Thermo-responsive cellulose-based architectures: tailoring LCST using poly(ethylene glycol) methacrylates†‡
Christian
Porsch
,
Susanne
Hansson
,
Niklas
Nordgren
and
Eva
Malmström
*
KTH Royal Institute of Technology, School of Chemical Engineering, Dept. of Fibre and Polymer Technology, SE-100 44, Stockholm, Sweden. E-mail: mavem@kth.se; Fax: +46 8 790 8283; Tel: +46 8 790 7225
First published on 16th February 2011
Abstract
There is a growing interest in designing advanced macromolecular architectures applicable for instance in drug delivery systems. Employing cellulose in these systems is particularly favorable due to attractive properties such as biocompatibility and low price. Additionally, thermo-responsive polymers of poly(ethylene glycol) methacrylates are promising in this field owing to their biocompatibility and non-toxicity. In the present study, amphiphilic thermo-responsive homo- and copolymers of oligo(ethylene glycol) methyl ether methacrylate (OEGMA300) and di(ethylene glycol) methyl ether methacrylate (DEGMA) were synthesized via ARGET ATRP. Both linear copolymers of DEGMA/OEGMA300 as well as comb architectures with copolymers of DEGMA/OEGMA300 grafted from hydroxypropyl cellulose were produced. The lower critical solution temperature of the linear copolymers was readily tailored by altering the monomer feed ratio. The grafting of the thermo-responsive polymers from hydroxypropyl cellulose resulted in a consistent decrease of the lower critical solution temperature compared to the linear analogues; however, interestingly the ability to tune the transition temperature remained. Moreover, the amphiphilic comb architectures formed polymeric micelles with low critical micelle concentrations. Consequently, these advanced architectures combine the favorable properties of hydroxypropyl cellulose with the interesting thermo-responsive and stealth properties of poly(ethylene glycol) methacrylates, and may, therefore, find potential applications in biomedicine.
Introduction
Cellulose is the most abundant polysaccharide, and an important sustainable raw material.1 During the last decades, cellulose and its derivatives have attracted significant interest in many fields, as a result of increased environmental concerns. Additionally, cellulose exhibits many attractive properties, such as renewability, biocompatibility, and good availability in nature. However, due to the hydrophilic nature of cellulose, modification is often required to tailor the final properties for specific applications. This can be accomplished by grafting polymers onto the cellulose surface, thus enhancing existing properties or adding new desired ones. Our group has extensively been grafting various polymers from a number of different cellulose substrates, for example: filter paper,2–5 cellulose microspheres,6 microcrystalline cellulose (MCC),7 microfibrillated cellulose (MFC),8 and hydroxypropyl cellulose (HPC).9HPC is a linear cellulose derivative consisting of a glucose-unit backbone, which has been propoxylated (see Fig. 1). Owing to the decreased polarity, HPC is fairly soluble, which in combination with the superb inherent properties of cellulose mentioned above makes it of great interest in biomedical applications. Recently, Malmström et al.10,11 reported how HPC can be grafted with PCL-block-PAA arms, obtaining nanocontainers suitable for guest molecule encapsulation. | ||
| Fig. 1 Repeating unit of hydroxypropyl cellulose (HPC) and the monomers used in the present study. | ||
The ability to tailor material properties by employing advanced macromolecular architectures has gained increased attention in biomedicine.12 Earlier, interesting architectures like graft copolymers suffered from poor control during polymerization of the grafts, which obstructed their use in many applications. However, the progress of controlled radical polymerization techniques has enabled synthesis of products with well-defined architectures, pre-determined molecular weights, and low polydispersity indices.13 The most common controlled radical polymerization technique, atom transfer radical polymerization (ATRP), was introduced in 1995 independently by the groups of Matyjaszewski14 and Sawamoto,15 and has proved to be a versatile method for preparing cellulose-based graft copolymers.16–19 In ATRP, a catalyst complex ensures a low concentration of radicals, thus suppressing irreversible termination reactions. However, due to ATRP's sensitivity to air and need for high amounts of catalyst, Matyjaszewski and Pintauer20 developed an improved technique: activators regenerated by electron transfer (ARGET) ATRP. In contrast to conventional ATRP, a reducing agent is employed in ARGET ATRP, which significantly reduces the amount of catalyst required to obtain a controlled polymerization. The technique also allows the polymerization to be conducted in the presence of small amounts of air. Consequently, less effort can be devoted to deoxygenation, which simplifies the reaction procedure.
Cellulose-based, amphiphilic graft copolymers are investigated as components in polymeric micelles, which can be used as carriers in drug delivery systems.16,21,22 Above the critical micelle concentration (CMC) these structures may associate into a core–shell morphology, where the hydrophobic core enables solubilization of a hydrophobic drug, and the hydrophilic shell ensures micelle solubility in the water solution. Additional benefits of polymeric micelles as drug delivery systems are for example long circulation times, improved biodistribution, and enhanced possibility to cross physiological barriers.
Thermo-responsive polymers have the ability to go through a phase transition in aqueous solution, switching from a hydrophilic to hydrophobic character upon heating. This phase transition, referred to as the lower critical solution temperature (LCST), is present due to inter- and intramolecular hydrogen bonds between water molecules and the polymer chains. Recently, Hu et al. demonstrated that HPC exhibits thermo-responsive properties with a phase transition temperature of 41 °C.23 Further, the most widely studied thermo-responsive polymer is poly(N-isopropylacrylamide) (PNIPAM), mainly due to the fact that its LCST (∼32 °C) is close to the physiological temperature.24 However, recently published results by Vihola et al.25 suggest that acrylamide-based polymers show cytotoxicity at the physiological temperature, which is believed to be related to the presence of unreacted monomeric residues. A promising group of thermo-responsive polymers is thus poly(ethylene glycol) methacrylates (PEGMAs), which have shown to be non-toxic25,26 and biocompatible.27–30 Furthermore, the LCST of these polymers can be tailored by changing the length of the ethylene glycol segment in the side chain.31 Longer ethylene glycol segments result in a more hydrophilic nature of the polymer, thus increasing the LCST. As a result, the LCST can be tuned in the range of 26–90 °C by the appropriate choice of monomer. However, none of the available PEGMAs shows an LCST close to the physiological temperature, which has been obstructing their possibility to compete with PNIPAM in biomedical applications. In 2006, Lutz et al.29,32 elegantly showed that the LCST of PEGMAs could be finely adjusted by random copolymerization of di(ethylene glycol) methyl ether methacrylate (DEGMA) and oligo(ethylene glycol) methyl ether methacrylate (OEGMA475) viaATRP; thus, widening the applicability of these polymers. Later, Schubert et al.31 showed that the tailoring of the LCST also can be achieved by using RAFT polymerization. These progresses in tailoring the responsive behavior of oligo(ethylene glycol) methacrylate-based polymers, combined with their biocompatibility and desirable stealth properties,27 have resulted in extensive research during the last years.28,33
In the present work, ARGET ATRP is employed as a straightforward technique for preparation of thermo-responsive copolymers of OEGMA300 and DEGMA with tunable LCSTs. Furthermore, comb copolymers of HPC and the thermo-responsive copolymers are synthesized, employing surface-initiated ARGET ATRP. These interesting architectures are an additional step towards the understanding of the fascinating solution properties, and potential applicabilities, of advanced poly(ethylene glycol)methacrylate structures.
Experimental section
Materials
Hydroxypropyl cellulose (HPC, Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Mn = 10000 g mol−1 according to manufacturer (Aldrich); Mw = 74000 g mol−1, Mn = 34000 g mol−1 was obtained using SEC with THF as a mobile phase; molar substitution of propoxy groups (MS) was determined to be 2.9 using 1H-NMR (Fig. S1, ESI‡)). 4-(Dimethylamino)pyridine (DMAP, 99%), ethyl 2-bromoisobutyrate (EBiB, 98%), 2-bromisobutyryl bromide (BiB, 98%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), 2,2′-bipyridyl (Bipy, >99%), ascorbic acid (AsAc, >99%) and α-bromoisobutyric acid were purchased from Aldrich. Copper(II) bromide (Cu(II)Br2, 99%) and N, N′-dicyclohexylcarbodiimide (DCC, 99%) were purchased from Acros Organics. The monomers oligo(ethylene glycol) methyl ether methacrylate (OEGMA300, Mw 300 g mol−1, Aldrich) and di(ethylene glycol) methyl ether methacrylate (DEGMA, Mw 188 g mol−1, Aldrich) were prior to use activated by passing it through a column of neutral aluminium oxide. Phosphate buffered saline (PBS) was prepared according to the recipe in the ESI S2‡.
000 g mol−1, Mn = 10000 g mol−1 according to manufacturer (Aldrich); Mw = 74000 g mol−1, Mn = 34000 g mol−1 was obtained using SEC with THF as a mobile phase; molar substitution of propoxy groups (MS) was determined to be 2.9 using 1H-NMR (Fig. S1, ESI‡)). 4-(Dimethylamino)pyridine (DMAP, 99%), ethyl 2-bromoisobutyrate (EBiB, 98%), 2-bromisobutyryl bromide (BiB, 98%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), 2,2′-bipyridyl (Bipy, >99%), ascorbic acid (AsAc, >99%) and α-bromoisobutyric acid were purchased from Aldrich. Copper(II) bromide (Cu(II)Br2, 99%) and N, N′-dicyclohexylcarbodiimide (DCC, 99%) were purchased from Acros Organics. The monomers oligo(ethylene glycol) methyl ether methacrylate (OEGMA300, Mw 300 g mol−1, Aldrich) and di(ethylene glycol) methyl ether methacrylate (DEGMA, Mw 188 g mol−1, Aldrich) were prior to use activated by passing it through a column of neutral aluminium oxide. Phosphate buffered saline (PBS) was prepared according to the recipe in the ESI S2‡.
Instrumentation
000 Da) from Tosoh Biosep, a VE 5200 GPCautosampler, a VE 1121 GPCsolventpump, and a VE 5710 GPC degasser (all from Viscotek Corp.). A calibration method was created using broad and narrow linear polystyrene standards. Corrections for the flow rate fluctuations were made using toluene as an internal standard. Viscotek OmniSEC version 4.0 software was used to process the data.
General procedure for polymerization of linear polymers
A typical polymerization of the linear polymers was conducted as described below, and the comonomer composition was altered to achieve different copolymers. The targeted monomer composition (20 mmol) was added to a 25 ml round bottom flask equipped with a magnetic stirrer. Anisole (50 wt%), PMDETA (8.7 mg, 50 μmol) and AsAc (8.7 mg, 50 μmol) were added, and allowed to dissolve while stirring. The reaction mixture was put on a water/ice bath (0 °C), EBiB (7.4 μl, 50 μmol) was added, and 5 minutes of purging with argon was conducted. Subsequently, Cu(II)Br2 (1.1 mg, 5 μmol) was added followed by additional 15 minutes of purging with argon. The polymerization was performed at 40 °C and NMR aliquots were withdrawn under argon flow to monitor the reaction (Fig. S12, ESI‡). The reaction was terminated by removal of the flask from the oil bath, exposure to air, and dilution with THF. Thereafter, the reaction mixture was allowed to pass through a short column of neutral aluminium oxide to remove the copper complex. Subsequently, the solution was concentrated through rotary evaporation and precipitated in diethyl ether cooled with dry ice. The solvent was decanted off and the sticky product was dried under vacuum. 1H-NMR (CDCl3): δ 0.91 (br), 1.06 (br), 1.61 (s), 1.84 (br), 1.94 (br), 3.42 (s), 3.59 (br), 3.66–3.69 (br), 4.13 (br) ppm. Typical yields of approximately 25% were achieved.Synthesis of 2-bromoisobutyryl anhydride
The procedure used was adopted from Malmström et al.102-Bromoisobutyric acid (10.0 g, 59.9 mmol) was dissolved in CH2Cl2 (DCM; 50 ml). N,N′-Dicyclohexylcarbodiimide (DCC; 6.80 g, 32.9 mmol) was dissolved in DCM (25 ml) and added to the solution. A milky white mixture was formed and was stirred at ambient temperature overnight. The precipitate was filtered off, the filtrate was concentrated through rotary evaporation, and precipitated in n-heptane cooled with dry ice. The product was filtered, washed with cold n-heptane and finally dried under vacuum. The purity of the white powder product was confirmed by NMR and achieved in a yield of 55%. 1H-NMR (CDCl3): δ 2.03 ppm. 13C-NMR (CDCl3): δ 30.12, 55.10, 165.84 ppm.Synthesis of hydroxypropyl cellulose macroinitiators (HPC-Is)
Two separate routes were employed to synthesize the HPC macroinitiator (Scheme 1, top). In one route 2-bromoisobutyryl bromide was utilized to achieve the pendant α-bromoesters, and in the other 2-bromoisobutyryl anhydride was employed.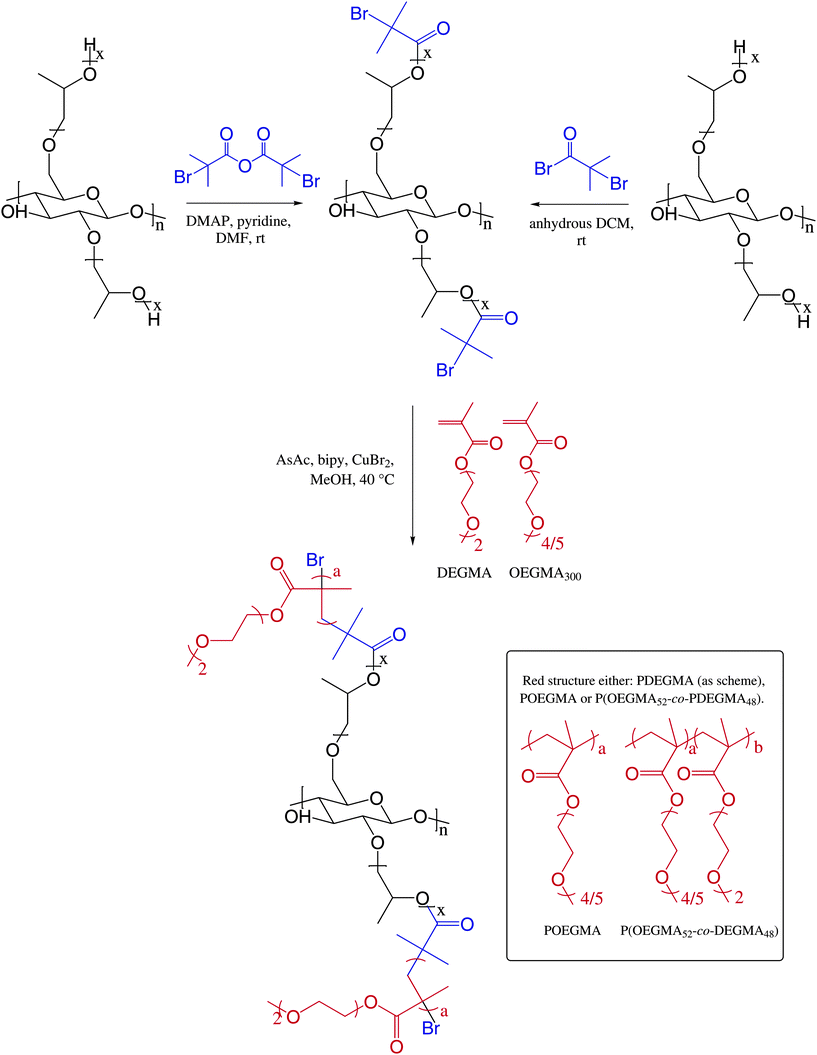 | ||
| Scheme 1 Schematic illustration of the comb polymers synthesized in this study. | ||
Synthesis of HPC-I using 2-bromoisobutyryl anhydride (HPC-I1.4)
The synthesis was conducted as described earlier in our group,10 and a light brown polymer was obtained in 85% yield. 1H-NMR (CDCl3): δ 1.16 (br), 1.28 (br), 1.94 (s), 2.70–4.20 (br), 3.60 (value of the sharpest peak inside broad peak), 4.35 (br), 5.05 (br) ppm. 13C-NMR (CDCl3): δ 16.39, 17.49, 30.75, 34.80, 56.14, 71.39, 73.44, 75.05, 99.99, 170.75–171.00 ppm.Synthesis of HPC-I using 2-bromoisobutyryl bromide (HPC0.6)
The synthesis procedure was adopted from Xu et al.18Hydroxypropyl cellulose (HPC; 0.5 g) was completely dissolved in anhydrous DCM (25 ml), and the flask subsequently put on a water/ice-bath (0 °C). 2-Bromoisobutyryl bromide (BiB; 0.81 mmol) was dissolved in 5 ml anhydrous DCM, and cooled in the water/ice-bath. Thereafter, the solution was slowly added to the HPC solution over a time period of ten minutes, the water/ice-bath was removed, and the reaction was allowed to proceed for 150 min at room temperature. The reaction was ended by precipitation in 300 ml diethyl ether cooled with dry ice. The polymer was filtered off and washed with cold diethyl ether. The pure product was dried under vacuum overnight, and a yield of 40% was achieved. 1H-NMR (MeOD): δ 1.16 (br), 1.93 (s), 3.00–4.10 (br), 4.50 (br), 4.99 (s) ppm. 13C-NMR (MeOD): δ 16.18, 18.47, 29.83, 55.46, 66.04, 66.39, 75.01, 76.61, 78.91, 83.00, 102.33, 172.14 ppm.General procedure for polymerization from the macroinitiator (HPC-I)
HPC-I1.4 or HPC-I0.6 (50 mg, 0.08 mmol and 0.04 mmol of Br, respectively) was added to a round bottom flask, and completely dissolved in anisole or methanol (80 wt%), respectively. Bipy (15.6 mg, 0.10 mmol) and AsAc (17.3 mg, 0.10 mmol) were added, and allowed to dissolve during gentle stirring. The flask was put in a water/ice bath, and the desired monomer mixture of OEGMA300 and DEGMA (34 mmol in total) was added. The solution was purged with argon for 5 minutes, Cu(II)Br2 (2.2 mg, 10 μmol) was added, and additionally 15 minutes of purging was performed. The reactions were carried out in a 40 °C tempered oil bath until the viscosity of the reaction mixture had increased significantly. The reaction was terminated by withdrawal from the oil bath, air exposure, and the solution was diluted with THF. The product was purified through 48 h of dialysis (MWCO 6–8000) with THF that was changed after 2, 6 and 36 h. The product was concentrated through rotary evaporation, dried under vacuum and typical yields of approximately 32% were obtained. 1H-NMR (CDCl3): δ 0.91 (br), 1.06 (br), 1.45 (s), 1.66 (s), 1.83 (br), 1.92 (br), 3.42 (s), 3.59 (s), 3.65–3.71 (br), 4.13 (br) ppm. 13C-NMR (CDCl3): δ 17.08, 18.85, 25.59, 30.29, 44.78, 53.44, 59.01, 63.78, 67.95, 70.20, 71.91, 176.16–177.04 ppm.Results and discussion
ARGET ATRP of linear homo- and copolymers
In the first part of this work, ARGET ATRP was employed to achieve controlled polymerizations of homo- and copolymers of OEGMA300 and DEGMA. It was found that ARGET ATRP provides a simple and attractive polymerization of polar, functional vinyl monomers as OEGMA300 and DEGMA. The controlled polymerizations of the monomers into linear polymers were easily conducted by mixing the desired monomer composition, anisole, PMDETA, AsAc, and EBiB, followed by 5 minutes degassing. The polymerization was initiated by the addition of CuBr2, 15 min degassing and temperature set to 40 °C. Copolymers with different compositions of OEGMA300 and DEGMA were prepared by aiming at different initial monomer feeds, and polymers of OEGMA300, DEGMA and OEGMA300-co-DEGMA were readily synthesized. The achieved products were confirmed with 1H-NMR spectroscopy, and their characteristics are reported in Table 1. The copolymer compositions were calculated by comparing the overall integration of the methoxyprotons with the overall integration of the ethylene glycolprotons (see Fig. S2, ESI‡). A Good agreement was found between feed amounts and final composition of OEGMA and DEGMA in the copolymers. This confirms that synthesizing P(OEGMA-co-DEGMA)copolymers employing ARGET ATRP under reported conditions enables the production of pre-determined polymer compositions.| Polymer | f OEGMA (mol%) | Conv.b (%) | M n SEC c/g mol−1 | M n NMR b/g mol−1 | PDIc | R h /nm | LCST e/°C | |
|---|---|---|---|---|---|---|---|---|
| Feed | Finalb | |||||||
| a P(OEGMAx-co-DEGMAy) where x = mol% of OEGMA and y = mol% of DEGMA in the final polymer. b Determined by 1H-NMR. c Assessed by THFSEC (polystyrene calibration). d Assessed by DLS measurements (conc. 100 mg l−1) at a temperature 1 °C below LCST of the polymer. A characteristic size distribution from DLS is shown in Fig. S13, ESI2. e Assessed by DLS measurements (conc. 100 mg l−1). | ||||||||
| PDEGMA | 0 | 0 | 45 | 30400 |
50800 |
1.2 | 7 | 27 |
| P(OEGMA15-co-DEGMA85) a | 13 | 15 | 46 | 25800 |
36800 |
1.2 | 6 | 33 |
| P(OEGMA19-co-DEGMA81)a | 19 | 19 | 51 | 33000 |
41500 |
1.3 | 6 | 36 |
| P(OEGMA27-co-DEGMA73) a | 24 | 27 | 63 | 53400 |
53000 |
1.6 | 12 | 38 |
| P(OEGMA37-co-DEGMA63) a | 36 | 37 | 75 | 58100 |
66700 |
1.3 | 11 | 42 |
| P(OEGMA54-co-DEGMA46) a | 50 | 54 | 18 | 21200 |
17600 |
1.1 | 6 | 55 |
| P(OEGMA73-co-DEGMA27) a | 70 | 73 | 41 | 30000 |
43700 |
1.3 | 9 | 59 |
| POEGMA | 100 | 100 | 13 | 14100 |
15600 |
1.1 | 4 | 73 |
Results from size exclusion chromatography (SEC) in THF, calibrated with linear polystyrene standards with toluene as the internal standard, suggest relatively good control of the polymerizations with polydispersity indices between 1.1 and 1.6 (Table 1). Lower molecular weights were received from SEC than expected theoretical values calculated from 1H-NMR. This is most likely an effect of the brush-like structure of POEGMA and PDEGMA, where SEC underestimates their molecular weight since calibration is performed with linear polystyrene standards. In addition, further attempts to determine the molecular weights and molecular weight distributions of the polymers were conducted using MALDI-TOF (see Fig. S3, ESI‡). However, although several different matrices were employed, all attempts failed.
Thermo-responsive behavior of linear polymers
The solution properties of the linear polymers in water were studied with dynamic light scattering (DLS), using polymer concentrations of 100 mg l−1. As expected, all synthesized polymers showed water solubility at room temperature. Below LCST, all polymers were present as associated structures with a diameter of 4–12 nm. However, at LCST they displayed a clear phase transition upon heating, going from soluble to insoluble in the aqueous solution. The phase transition at LCST occurs when hydrogen bonds between water and the ethylene glycol segments in the polymer side chains are broken. The more favorable polymer–polymer interaction then results in a structural collapse and subsequently, a more hydrophobic polymer chain. Consequently, the collapsed chains start to aggregate, thus going from 4–12 nm to, in most cases, several hundred nanometres. As expected, further increase of the temperature above LCST resulted in additional increase in size due to further aggregation (see Fig. S3, ESI‡).The obtained LCSTs, reported in Table 1, correlate well with earlier established results for these polymers synthesized by other controlled polymerization techniques.31,34 As expected, the PDEGMAhomopolymer showed the lowest value of LCST at 27 °C. Moreover, as shown recently by Lutz et al.,34 the transition temperatures for P(OEGMA-co-DEGMA)copolymers increase with increasing molar fraction of OEGMA300 in the copolymer (Fig. 2; ■). Consequently, ARGET ATRP proved to enable a tunable responsive behavior of PEGMAs comparable to other controlled polymerization techniques. However, noteworthy is that the obtained LCST for the POEGMAhomopolymer (73 °C) is about 8–10 °C higher than earlier reported results.34,35 This is believed to be an effect of the lower molecular weight of this specific polymer sample. Results demonstrated by Lutz et al.29 and Chu et al.36 indicate that shorter ethylene glycol-based polymers may result in increased transition temperatures in comparison to longer ones.
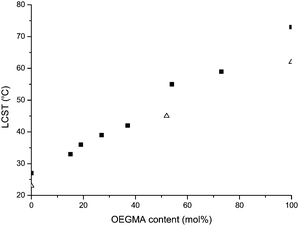 | ||
| Fig. 2 LCST as a function of OEGMA content in P(OEGMA-co-DEGMA)polymers (■) and HPC-g-P(OEGMA-co-DEGMA) polymers (△). | ||
As stated above, the achieved results confirm that ARGET ATRP is a viable technique for producing tunable responsive PEGMAs, in similarity with other controlled radical polymerization techniques reported by others. However, as discussed earlier, ARGET ATRP provides advantages compared to ordinary ATRP, such as more simple reaction conditions, and improved environmental conditions due to the reduced amount of catalyst used. To the authors' best knowledge, this is the first time tunable LCSTs of P(OEGMA-co-DEGMA) have been reported using ARGET ATRP.
Synthesis of HPC-macroinitiators
Two different macroinitiators with different initiator densities were synthesized employing routes adopted from Malmström et al.10 and Xu et al.,18 respectively (Scheme 1, top). The successful formation of the initiating moieties was confirmed by FT-IR and 1H-NMR spectroscopies, as well as ICP-SMS analysis.37 In the 1H-NMRspectra of HPC-I1.4 (Fig. S8, ESI‡) and HPC-I0.6 (Fig. S9, ESI‡), the appearance of a singlet at 1.94 ppm originates from the new methylprotons originating from the initiating moiety, thus corroborating successful esterification. FT-IR proved to be a very powerful tool to confirm successful reactions, and spectra of native HPC (a), HPC-I0.6, and HPC-I1.4 are presented in Fig. 3. The reduction of the peak at 3411 cm−1, corresponding to the available hydroxylgroups of HPC, shows that both synthetic routes resulted in successful functionalization. Additionally, the appearance of a peak related to the carbonyl stretching of the initiator at 1729 cm−1 further confirms a successful reaction. The spectrum for HPC-I0.6 shows a weaker carbonyl signal than HPC-I1.4, as well as more unreacted hydroxylgroups, thus indicating that the route employing 2-bromoisobutyric acid is not as effective, and provides a macroinitiator with a lower density of initiating moieties. This result is assumed to be an effect of the solvent, rather than the lower reactivity of the bromo acid compared to the anhydride. DMF is a more efficient solvent than DCM, thus suggesting that the hydroxylgroups are more available for substitution. Consequently, DMF facilitates the esterification better than DCM. The lower amount of initiator in HPC-I0.6 was further confirmed using ICP-SMS analysis, which determined the bromine content in HPC-I1.4 to be 20.1 wt% and in HPC-I0.6 to be 11.4 wt%. This was used to calculate the total degree of substitution (DS) of the macroinitiators (see the ESI S1‡), and 1.41 and 0.59 of the 3 available hydroxylgroups of each glucose unit were converted for HPC-I1.4 and HPC-I0.6, respectively. This states that two macroinitiators with different initiator densities were achieved, thus enabling synthesis of comb copolymers with different grafting densities.
ARGET ATRP from the macroinitiators
By employing ARGET ATRP, thermo-responsive polymers of OEGMA300, DEGMA and OEGMA300-co-DEGMA were grafted from HPC, forming comb polymer architectures. The HPC-Is were, prior to the reaction, allowed to dissolve completely in the solvent to prevent uneven and uncontrolled grafting. Grafting from the HPC-I1.4 macroinitiator was, as for the linear polymers, conducted in anisole. However, due to the lower amount of ATRP-initiators on HPC-I0.6, the more polar character made it insoluble in anisole. Instead, methanol was used as solvent in these polymerizations. All reactions were performed in highly diluted systems. Due to the high number of initiating sites, dilution is important to keep the local concentration of radicals low.10 By doing so, undesired termination and crosslinking reactions are avoided. In addition, due to the diluting effect of the monomer, the reactions were stopped at low conversions (typically 20–25%), since higher monomer conversions increase the probability of radical coupling reactions.Characteristics of the synthesized comb copolymers are reported in Table 2. The polymerizations performed from the HPC-I0.6 macroinitiator in methanol proceeded in a repeatable manner, thus forming HPC-based comb copolymers in 4 hours. However, the polymerizations using the HPC-I1.4 macroinitiator were less reproducible and showed tendencies of undesired intermolecular coupling and gelation, even at much diluted systems. This corroborates that the higher concentration of radicals, due to the significantly higher density of initiators of HPC-I1.4, increases the probability of radical coupling reactions. As an effect, HPC-I1.4 was successfully grafted only with PDEGMA, which most likely is due to the shorter side chains of PDEGMA compared to both POEGMA and P(OEGMA-co-DEGMA). Although only one copolymer was synthesized from HPC-I1.4, the more substituted HPC backbone is interesting in order to understand how the architectural difference affects the characteristics of the polymer.
| Polymer | f OEGMA (mol%) | Conv.a (%) | R h /nm | LCST c/°C | CMC/μg ml−1 | |
|---|---|---|---|---|---|---|
| Fluorescenced | Surface tensione | |||||
| a Final OEGMA content determined by 1H-NMR. b Assessed by DLS measurements (conc. 500 mg l−1) at a temperature 1 °C below LCST of the polymer. A characteristic size distribution from DLS is shown in Fig. S14, ESI2. Since DLS measurements are performed on carbohydrates the size distributions are somewhat broad. Therefore, the reported values are rounded off to tenth nanometres. c Assessed by DLS measurements (conc. 500 mg l−1). d Determined by the fluorescent probe technique. e Determined by surface tension measurements. | ||||||
| HPC1.4-g-PDEGMA | 0 | 20 | 40 | 22 | 0.12 | 11.3 |
| HPC0.6-g-PDEGMA | 0 | 21 | 20 | 23 | 0.65 | 10.5 |
| HPC0.6-g-P(OEGMA52-co-DEGMA48) | 52 | 28 | 30 | 45 | 0.55 | 11.9 |
| HPC0.6-g-POEGMA | 100 | 35 | 30 | 62 | ||
Successful grafting from the macroinitiators was confirmed with 1H-NMR and FT-IR spectroscopies. In the 1H-NMRspectrum of HPC0.6-g-PDEGMA (see the ESI S11‡), peaks from PDEGMA can easily be observed, confirming successful grafting of the polymers onto the cellulose backbone. However, due to the very small amount of HPC backbone compared to polymer grafts, as well as a concealing effect, no peaks originating from HPC could be detected. This has also been reported earlier.10,38 Grafting was further confirmed by FT-IR analysis (Fig. 4). The shift of the peak corresponding to the carbonyl stretching from 1732 cm−1 to 1725 cm−1, and its somewhat broadened appearance in the HPC0.6-g-PDEGMAspectrum, compared to the spectrum of the HPC-I0.6 macroinitiator, is most certainly due to the presence of new ester moieties in the new material. Furthermore, the clear change of appearance in the region around 2900 cm−1 is a consequence of the dominance of methylenes in the ethylene glycol segments of HPC0.6-g-PDEGMA.
 | ||
| Fig. 4 FT-IR of HPC-I0.6 (a) and HPC0.6-g-PDEGMA (b). | ||
Thermo-responsive behavior of comb polymers
The solution properties of the comb copolymers were determined by DLS, fluorescence spectroscopy and surface tension measurements, and are reported in Table 2. DLS was employed to obtain information about the polymers' response to changes in temperature, and to understand how the new architectures affect the phase transition temperature compared to their linear analogues. DLS measurements were performed in aqueous solutions, and all comb polymers were water soluble. For the comb copolymers, polymer concentrations of 500 mg l−1, instead of 100 mg l−1 as for the linear polymers, were required for DLS measurements. As a consequence, to enable accurate comparisons, DLS measurements of linear PDEGMA, P(OEGMA54-co-DEGMA46) and POEGMA in Fig. 5 were performed at a 500 mg l−1 concentration. | ||
| Fig. 5 Size as a function of temperature for HPC-based comb polymers and their linear analogues: PDEGMA (■), HPC1.4-g-PDEGMA (★), HPC0.6-g-PDEGMA (□), P(OEGMA54-co-DEGMA46) (▲), HPC0.6-g-P(OEGMA52-co-DEGMA48) (△), POEGMA (●), and HPC0.6-g-POEGMA (○). Insets: (a) polymer sample below LCST in aqueous solution and (b) polymer sample above LCST in aqueous solution. | ||
In the fully soluble state, i.e. below the LCST of the grafts, the comb polymers formed self-assemblies with sizes in the range 20–40 nm. This correlates well with earlier reported results for similar structures,21,22 and shows that the comb polymers form considerably larger micellar assemblies than the linear polymers. Moreover, at the LCST the comb polymers show the same distinct and instant increase in size as the linear polymers, resulting in a collapse and the formation of micellar aggregates (Fig. 5). However, the observed LCSTs were all lower than for the linear analogues (Table 2). This decrease in LCST has previously been observed for similar systems,17 and is most likely due to the increased hydrophobic character of the comb polymer structures. Additionally, the restricted mobility of the polymer chains when grafted to the cellulose backbone is believed to further increase this effect. However, it is noteworthy to consider the slightly lower OEGMA300 content in HPC0.6-g-P(OEGMA52-co-DEGMA48) compared to P(OEGMA54-co-DEGMA46), which surely contributes to the lowered LCST in this case. In addition, the significantly higher molecular weights of the comb polymers may contribute to the lowered LCSTs. The fact that the lowering of the LCST for HPC0.6-g-PDEGMA is less pronounced, in relation to its linear analogue, than observed in the case of both HPC0.6-g-P(OEGMA52-co-DEGMA48) and HPC0.6-g-POEGMA, further indicates the relevance of the molecular weight.
As observed when comparing HPC0.6-g-PDEGMA with the more dense HPC1.4-g-PDEGMA, the increase in grafting density has a rather small influence on the transition temperature, lowering the LCST by only 1 °C. However, this indicates that the degree of substitution influences the solution properties for these kind of structures. The effects of substitution on the aggregation behavior for ungrafted cellulose structures have recently been demonstrated by Bodvik et al.39 In addition, a striking result is that although the LCST is generally lowered for the HPC-grafted polymers, the ability to tune the transition temperature with OEGMA content remains (see Fig. 2; △). This suggests that the attractive behavior of PEGMAs favourably can be utilized in advanced macromolecular designs in the same way as for the linear polymers.
Self-assembly of comb polymers
One potential application for these advanced architectures is as carriers in drug delivery systems. In the present study, micellization of the HPC-based copolymers was studied using the fluorescent probe technique and surface tension measurements. Pyrene used as fluorescent probe exhibits different fluorescence characteristics depending on the polarity of the solubilizing medium. This can favorably be employed to determine the critical micelle concentration (CMC) of a polymer in solution.40 Furthermore, since earlier studies41,42 indicate discrepancies between CMCs obtained with different methods, surface tension measurements were used as a complementary technique to determine CMC of the polymers. Surface active amphiphilic polymers decrease the surface tension of an aqueous solution with increasing polymer concentration. At the CMC the air–water interface is saturated with amphiphilic molecules and micelles start to form in the bulk liquid.43 Consequently, the surface tension is almost constant for polymer concentrations above the CMC since the amphiphilic molecules will then end up in micelles.Results from fluorescent spectroscopy and surface tension measurements show that HPC1.4-g-PDEGMA, HPC0.6-g-PDEGMA and HPC0.6-g-P(OEGMA52-co-DEGMA48) form micelles above the CMC, as reported in Table 2. Fig. 6 shows the change in fluorescence characteristics of pyrene as a function of HPC1.4-g-PDEGMA concentration. The observed decrease in intensity ratio (I1/I3) originates from the entrapment of pyrene in the hydrophobic core of the formed polymeric micelles. The obtained CMCs from the fluorescent probe technique are in the range 0.12–0.65 μg ml−1. This is in good agreement with reported values for similar architectures,21 and indicates that the synthesized materials self-assemble into micelles at low concentrations, which is essential for possible use as drug carriers. The difference in grafting densities of HPC1.4-g-PDEGMA and HPC0.6-g-PDEGMA resulted in an increase of CMC from 0.12 μg ml−1 to 0.65 μg ml−1, thus demonstrating that the micelle formation is affected by the polymer architecture. The longer grafts in HPC0.6-g-PDEGMA seem to facilitate micelle formation more than the shorter and more dense grafts of HPC1.4-g-PDEGMA. A recent study44 from our group suggests that the architecture, as well as the molecular weight, clearly affects the critical micelle concentration of advanced amphiphilic architectures.
 | ||
| Fig. 6 Change in the fluorescence characteristics of pyrene as a function of HPC1.4-g-PDEGMA concentration. | ||
In Fig. 7, surface tension is shown as a function of HPC1.4-g-PDEGMA concentration. An increased concentration of the comb polymer lowers the surface tension of the solution, corroborating surface active properties. The stabilization of the surface tension at increased concentration verifies the formation of micelles, i.e. CMC is reached. The CMCs determined by surface tension measurements, ranging from 10.5–11.9 μg ml−1, are consistently higher compared to the values obtained using fluorescence spectroscopy. This difference between the methods has been observed earlier, for both low42 and high41 molecular weight surfactants. However, important to bear in mind is that the polymers discussed in this study exhibit very elaborate architectures, resulting in a more complex association behavior compared to ideal low molecular weight surfactant systems. Furthermore, factors such as polydispersity and physical restraints of the architecture most certainly affect their assembly. In addition, the pyrene–copolymer interaction may affect the formation of micelles, thereby influencing the obtained values of CMC. This effect has earlier been observed for low molecular weight surfactant systems when using pyrene as a probe in fluorescent measurements.42
 | ||
| Fig. 7 Change in surface tension as a function of concentration of HPC1.4-g-PDEGMA solutions. | ||
In the case of HPC0.6-g-POEGMA no CMC was observed neither by fluorescence spectroscopy nor surface tension measurements (in the investigated concentration range). However, the hydrodynamic radius obtained from DLS (30 nm) suggests that HPC0.6-g-POEGMA forms self-assembled structures in solution. In addition, the surface tension of the solution was lowered, indicating that HPC0.6-g-POEGMA exhibits surface active properties.
Conclusions
Linear homo- and copolymers of oligo(ethylene glycol) methyl ether methacrylate (OEGMA300) and di(ethylene glycol) methyl ether methacrylate (DEGMA) were successfully synthesized via ARGET ATRP. By altering the initial monomer feed ratio, copolymers with tunable LCSTs were produced, thus enabling polymers with LCSTs in between 27 and 73 °C. This widens the use of these benign and non-toxic materials in different fields. Furthermore, this group of thermo-responsive polymers was grafted from hydroxypropyl cellulose, resulting in complex macromolecular comb architectures. The comb architecture preserved the thermo-responsive behavior, however, the LCST was consistently lowered compared to the linear analogues. This is believed to be an effect of the distorted hydrophilic/hydrophobic balance of the structures and the restrained mobility of the polymer grafts. Additionally, the significantly higher molecular weight is believed to contribute to this effect. Interestingly, although grafting of PEGMAs onto hydroxypropyl cellulose resulted in lower LCSTs, the ability to tune the transition temperature with the OEGMA content of these architectures remained. Furthermore, the amphiphilic comb polymers' ability to form polymeric micelles was examined by dynamic light scattering, fluorescence spectroscopy and surface tension measurements. The results show that, above their CMCs, HPC1.4-g-PDEGMA, HPC0.6-g-PDEGMA and HPC0.6-g-P(OEGMA52-co-DEGMA48) associates into micellar structures in the size range of 20–40 nm. Conclusively, the obtained results imply that these architectures, which combine the beneficial properties of the biocompatible HPC with the thermo-responsive and stealth properties of poly(ethylene glycol) methacrylates, may be of interest for biomedical applications, which also will be the topic of future studies to be conducted in our laboratory.References
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS.
- A. Carlmark and E. Malmström, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS.
- S. Hansson, E. Östmark, A. Carlmark and E. Malmström, ACS Appl. Mater. Interfaces, 2009, 1, 2651–2659 CrossRef CAS.
- J. Lindqvist, D. Nyström, E. Östmark, P. Antoni, A. Carlmark, M. Johansson, A. Hult and E. Malmström, Biomacromolecules, 2008, 9, 2139–2145 CrossRef CAS.
- R. Westlund, A. Carlmark, A. Hult, E. Malmström and I. M. Saez, Soft Matter, 2007, 3, 866–871 RSC.
- N. Nordgren, H. Lönnberg, A. Hult, E. Malmström and M. W. Rutland, ACS Appl. Mater. Interfaces, 2009, 1, 2098–2103 CrossRef CAS.
- J. Lindqvist and E. Malmström, J. Appl. Polym. Sci., 2006, 100, 4155–4162 CrossRef CAS.
- H. Lönnberg, L. Fogelström, L. Berglund, E. Malmström and A. Hult, Eur. Polym. J., 2008, 44, 2991–2997 CrossRef.
- E. Östmark, J. Lindqvist, D. Nyström and E. Malmström, Biomacromolecules, 2007, 8, 3815–3822 CrossRef.
- E. Östmark, S. Harrisson, K. L. Wooley and E. E. Malmström, Biomacromolecules, 2007, 8, 1138–1148 CrossRef CAS.
- E. Östmark, D. Nyström and E. Malmström, Macromolecules, 2008, 41, 4405–4415 CrossRef.
- S. M. Grayson and W. T. Godbey, J. Drug Targeting, 2008, 16, 329–356 CrossRef CAS.
- M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel and J. Bernard, Macromol. Rapid Commun., 2010, 31, 1751–1772 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- X. Li, M. Yin, G. Zhang and F. Zhang, Chin. J. Chem. Eng., 2009, 17, 145–149 Search PubMed.
- Y. Li, R. Liu, W. Liu, H. Kang, M. Wu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6907–6915 CrossRef CAS.
- F. J. Xu, Y. Ping, J. Ma, G. P. Tang, W. T. Yang, J. Li, E. T. Kang and K. G. Neoh, Bioconjugate Chem., 2009, 20, 1449–1458 CrossRef CAS.
- F. J. Xu, Y. Zhu, F. S. Liu, J. Nie, J. Ma and W. T. Yang, Bioconjugate Chem., 2010, 21, 456–464 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- J.-J. Tan, Y.-X. Li, R.-G. Liu, H.-L. Kang, D.-Q. Wang, L. Ma, W.-Y. Liu, M. Wu and Y. Huang, Carbohydr. Polym., 2010, 81, 213–218 CrossRef CAS.
- Q. Yan, J. Yuan, F. Zhang, X. Sui, X. Xie, Y. Yin, S. Wang and Y. Wei, Biomacromolecules, 2009, 10, 2033–2042 CrossRef CAS.
- J. Gao, G. Haidar, X. Lu and Z. Hu, Macromolecules, 2001, 34, 2242–2247 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- J.-F. Lutz, J. Andrieu, S. Uezguen, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- R. B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Adv. Drug Delivery Rev., 2003, 55, 217–250 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- Z.-B. Hu, T. Cai and C.-L. Chi, Soft Matter, 2010, 6, 2115–2132 RSC.
- J.-F. Lutz, A. Hoth and K. Schade, Des. Monomers Polym., 2009, 12, 343–353 Search PubMed.
- S. J. Holder, G. G. Durand, C.-T. Yeoh, E. Illi, N. J. Hardy and T. H. Richardson, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7739–7756 CrossRef CAS.
- T. Liu, Z. Zhou, C. Wu, V. M. Nace and B. Chu, Macromolecules, 1997, 30, 7624–7626 CrossRef CAS.
- Conducted at ALS Scandinavia AB; Aurorum 10, 97 775 Luleå, Sweden.
- D. Shen, H. Yu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4099–4108 CrossRef CAS.
- R. Bodvik, A. Dedinaite, L. Karlson, M. Bergström, P. Bäverbäck, J. S. Pedersen, K. Edwards, G. Karlsson, I. Varga and P. M. Claesson, Colloids Surf., A, 2010, 354, 162–171 CrossRef CAS.
- K. Kalyanasundaram, F. Grieser and J. K. Thomas, Chem. Phys. Lett., 1977, 51, 501–505 CrossRef CAS.
- E. Amado, C. Augsten, K. Maeder, A. Blume and J. Kressler, Macromolecules, 2006, 39, 9486–9496 CrossRef CAS.
- E. D. Goddard, N. J. Turro and P. L. Kuo, Langmuir, 1985, 1, 352–355 CrossRef.
- B. Jönsson, B. Lindman, K. Holmberg and B. Kronberg, Surfactants and Polymers in Aqueous Solution, John Wiley & Sons, 2003 Search PubMed.
- P. Lundberg, M. V. Walter, M. I. Montañez, D. Hult, A. Hult, A. Nyström and M. Malkoch, Polym. Chem., 2011, 2, 394–402 RSC.
Footnotes |
| † This work was supported by AB Willhelm Beckers Jubileumsfond, the Swedish Research Council, and the Swedish Foundation for Strategic Research (via Biomime, the Swedish Center for Biomimetic Fiber Engineering). |
| ‡ Electronic supplementary information (ESI) available: Calculations of the molar substitution of propoxy groups and the total degree of substitution of the initiators on HPC, NMR spectra, calculation of copolymer compositions, kinetic plots, CMC and DLS measurements. See DOI: 10.1039/c0py00417k |
| This journal is © The Royal Society of Chemistry 2011 |