thirty years, still remains a great challenge. In this paper, we will briefly recall some previous works that have been carried out in the past few years. Then, we will show how the introduction of the silyl chemistry into PIS can open new opportunities in sunlight induced polymerization reactions of low viscosity monomers.
Experimental
(i) Compounds
The investigated monomers for CP are shown in Scheme 1. Epoxidized soybean oil (ESO) was obtained from Arkema (Ecepox; epoxy content: 3.7 M kg−1) and limonene dioxide (LDO) from Millennium Speciality Chemicals. (3,4-Epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX, UVACURE 1500) was a gift of Cytec. Epoxidized linseed oil (ELO) was a gift from Akcros chemicals. Tris-(trimethylsilyl)silane (TTMSS) and diphenyl iodonium hexafluorophosphate (Ph2I+) were obtained from Aldrich and phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO) or 2,4,6-trimethylbenzoyl-diphenyl phosphine (TPO) from Ciba-Basel.
 |
| | Scheme 1 Investigated monomers | |
For the FRP process, different monomers were selected: trimethylolpropane triacrylate (TMPTA), ethoxylated pentaerythritol tetraacrylate (EPT) and an epoxy acrylate (Ebecryl 605) (from Cytec).
The films deposited on a BaF2 pellet were irradiated with sunlight. The solar irradiation was carried out in Mulhouse (France); an absolute irradiance measurement (Ocean Optics HR4000) leads to an estimated incident energy <5 mW cm−2 in the 390–800 nm range. For the polymerization of LDO, ESO and ELO (see below), the conversion of the epoxy can be observed at 800–850 cm−1 and the formation of the polyether network followed at about 1050–1100 cm−1. For the FRP process, the acrylate conversion is followed at about 1635 cm−1. These procedures were already described in detail in ref. 4.
Results and discussions
Backgrounds
Visible light induced FRP, CP or FRPCP is well documented as it can be seen e.g. in ref. 5–15 but works devoted to the sunlight curing are rather scant. Sunlight curing is, however, a green technology that presents the great advantage to combine both radiation curing and a sun-assisted processing, together with no volatile organic compound (VOC) release, no energy consumption, no irradiation device, possibility of curing large dimension pieces or surfaces.
A lot of work (e.g. in ref. 16–22) have been carried out in the 80's mainly by large industrial companies to develop paints for crack-bridging applications and anti-soiling properties in outdoor conditions of illumination. They were intended for the protection, the renovation or the decoration of facades, roofs, walls and terraces in order to improve the water proofing and the water resistance of the substrate together with a less soiling. Soft polymers are known to avoid cracks but they do not avoid soil. The developed sunlight photosensitive paints are mostly based e.g. on an elastomeric latex containing a suitable proportion of hard monomer (methyl methacrylate)/soft monomer (butyl acrylate), a photocrosslinkable binder (typically dicyclopentadienylacrylate (DCPA)) and a PIS (typically benzophenone (BP) alone). They ensure a surface photopolymerization of the paint which decreases the stickiness and the soiling by dust. The elastomeric character of the latex, as in a conventional anti-soiling coatings,21 preserves the flexibility. The photocrosslinking process is achieved under air through an oxidation/peroxyldecomposition mechanism after the primary hydrogen abstraction reaction between BP and the polymer backbone.22
The photocrosslinking of waterborne latexes was also achieved and gave tack-free coatings under air upon visible lights or/and sunlight exposure using a latex based on ethylene (E)–vinyl acetate (VA)–aceto acetoxyethyl methacrylate, a binder (TMPTA or polyethylene glycol diacrylate (PEGDA)) and a α-diketone. Using camphorquinone (CQ) leads to a better efficiency than incorporating a bisphosphine oxide derivative BAPO as PI.23
Interpenetrating polymer networks (IPN) usable as protective coatings and glues were readily obtained under air by sunlight FRP curing of acrylate monomers dispersed in a solid matrix (such as polymethyl methacrylate or in a styrene-butadiene rubber) in order to increase the matrix viscosity and reduce the oxygen inhibition.24 The creation of an IPN formed from an epoxidized natural rubber and a vinyl ether (or an acrylate or an epoxide),25 the fabrication of glass fiber reinforced composites containing epoxidized vegetable oils (but in laminated conditions)26 as well as the manufacture of hard and rigidified four layer glass cloth laminate containing an epoxidized linseed oil, curcumin (as a PS) and an iodonium salt 13b were also claimed using sunlight through a CP reaction. Systems based on quinoxalines were also recently proposed for cationic polymerization.13c
Other papers mention the possibility of various clearcoats based on multiacrylate resins,27thiol–ene systems,28 cycloaliphatic epoxy compounds29 or acrylate/SBS matrix30 to be cured under UV, visible light or eventually sunlight. Polymer–clay composites can also polymerize by FRP under artificial lights and sunlight.31
Except a few of them which clearly opened up promising research directions (see e.g.ref. 13,21,23–26 and 30), the above mentioned works suffer from the oxygen inhibition and the low photosensitivity of the formulations upon sunlight exposure.
Very recent studies have proposed a new way to cure coatings under sun, under air, even in relatively low viscosity matrix (as exemplified by the FRPCP of renewable epoxy monomers32) thanks to a specific photoinitiating system PIS based on the silyl radical (R3Si˙) chemistry.6c,32−35 Such a typical PIS for FRPCP consists of three compounds: an absorbing molecule (AM) (4a), a silane R3SiH and an iodonium salt Ph2I+. The AM compound should lead to silyl radicals (4c) through (4b) and (d). The R3Si˙ radicals and the other radicals R′˙ overcome the oxygen inhibition through the oxygen consumption (5) and the scavenging of the peroxyls (6a and b). The generated peroxides or hydroperoxides cleave under light irradiation (6c) leading to new radicals which in turn can abstract a hydrogen atom from R3Si–H (6d). New silyls are thus formed and the total amount of interesting R3Si˙ increases so that oxygen has a positive effect on the silyl radical production. Finally, the same cationic structure R3Si+ is formed (7). This mechanistic scheme (4a)–(7) has been well established in our previous works.32| | | R3SiOO˙ + R3SiH → R3Si˙ + R3SiOOH | (6a) |
| | | R′OO˙ + R3SiH → R3Si˙ + R′OOH | (6b) |
| | | R3SiOOH → R3SiO˙ + OH˙ (hν) | (6c) |
| | | RO˙ + R3SiH → R3Si˙ + ROH | (6d) |
| | | R3Si˙ + Ph2I+ → R3Si+ + PhI + Ph˙ | (7) |
In our approach, the onium salt does not play any role except to oxidize the silyl radical into a silylium cation. The same cation is formed whatever AM. A suitable selection of AM obviously allows to easily tune the absorption from the near UV to the visible wavelength range and to match the sun emission spectrum. The FRPCP using the silyl chemistry might be still even more interesting than the FRPCP developed so far as there is now no change of the initiating cation. Moreover, the higher efficiency of the AM/silane/onium salt (the counter anion being hexafluorophosphate) system compared to AM/onium salt avoids the use of an onium salt containing a counter anion more efficient for the propagation of CP but which is also either more expensive such as B(C6F5)4− (ref. 36) or toxic such as SbF6− (e.g. the presence of Sb containing residue in the cured coating).38
Fig. 1 shows that the polymerization of low viscosity limonene dioxide under sunlight in the presence of the isopropylthioxanthone (ITX/Ph2I+) photoinitiating system is significantly improved upon addition of a silane (here tris(trimethylsilyl)silane (TTMSS); see curve b vs. curve c in Fig. 1). Other silanes were investigated but the best polymerization profiles are obtained with TTMSS.32,33 Therefore, for the present low intensity applications (i.e. sunlight), TTMSS was selected here. With ITX/Ph2I+/TTMSS, tack free coatings are cured in less than 10 min. With other systems based on TPO or BAPO instead of ITX, very fast sunlight induced polymerizations were obtained. As shown in Fig. 1B and C, with the BAPO/TTMSS/Ph2I+ photoinitiating systems, tack free coatings were produced in 12 min and 45 min with LDO and ESO (Scheme 1), respectively. A very fast polymerization of ELO is also found (Fig. 1D) and a tack free coating is also obtained for sunlight irradiation. In the same irradiation conditions but in the absence of TTMSS, tack free coatings were only obtained at noticeably longer irradiation times: 45 min and >3 hours for LDO and ESO (or ELO), respectively. This demonstrates the favorable effect of TTMSS on the polymerization under sunlight.
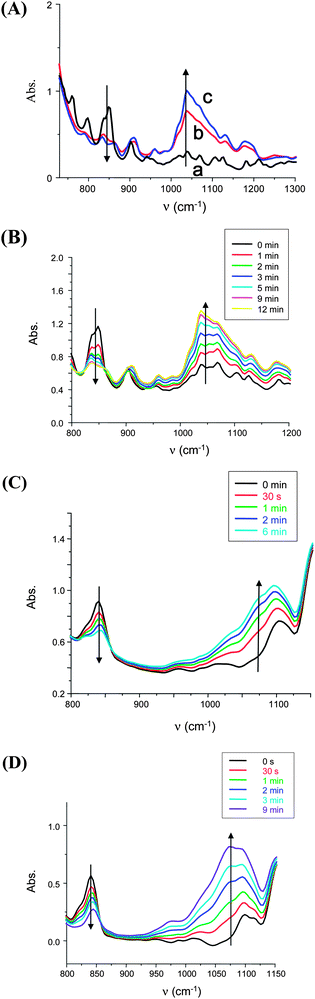 |
| | Fig. 1
IR spectra recorded during the photopolymerization reaction of various monomers (film thickness: ∼25 µm) under sunlight irradiation. Estimated light intensity: <5 mW cm−2 in the range 390–800 nm; experiments in Mulhouse-France. (A) Limonene dioxide (LDO) in the presence of the ITX/Ph2I+ (1%/1%, w/w) at (a) t = 0; (b) t = 10 min without TTMSS and (c) at t = 10 min upon addition of TTMSS (3%, w/w); the bands at about 840 and 1050 cm−1 are associated to the epoxy and ethergroup, respectively. (B) Limonene dioxide in the presence of the BAPO/TTMSS/Ph2I+ (1%/3%/2%, w/w) at t = 0 to 12 min. (C) Epoxidized soybean oil (ESO) in the presence of the BAPO/TTMSS/Ph2I+ (1%/3%/2%, w/w) at t = 0 to 6 min. (D) Epoxidized linseed oil (ELO) in the presence of the BAPO/TTMSS/Ph2I+ (1%/3%/2%, w/w) at t = 0 to 9 min. | |
Some other examples of recently developed AM/silane/Ph2I+ photoinitiating systems (where AM is a conventional PI) leading to tack free coatings under sunlight are gathered in Table 1. Different epoxy monomers were investigated.32 The obtained results highlight the versatility of the proposed approach.
Table 1
PI/TTMSS/Ph2I+PF6− combinations leading to an efficient photopolymerization reaction under air and upon sunlight exposure (tack free coating within 3 hours of irradiation; for different systems, tack free coatings are obtained for quite short sunlight irradiation—see text)
The developments of new photoinitiating systems are in progress. They are based on less usual PIs, for example acridinediones (e.g.3,4,6,7,9,10-hexahydro-(1,8) (2H,5H) acridinedione (AD))34 or transition metal carbonyls (e.g.Mn2CO10).33 They exhibit a good to excellent light absorption at λ > 390 nm and appear highly efficient in FRPCP when combined with silanes (Fig. 2). Tack free coatings are also obtained under sunlight exposure within one hour of irradiation. More recent works show that sunlight curing of EPOX or LDO can be achieved using tris(2,2′-bipyrydine)ruthenium (II)/TTMSS/Ph2I+PF6− (9 min of exposure),39tris(1,10-phenanthroline)ruthenium (II)/TTMSS/Ph2I+PF6− or tris(2-phenylpirydine)iridium/TTMSS/Ph2I+PF6− (<10 min of exposure), as PIS.
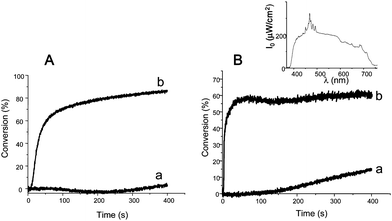 |
| | Fig. 2
Polymerization profiles of EPOX (thickness ≈ 20 µm) under air upon a Xenon lamp irradiation (λ > 390 nm; estimated light intensity ≈ 60 mW cm−2 in the range 390–800 nm range) (A) in the presence of (a) AD/Ph2I+ (3%/2%, w/w); (b) AD/TTMSS/Ph2I+ (3%/3%/2%, w/w) and (B) in the presence of (a) Mn2CO10/Ph2I+ (1%/1%, w/w) and (b) Mn2CO10/TTMSS/Ph2I+ (1%/3%/1%, w/w). Inset: emission spectrum of the xenon lamp. | |
| | | R3Si˙ + monomer (M) → R3Si–M˙ | (8) |
In previous works, phosphine oxide derivatives were proposed for the sunlight induced polymerization of very viscous media.25 In such a matrix (see above), the oxygen inhibition can be easily overcome even for a low excitation light intensity. In our present study, BAPO was used as PI for the photopolymerization of an epoxy acrylate (Ebecryl 605). Interestingly, Fig. 3 shows that the addition of TTMSS still significantly improves the polymerizationi.e. tack free coatings are obtained after 12 min vs. 1 hour using BAPO/TTMSS (2%/3%, w/w) instead of BAPO alone (2% w/w), respectively. For other phosphine oxides (e.g.TPO) a similar effect of TTMSS is found i.e. tack free coatings are obtained for 12 min and 45 min of sunlight irradiation using TPO/TTMSS and TPO, respectively (Fig. 3C).
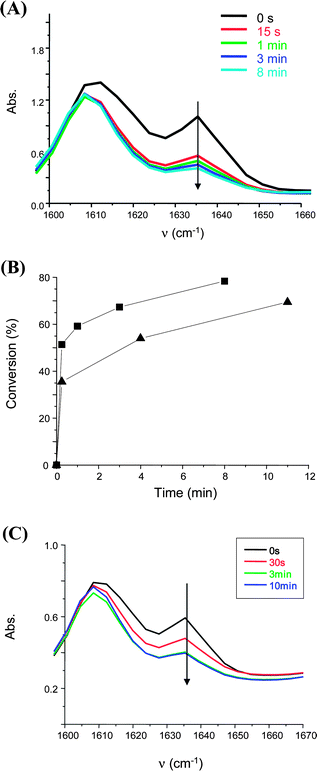 |
| | Fig. 3 Radical photopolymerization of an epoxy acrylate (Ebecryl 605) film. Photoinitiating system: BAPO (2%, w/w)/TTMSS (3%, w/w). Under air. Sunlight exposure in Mulhouse France; May 2010. (A) Evolution of the acrylateIR band. (B) Photopolymerization profile. Photoinitiating systems: (squares) BAPO/TTMSS (2%/3%, w/w) and (triangles) BAPO (2%, w/w). (C) Photopolymerization profile. Photoinitiating system: TPO (2%, w/w)/TTMSS (3%, w/w). | |
Up to now, under sunlight, however, tack free coatings cannot be obtained with low viscosity monomers such as trimethylolpropane triacrylate (TMPTA) or ethoxylated pentaerythritol tetraacrylate (EPT).
Conclusions
Sunlight induced polymerization and crosslinking might be a promising way for a practical low cost outdoor curing process, specially designed for large dimension items. Usual synthetic monomers, oligomers, prepolymers, polymers as well as renewable monomers and naturally derived materials can be used. Various photopolymerizable systems such as conventional cationic or radical bulk formulations, mixed radical/cationic matrices, water-borne latexes and dispersions are usable. The nature of the matrix, its viscosity, its reactivity towards the photoinitiating species, the presence of a pigment governs for a large part the possibility of an efficient curing under air and upon a low light intensity. FRP, CP and FRPCP are operative. Research works on the design of sunlight photosensitive matrixes for applications such as those presented in ref. 13a (orthopedic casts, assembly of catheters, in-field repair of equipment, fiber reinforced composite boats, waterstorage and delivery systems, building panels) as well as for the development of e.g. anti-soiling coatings, paints, protective coatings, strongly adhesive glass laminates, quick setting glues for the bonding of glass to a substrate should presumably know a growing interest in the coming years.
The new chemistry involving the silyl chemistry should really allow new opportunities in FRP and FRPCP. Formerly developed matrices could know a revival of interest. For example, one might expect that the AM/silane two-component photoinitiating systems are usable for the radical sunlight curing of paints based on latex/DCPA or latex/PEGDA formulations. In the same way, curing under standard household fluorescent bulbs for indoor applications under air also requires highly reactive (under low light intensities) and well designed PIS that are rather similar to those developed here for sunlight curing: works are under progress in this area.39
References
-
(a)
Lasers in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, CRC Press, Boca Raton, 1990 Search PubMed;
(b)
Radiation Curing of Polymeric Materials, ed. C. E. Hoyle and J. F. Kinstle, ACS Symposium Series 417, 1990 Search PubMed;
(c)
Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier Science Publishers Ltd, London, 1993 Search PubMed;
(d)
Chemistry and Technology of UV and EB Formulation for Coatings, Inks and Paints, ed. P. K. T Oldring, Sita Technology, London, 1994 Search PubMed;
(e)
Current Trends in Polymer Photochemistry, ed. N. S. Allen, M. Edge, I. R. Bellobono and E. Selli, Ellis Horwood, New-York, 1995 Search PubMed;
(f)
J. P. Fouassier, Photoinitiation, Photopolymerization and Photocuring: Fundamental and Applications, Hanser Publishers, New York, 1995 Search PubMed;
(g)
J. V. Crivello, Photoinitiators for Free Radical, Cationic and Anionic Photopolymerization, ed. G. Bradley, New York, 2nd edn, 1998 Search PubMed;
(h)
D. C. Neckers and W. Jager, UV and EB at the Millenium, Sita Technology, London, 1999 Search PubMed;
(i)
S. Davidson, Exploring the Science, Technology and Application of UV and EB Curing, Sita Technology Ltd, London, 1999 Search PubMed;
(j)
K. Dietliker, A Compilation of Photoinitiators Commercially Available for UV today, Sita Technology Ltd, Edinburgh, London, 2002 Search PubMed;
(k)
Photoinitiated Polymerization, ed. K. D. Belfied and J. V. Crivello, ACS Symposium series 847, Washington, DC, 2003 Search PubMed;
(l)
Photochemistry and UV Curing, ed. J. P. Fouassier, Research Signpost, Trivandrum, India, 2006 Search PubMed;
(m)
R. Schwalm, UV Coatings: Basics, Recent Developments and New Applications, Elsevier, Oxford, UK, 2007 Search PubMed;
(n)
W. Schnabel, Polymers and Light: Fundamentals and Technical Applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
-
(a)
J. P. Fouassier, X. Allonas and J. Lalevée, in Macromolecular Engineering: from Precise Macromolecular Synthesis to Macroscopic Materials Properties and Applications, ed. K. Matyjaszewski, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, 2007, vol. 1, pp. 643–672 Search PubMed;
(b)
X. Allonas, C. Crouxte-Barghorn, J. P. Fouassier, J. Lalevée, J. P. Malval and F. Morlet-Savary, in Lasers in Chemistry, ed. M. Lackner, Wiley-VCH., Weinheim, 2008, vol. 2, pp. 1001–1027 Search PubMed;
(c) C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
-
(a) A. Ledwith, Polymer, 1978, 19, 1217–1219 CrossRef CAS;
(b) H. Baumann and H. J. Timpe, Z. Chem., 1984, 24, 18–19 CAS;
(c) Y. Yagci and W. Schnabel, Makromol. Chem., Rapid Commun., 1987, 8, 209–213 CrossRef CAS;
(d) Y. Yagci, I. Kminek and W. Schnabel, Polymer, 1993, 34, 426–428 CrossRef CAS;
(e) Y. Yagci and A. Ledwith, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1911–1918 CrossRef CAS;
(f) J. V. Crivello and M. Sangermano, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 343–356 CrossRef CAS;
(g) C. Dursun, M. Degirmenci, Y. Yagci, S. Jockusch and N. J. Turro, Polymer, 2003, 44, 7389–7396 CrossRef CAS;
(h) J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 474–483 Search PubMed;
(i) N. Arsu, G. Hizai and Y. Yagci, Macromol. Rep., 1995, 1257–1262 Search PubMed;
(j) Y. Yagci and I. Reetz, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef CAS.
-
(a) M. El-Roz, M.-A. Tehfe, J. Lalevée, B. Graff, X. Allonas and J.-P. Fouassier, Macromolecules, 2010, 43, 2219–2227 CrossRef CAS;
(b) J. Lalevée, X. Allonas, S. Jradi and J.-P. Fouassier, Macromolecules, 2006, 39, 1872–1879 CrossRef CAS.
- G. K. Oster, G. Oster and G. Pratti, J. Am. Chem. Soc., 1957, 79, 595–599 CrossRef CAS.
-
(a) M.-A. Tehfe, N. Blanchard, C. Fries, J. Lalevée, M. El-Roz, X. Allonas and J. P. Fouassier, Makromol. Chem., Rapid Commun., 2009, 31, 473–478;
(b) J. Lalevée, M. El-Roz, M.-A. Tehfe, X. Allonas and J.-P. Fouassier, Macromolecules, 2009, 42, 8669–8674 CrossRef;
(c) J. Lalevée, M. El-Roz, F. Morlet-Savary, X. Allonas and J.-P. Fouassier, Macromol. Chem. Phys., 2009, 210, 311–319 CrossRef CAS;
(d) M. El-Roz, F. Morlet-Savary, J. Lalevée, X. Allonas and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7369–7375 CrossRef CAS;
(e) J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS;
(f) X. Allonas, H. Obeid, J. P. Fouassier, M. Kaji, Y. Ichihashi and Y. Murakami, J. Photopolym. Sci. Technol., 2003, 16, 123–128 CrossRef CAS;
(g) D. Burget, C. Mallein and J. P. Fouassier, Polymer, 2003, 44, 7671–7678 CrossRef CAS;
(h) D. Burget, J. P. Fouassier, F. Amat-Guerri, R. Sastre and R. Mallavia, Acta Polym., 1999, 50, 337–346 CrossRef CAS;
(i) E. Chesneau and J. P. Fouassier, Angew. Makromol. Chem., 1985, 135, 41–64 CrossRef CAS;
(j) K. Yamashita, S. Imahashi, F. Morlet-Savary and J. P. Fouassier, Polymer, 1996, 38, 1415–1421.
-
(a)
H. J. Timpe, in Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier, Elsevier, Barking UK, 1993, vol. 2, pp. 529–554 Search PubMed;
(b)
A. F. Cunningham and V. Desobry, in Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier and J. F. Rabek, Elsevier, Barking UK, 1993, vol. 2, pp. 323–374 Search PubMed.
- C. K. Nguyen, C. E. Hoyle, T. Y. Lee and S. Joensson, Eur. Polym. J., 2007, 43, 172–177 CrossRef CAS.
- K. S. Padon and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3336–3346 CrossRef CAS.
- J. Kabatc, M. Pietrzak and J. Paczkowski, J. Chem. Soc., Perkin Trans. 2, 2002, 287–295 RSC.
-
(a) J. Jakubiak and J. F. Rabek, Polimery, 1999, 44, 447–461 Search PubMed;
(b)
J. F. Rabek, L. A. Linden, J. F. Fouassier, J. Nie, E. Andrzejewska, J. Paczkowski, J. Jakubiak, A. Wrzyszczynski and A. Sionkowska, Trends in Photochemistry and Photobiology: Photosensitive Systems for Photopolymerization Reactions, ed. J. P. Fouassier, Research Trends, Trivandum, India, 1999, vol. 5, pp. 51–62 Search PubMed;
(c) J. Nie, E. Andrzejewska, J. F. Rabek, L. A. Linden, J. P. Fouassier, J. Paczkowski, F. Scigalski and A. Wrzyszczynski, Macromol. Chem. Phys., 1999, 200, 1692–1701 CrossRef CAS;
(d) T. Urano, J. Photopolym. Sci. Technol., 2003, 16, 129–156 CrossRef CAS;
(e) T. Takahashi, H. Watanabe, N. Miyagawa, S. Takahara and T. Yamaoka, Polym. Adv. Technol., 2002, 13, 33–39 CrossRef CAS;
(f)
M. Riediker, K. Meier and H. Zweifel, Ciba Geigy, Eur. Pat., 186626, 1986;
(g) D. Eaton, Photogr. Sci. Eng., 1979, 23, 150–154 Search PubMed;
(h) A. D. liu, A. D. Trifunac and V. Krongrauz, J. Phys. Chem., 1991, 95, 5822–5827 CrossRef CAS.
-
(a) B. Ganster, U. K. Fischer, N. Moszner and R. Liska, Macromol. Rapid Commun., 2008, 29, 57–62 CrossRef CAS;
(b) B. Ganster, U. K. Fischer, N. Moszner and R. Liska, Macromolecules, 2008, 41, 2394–2400 CrossRef CAS;
(c) Y. Y. Durmaz, N. Moszner and Y. Yagci, Macromolecules, 2008, 41, 6714–6718 CrossRef CAS.
-
(a) J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866–875 CrossRef CAS;
(b) J. V. Crivello and U. Bulut, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5217–5321 CrossRef CAS;
(c) U. Bulut, G. E. Gunbas and L. Topare, J. Polym. Sci., Part A: Polym. Chem., 2009, 48, 209–213.
-
(a) M. Degirmenci, A. Onen, Y. Yagci and S. P. Pappas, Polym. Bull., 2001, 46, 443–449 CrossRef CAS;
(b) D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- J. Yang and D. C. Neckers, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3836–3841 CrossRef CAS.
-
B. H. Trevor, Du Pont de nemours, US Pat., 3320198, 1967.
-
Fr Pat., 1551563, 1968;
Chem. Abstr., 1969, 71, 40371u, Hoescht AG Search PubMed.
-
L. S. Frankel and D. H. Clemens, Rohm and Hass, Eur. Pat., 10000, 1979.
-
R. Reeb, Rhone poulenc, Fr. Pat., 2697530, 1994.
-
G. Rehmer, M. G. Rau, E. Wistuba and A. Bottcher, BASF AG, Eur. Pat., 417568, 1991.
-
W. D. Emmons and K. Nyi, Rohm and Hass, US. Pat., 4261872, 1981.
- G. Eiselé, J. P. Fouassier and R. Reeb, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 2333–2345 CrossRef CAS.
- C. Bibaut-Renauld, D. Burget, J. P. Fouassier, C. G. Varelas, J. Thomatos, G. Tsagaropoulos, L. O. Ryrfors and O. J. Karlsson, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3171–3181 CrossRef CAS.
- C. Decker and T. Bendaikha, J. Appl. Polym. Sci., 1998, 70, 2269–2282 CrossRef CAS.
- C. Decker, T. Nguyen Thi Viet and H. Le Xuan, Eur. Polym. J., 1996, 32, 1319–1326 CrossRef CAS.
- J. V. Crivello, R. Narayan and S. S. Sterstein, J. Appl. Polym. Sci., 1997, 64, 2073–2079 CrossRef CAS.
- W. Chiang and W. T. Lin, J. Appl. Polym. Sci., 1994, 51, 1901–1909 CrossRef CAS.
- W. Chiang and F. C. Ding, J. Appl. Polym. Sci., 2002, 86, 1878–1885 CrossRef CAS.
- H. L. Zang, J. L. Massingill and J. T. Woo, J. Coat. Technol., 2000, 72, 79–81 CrossRef.
- C. Decker, K. Zahouily, D. Decker and T. Nguyen Thi Viet, Polymer, 2001, 42, 7551–7560 CrossRef CAS.
- B. Paczkowska, S. Strzelec, L. A. Linden and J. Packowski, Appl. Clay Sci., 2004, 25, 221–227 CrossRef CAS.
- M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS.
- M. A. Tehfe, J. Lalevée, D. Gigmes and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1830–1837 CrossRef CAS.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, N. Blanchard, C. Fries, B. Graff, X. Allonas, F. Louërat and J.-P. Fouassier, Eur. Polym. J., 2010, 46, 2138–2144 CrossRef CAS.
-
(a) J. Lalevée, M. El-Roz, X. Allonas and J.-P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2008–2014 CrossRef CAS;
(b) J. Lalevée, N. Blanchard, B. Graff, X. Allonas and J.-P. Fouassier, J. Organomet. Chem., 2008, 693, 3643–3649 CrossRef CAS.
-
F. Castellanos, J. Cavezzan, J. P. Fouassier and C. Priou, Rhodia, US. Pat. 5668192, 1998.
-
(a) J. Lalevée, X. Allonas and J.-P. Fouassier, J. Org. Chem., 2007, 72, 6434–6439 CrossRef CAS;
(b) M. El-Roz, J. Lalevée, X. Allonas and J.-P. Fouassier, Macromolecules, 2009, 42, 8725–8732 CrossRef CAS.
- K. Ren, J. H. Malpert, H. Li, H. Gu and D. C. Neckers, Macromolecules, 2002, 35, 1632–1637 CrossRef CAS.
- J. Lalevée, N. Blanchard, M.-A. Tehfe, F. Morlet-Savary and J.-P. Fouassier, Macromolecules, 2010, 43, 10191–10195 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.