Synthesis of thermoresponsive oxazolone end-functional polymers for reactions with amines using thiol-Michael addition “click” chemistry†
The Hien
Ho
,
Martin
Levere
,
Jean-Claude
Soutif
,
Véronique
Montembault
,
Sagrario
Pascual
and
Laurent
Fontaine
*
UCO2M, UMR CNRS 6011, Université du Maine, Avenue Olivier Messiaen, 72085, Le Mans, France. E-mail: laurent.fontaine@univ-lemans.fr; Fax: +33 (0)2 4383 3754; Tel: +33 (0)2 4383 3325
First published on 16th May 2011
Abstract
Well-defined poly(N-isopropylacrylamide) (PNIPAM) polymers with an oxazolone ring at the chain end have been synthesized by combining controlled radical polymerization and thiol-Michael addition “click” chemistry. First, PNIPAM was synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization to afford polymers of controlled molecular weight and molecular weight distribution (Mn (1H NMR) = 3200 g mol−1; PDISEC = 1.05). The chain end was quantitatively converted to a thiol by aminolysis. Then, the functional monomer vinyl azlactone (VDM) was quantitatively “clicked” onto the chain end using a thiol-Michael addition reaction. The polymers were reacted with a model amine in order to demonstrate the potential of these polymers for bioconjugation.
Oxazolones are five-membered rings that undergo reactions with nucleophiles. The monomer vinyl azlactone (VDM, 2-vinyl-4,4-dimethyl-5-oxazolone) was developed by 3M and features electrophilic groups that may undergo reactions under the appropriate choice of catalyst.1 The C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vinyl group may be polymerized via a free-radical or controlled radical methodology,2 and after polymerization pendant azlactone rings remain that readily react with nucleophiles, in particular primary amines1–5 at the CO bond. The oxazolone ring vinyl azlactone is resistant to hydrolysis5 when compared with succinimide monomers. Azlactone rings are thus considered promising candidates to be investigated for use as chemical linkers in bioconjugation. The covalent attachment of polymers to bio(macro)molecules and drugs in order to optimize the properties of the bio(macro)molecule for medicinal purposes6 is an area of considerable current research. The development of the controlled radical methodologies, such as Nitroxide Mediated Polymerization7 (NMP), Atom Transfer Radical Polymerization8 (ATRP) and Reversible Addition–Fragmentation Chain Transfer9 (RAFT) polymerization, has facilitated the synthesis of well-defined polymers with a range of end-functionalities that may be used for bioconjugation to bio(macro)molecules.6 Among the controlled radical techniques listed, RAFT is arguably the most appropriate method for the synthesis of macromolecules for biomedicinal purposes due to the metal-free nature of the reaction, the wide range of monomers that may be used and the large number of functionalities that can be incorporated at the chain end. Moreover, various strategies have been described for end-group transformation of RAFT polymers.10N-Isopropylacrylamide (NIPAM) is among the most widely studied monomers in polymer science due in most part to the readily accessible LCST around 32 °C of PNIPAM in water, just below the physiological temperature (37 °C).11 A number of thermoresponsive polymers based on PNIPAM have thus been obtained using RAFT polymerization.12
C vinyl group may be polymerized via a free-radical or controlled radical methodology,2 and after polymerization pendant azlactone rings remain that readily react with nucleophiles, in particular primary amines1–5 at the CO bond. The oxazolone ring vinyl azlactone is resistant to hydrolysis5 when compared with succinimide monomers. Azlactone rings are thus considered promising candidates to be investigated for use as chemical linkers in bioconjugation. The covalent attachment of polymers to bio(macro)molecules and drugs in order to optimize the properties of the bio(macro)molecule for medicinal purposes6 is an area of considerable current research. The development of the controlled radical methodologies, such as Nitroxide Mediated Polymerization7 (NMP), Atom Transfer Radical Polymerization8 (ATRP) and Reversible Addition–Fragmentation Chain Transfer9 (RAFT) polymerization, has facilitated the synthesis of well-defined polymers with a range of end-functionalities that may be used for bioconjugation to bio(macro)molecules.6 Among the controlled radical techniques listed, RAFT is arguably the most appropriate method for the synthesis of macromolecules for biomedicinal purposes due to the metal-free nature of the reaction, the wide range of monomers that may be used and the large number of functionalities that can be incorporated at the chain end. Moreover, various strategies have been described for end-group transformation of RAFT polymers.10N-Isopropylacrylamide (NIPAM) is among the most widely studied monomers in polymer science due in most part to the readily accessible LCST around 32 °C of PNIPAM in water, just below the physiological temperature (37 °C).11 A number of thermoresponsive polymers based on PNIPAM have thus been obtained using RAFT polymerization.12
“Click” chemistry is a term given to near-perfect chemical transformations that display high conversion, is highly selective, produces non-hazardous by-products and may be separated from the reaction medium via non-chromatographic methods.13 Free-radical14 and thiol-Michael addition15 reactions between thiols and activated –ene groups have been largely studied as metal-free “click” reactions16 to synthesize a range of macromolecular structures and to conjugate –ene groups to proteins. Thiol-Michael addition reactions are usually performed using organic phosphines such as dimethylphenyl phosphine (DMPP) as a co-reagent. They have been used to synthesize biotin functionalized glycopolymers17a to mediate the reaction between thiol functional polymer and mannose-modified methacrylate,17b and to synthesize polymer–protein conjugates between free cysteine residues on proteins and macromonomers,17c and other functional methacrylates.17d,e
Recently the use of reagents that can control the polymerization and then be exploited for post-polymerization modification has been demonstrated.18 Such orthogonal “relay” reactions are elegantly achieved by using RAFT polymerization with a trithiocarbonate chain transfer agent and then reducing the chain ends to thiols for use in thiol–ene “click” chemistry.19 We have used such an orthogonal “relay” approach to synthesize polymers with an azlactone ring at the chain end by “clicking” vinyl azlactone to a thiol end-functional polymer and assessed their reactivity towards a model amine, 4-fluorobenzylamine.
A well-defined PNIPAM–CTA (PNIPAM with a chain transfer agent as the end-group, 1Scheme 1) was synthesized by RAFT polymerization in N,N-dimethylformamide at 70 °C using methyl-2-(n-butyltrithiocarbonyl) propanoate (MBTCCP) as a RAFT agent. After 5 h, the polymer was isolated and characterized by SEC, 1H NMR spectroscopy (Fig. S2 in the ESI†) and MALDI-TOF mass spectrometry. Average molecular weights were determined relative to polystyrene standards using SEC (Mn = 7850 g mol−1, Mw = 8300 g mol−1, PDI = 1.05). The number-average degree of polymerization was determined to be 26 from 1H NMR spectroscopy, leading to a molecular weight of 3190 g mol−1 for the polymer. This compared favourably with the data obtained from MALDI-TOF mass spectrometry analysis. A single series of signals separated by 113.12 units, corresponding to the molecular weight of the NIPAM repeat unit (calculated value = 113.16 g mol−1), was detected. The peak at m/z = 3103.24 g mol−1 in the MALDI-TOF spectrum corresponds to a polymer chain consisting of 25 NIPAM units, an ester at one chain end and a trithiocarbonate moiety (with butyl chain) at the other chain end and a sodium atom responsible for ionization (calculated value = 3104.35 g mol−1). Moreover, the presence of the trithiocarbonate moiety at the chain end was confirmed by the appearance of a peak at 309 nm in the SEC trace using UV detection, corresponding to the chromophoric CS bond of the chain transfer agent (Fig. S3 in the ESI†).
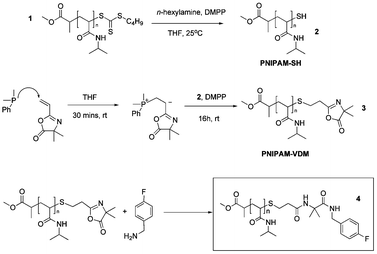 | ||
| Scheme 1 Full schematic for the synthesis of oxazolone end-functional PNIPAM and their reactivity towards 4-fluorobenzylamine. | ||
Introduction of orthogonal azlactone functionality was achieved via a two-step process. Step one is the transformation of the trithiocarbonate end-group to a thiol and step two is the modification of the thiol group formed by thiol-Michael addition “click” chemistry.
In the first step the trithiocarbonate chain end functionality was reduced to thiolviaaminolysis in the presence of an excess of dimethylphenyl phosphine (DMPP).16b The reduction to a thiol was confirmed by the absence of a peak at 309 nm in the SEC trace using UV detection corresponding to the loss of the CS bond from the polymer (Fig. S5 in the ESI†). Coupling between thiol groups to form disulfide bonds was avoided by DMPP as a mostly symmetrical monomodal peak shape was observed on the differential refractometer (dRI) trace of the SEC (Fig. S6 in the ESI†). The principal peaks of the MALDI-TOF spectrum decreased by 132.29 relative to PNIPAM–CTA, corresponding to the chemical modification of –S–(CS)–S–C4H9 fragment into –SH fragment at the chain end (calculated value = 132.25 g mol−1) of the polymer (Fig. S8 in the ESI†). A single series of peaks separated by m/z = 113.11, the molecular weight of the NIPAM repeat unit, was observed. The peak at m/z = 2971.73 corresponds to a polymer of 25 NIPAM units ionized by a sodium atom, with an ester at one chain end and a thiol group at the other chain end (calculated value = 2972.10 g mol−1). Comparison between the 1H NMR spectra of PNIPAM–SH and PNIPAM–CTA shows that peaks at 1.0 ppm and 3.4 ppm, corresponding to the methyl protons –S–(CH2)3–CH3 and the methylene protons (–S–CH2–(CH2)2–CH3) of the PNIPAM–CTA, respectively, disappear confirming that PNIPAM–SH, 2 (Scheme 1), is obtained.
In the second step the precipitated PNIPAM–SH polymer was reacted with VDM in the presence of DMPP to afford PNIPAM–VDM, 3, with azlactone end functionality (Scheme 1). When literature conditions were used for the thiol-Michael addition, i.e. a catalytic amount of DMPP16c ([thiol]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[VDM]0:[DMPP]0 = 5:5:1), a bimodal non-symmetric peak shape was observed on the dRI trace of the SEC, indicating that bisulfide products were formed.
:[VDM]0:[DMPP]0 = 5:5:1), a bimodal non-symmetric peak shape was observed on the dRI trace of the SEC, indicating that bisulfide products were formed.
This result means that VDM has a different behavior than (meth)acrylates during the thiol-Michael addition. In order to improve the thiol-Michael addition efficiency, an excess of DMPP in comparison with PNIPAM–SH and VDM was used ([thiol]0:[VDM]0:[DMPP]0 = 1:3.5:8.9). The SEC trace of the resulting polymer showed a symmetrical monomodal peak (Fig. S11 in the ESI†). The presence of the azlactone ring at the chain end of the new polymer was confirmed by infrared spectroscopy, with the appearance of a band at 1817 cm−1 corresponding to CO of the azlactone group (Fig. S12 in the ESI†). The chain end functionality was also investigated using MALDI-TOF mass spectrometry. The MALDI-TOF spectrum is shown in Fig. 1. A single distribution of peaks was observed, separated by 113.08, corresponding to the molecular weight of the NIPAM repeating unit. The peak at m/z = 3110.90 was assigned to a polymer consisting of 25 NIPAM units ionized by a sodium atom and featuring an ester at one chain end and vinyl azlactone connected via a sulfur atom at the other chain end (calculated value m/z = 3111.25). In addition, the m/z of the peaks increased by 139.52 relative to those in PNIPAM–SH, comparable with the molar mass of the VDM monomer (calculated value m/z = 139.15). The azlactone functionality was determined by 1H NMR spectroscopy by comparing the integration of CH3O– protons (at 3.65 ppm) of the ester group at one chain end and the integration of the –CH2S– protons (at 2.90 ppm) at the other chain end (Fig. 2). The results showed that the reaction is quantitative. Such results are surprising in comparison with previous studies using DMPP in thiol-Michael addition involving (meth)acrylates.16b,c Then, to get a better understanding of our results, a model reaction was performed between VDM and DMPP used in excess ([VDM]0:[DMPP]0 = 1:1.15) in THF. Analysis of 1H NMR spectra after 15 minutes, 1 hour and 4 hours showed a decrease in the ratio of the vinyl peaks at 5.9 ppm and 6.2 ppm, indicating the partial loss of the vinyl group. By contrast new signals are observed to form at 2.0 ppm and 2.1 ppm corresponding to the protons on the methyl groups attached to the phosphorus. This result shows that there is addition of DMPP onto the vinyl group leading to the formation of an ylide or zwitterion Michael adduct.1 The full results of these experiments are included as ESI†. It appears that this ylide or zwitterion is sufficiently basic to react with the thiol of the PNIPAM–SH producing the thiolate which is the nucleophile involved in the Michael addition. Thus, an excess of DMPP is necessary to ensure that all of the vinyl groups are converted to ylide or zwitterion so that no unwanted side reactions occur at the carbonyl group of the azlactone ring and also to prevent bisulfide formation.
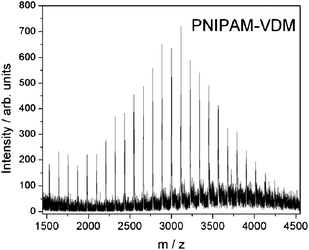 | ||
| Fig. 1 MALDI-TOF mass spectrum for PNIPAM-VDM after thiol-Michael addition “click” reaction. | ||
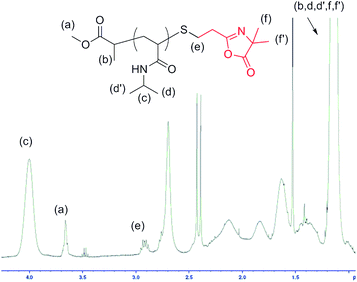 | ||
| Fig. 2 Partially assigned 1H NMR spectrum PNIPAM-VDM after thiol-Michael addition “click” reaction. | ||
The reactivity of the so-obtained azlactone functional polymer towards a labelled amine, 4-fluorobenzylamine, was investigated. The PNIPAM–VDM 3 was dissolved in THF and an excess of 4-fluorobenzylamine was added. The precipitated polymer, 4 (Scheme 1), was analyzed by SEC and 1H NMR spectroscopy. A peak was observed at 263 nm of the UV detector of the SEC, corresponding to the aromatic group of 4-fluorobenzylamine and demonstrating that the oxazolone functionality has reacted. Two new peaks corresponding to the aromatic protons of 4-fluorobenzylamine were clearly visible in the 1H NMR spectrum at 7.0 ppm and 7.4 ppm and integrations show that the reaction is quantitative (Fig. S17 in the ESI†).
In conclusion, a well-defined oxazolone functional PNIPAM was synthesized via an orthogonal RAFT and thiol-Michael addition approach for the first time. The polymer has a high oxazolone chain end functionality and it is reactive towards amines.
This work was funded by Région Pays de la Loire.
Note added after first publication
This article replaces the version published on 2nd April 2011, which contained errors in the notes and references section.Notes and references
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655 CrossRef CAS.
- (a) D. C. Tully, M. J. Roberts, B. H. Geierstanger and R. B. Grubbs, Macromolecules, 2003, 36, 4302 CrossRef CAS; (b) D. Fournier, S. Pascual and L. Fontaine, Macromolecules, 2004, 37, 330 CrossRef CAS; (c) B. S. Lokitz, J. M. Messman, J. P Hinestrosa, J. Alonzo, R. Verduzco, R. H. Brown, M. Osa, J. F. Anker and S. M. Kilbey, Macromolecules, 2009, 42, 9018 CrossRef CAS; (d) S. Pascual, T. Blin, P. J. Saikia, M. Thomas, P. Gosselin and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5053 CrossRef CAS.
- (a) S. M. Heilmann, J. K. Rasmussen, F. J. Palensky and H. K. Smith, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 1179 CrossRef CAS; (b) S. M. Heilmann, J. K. Rasmussen, F. J. Palensky and H. K. Smith, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1 CrossRef CAS.
- (a) L. Fontaine, T. Lemêle, J. C Brosse, G. Sennyey, J. P. Senet and D. Wattiez, Macromol. Chem. Phys., 2002, 203, 1377 CrossRef CAS; (b) D. Fournier, S. Pascual, V. Montembault, D. M. Haddleton and L. Fontaine, J. Comb. Chem., 2006, 8, 522 CrossRef CAS; (c) S. P. Cullen, I. C. Mandel and P. Gopalan, Langmuir, 2008, 24, 13701 CrossRef CAS; (d) B. Sun, X. Liu, M. E. Buck and D. M. Lynn, Chem. Commun., 2010, 46, 2016 RSC.
- J. M. Messman, B. S. Lokitz, J. M. Pickel and S. M. Kilbey, Macromolecules, 2009, 42, 3933 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563 RSC.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- (a) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC; (b) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- (a) Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley, 2008 Search PubMed; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H Thang, Polymer Int., 2011, 60, 9 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS; (b) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS; (c) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS; (d) K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS.
- (a) B. M. Cole, H. M. Foudoulakis and A. Bartolozzi, Synthesis, 2008, 13, 2023; (b) J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751 CrossRef CAS.
- (a) A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995 CrossRef CAS; (b) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC; (c) G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- (a) C. Boyer and T. P. Davis, Chem. Commun., 2009, 45, 6029 Search PubMed; (b) C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS; (c) M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 45, 5272 Search PubMed; (d) J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751 CrossRef CAS; (e) B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Descriptions of methods, SEC chromatograms, MALDI-TOF mass spectra, 1H NMR spectra, and infrared spectra. See DOI: 10.1039/c1py00071c |
| This journal is © The Royal Society of Chemistry 2011 |