Through-space conjugated polymers consisting of [2.2]paracyclophane
Yasuhiro
Morisaki
* and
Yoshiki
Chujo
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: ymo@chujo.synchem.kyoto-u.ac.jp; chujo@chujo.synchem.kyoto-u.ac.jp
First published on 4th March 2011
Abstract
In this review, the synthesis and fundamental properties of [2.2]paracyclophane-containing through-space conjugated polymers are introduced. By incorporating pseudo-para-, pseudo-ortho-, and pseudo-geminal-linked [2.2]paracyclophane skeletons into conjugated polymer backbones, through-space conjugated polymers consisting of stacked π-electron systems can be prepared. Recent reports in the last two years are especially focused on conjugated polymers, and the characteristic features of the through-space conjugated polymers constructed by [2.2]paracyclophane are reviewed.
 Yasuhiro Morisaki | Yasuhiro Morisaki was born in Osaka, Japan, in 1972. He received his PhD degree from Kyoto University in 2000 under the supervision of Professor Take-aki Mitsudo. After his postdoctoral research with Dr Yoshie Souma at Osaka National Research Institute for six months, he joined the Professor Chujo's group at Kyoto University in 2000. He has been working as an Assistant Professor (2000–2007) and a Lecturer (2008–present) at Kyoto University. He carried out postdoctoral work with Professor Rik R. Tykwinski at University of Alberta (2004–2005). His research interest includes conjugated polymers and optically active polymers. |
 Yoshiki Chujo | Yoshiki Chujo is a professor in the Department of Polymer Chemistry at Kyoto University. Chujo received his PhD from Kyoto University in 1980. He has then joined Nagoya University as an assistant professor in 1981. In 1983, as a postdoctoral research fellow, Chujo worked with Prof. J. McGrath at Virginia Tech. He returned to Kyoto University as a lecturer in 1986, was promoted to an associate professor in 1993, and a full professor in 1994. His current interests focus on polymer synthesis, inorganic polymers, and polymeric hybrid materials. |
1. Introduction
Recently, much attention has been focused on conjugated polymers1 such as poly(p-arylene)s (PAs),2 poly(p-arylenevinylene)s (PAVs),3 and poly(p-arylene-ethynylene)s (PAEs)4 as next-generation functional materials. They possess the inherent features of organic polymers, including lightweight, flexibility, processability, and film-forming properties. In addition, some of the conjugated polymers show non-linear optical properties, fluorescence, electroluminescence, conductivity, and so on; therefore, they are being applied to optoelectronic materials. The properties of the conjugated polymers strongly depend on the structure and characteristics of the constituent units in the polymer backbones, namely, the aromatic units play a pivotal role. The design of key aromatic components as well as carbon–carbon bond structures in the polymer side chain and main chain is important to achieve the desired properties. Thus, the synthesis of new conjugated polymers by incorporation of various aromatic compounds into the polymer backbones, and the exploration of their functionalities from various aspects, are essential to create 21st-century organic polymers in the field of material chemistry.Common conjugate polymers are polymers in which the aromatic units connect directly with carbon–carbon single, double, or triple bonds. Many of the conjugate polymers reported thus far generally consist of sp and/or sp2carbon frameworks. Conjugated polymers comprising stacked π-electron systems have rarely been prepared.5 In conjugated polymer materials, the charge and energy are transferred among π-electron systems as well as among through-bonds of the conjugated polymer backbone. For example, in organic photovoltaic solar cells6 consisting of conjugated polymers, excitons are delocalized among the cells. Furthermore, in organic electroluminescence (EL) devices,7 holes and electrons move in each layer by a hopping mechanism among π-electron systems. It has been recognized that in such optoelectronic devices, the enhancement of device performance requires the construction of ordered π-stacked structures of π-electron systems. However, construction of such π-stacked structure of the π-electron systems in a single polymer chain has not attracted much attention.
We have focused our attention on cyclophane compounds8 such as [2.2]paracyclophane9 and dithia[3.3]metaphanes10 as the aromatic units of conjugated polymers. In particular, [2.2]paracyclophane is an attractive aromatic compound because two benzene rings are fixed with two ethylene chains and π-stacked with a proximity of approximately 3 Å. We have incorporated [2.2]paracyclophane as a repeating unit into the conjugated polymer main chains and have revealed their optical and electrochemical properties.9[2.2]Paracyclophane realizes the construction of π-stacked structures of π-electron systems in the primary structure of the polymer chain.
This review deals with recent results on the synthesis and properties of [2.2]paracyclophane-containing through-space conjugated polymers. Conjugated and non-conjugated polymers containing other cyclophane skeletons are not included. π-Stacked polymers have been comprehensively reviewed, and interested readers are directed to the relevant literature.5 This review mainly covers our studies over the last two years. In addition, developments in this field and significant references, where necessary for discussion, are also briefly summarized.
2. Pseudo-para-disubstituted [2.2]paracyclophane-containing through-space conjugated polymers
Since the first report on the synthesis of [2.2]paracyclophane in 1949,11 its functionalizations and numerous number of its derivatives have been extensively reported.8 However, polymers containing cyclophane skeletons in their main chain12–16 and side chain17–20 are rarely synthesized despite the progress in the field of cyclophane chemistry. In addition, there have been few reports on the synthesis of conjugated polymers possessing [2.2]paracyclophane moieties as repeating units in the main chain.In the beginning of the last decade, the electrochemical polymerizations of pseudo-para-di(2-thienyl)[2.2]paracyclophane (1)15 and pseudo-para-di(2,2′-bithienyl)[2.2]paracyclophane (2)16 were reported by two independent groups, as shown in Scheme 1. Thin films of polymers P1 and P2 were formed on the electrode surface during cyclic voltammetry of solutions containing each polymer. The peak oxidation potentials of P1 and P2 both appeared at around 1.0 eV, against the saturated calomel (SCE) and Ag/Ag+ reference electrodes, respectively.
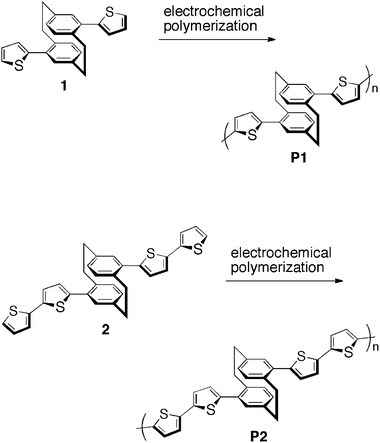 | ||
| Scheme 1 Synthesis of polymers P1 and P2 by electrochemical polymerization. | ||
Palladium-catalyzed coupling reactions have realized the synthesis of a wide variety of conjugated polymers. We prepared [2.2]paracyclophane-containing PAE-type conjugated polymer P3 by Sonogashira–Hagihara coupling21 of pseudo-para-dibromo[2.2]paracyclophane (3) with diethynylbenzene (4), as shown in Scheme 2.9a An appropriate monomer combination was important to obtain polymers with high molecular weight; the treatment of pseudo-para-diethynyl[2.2]paracyclophane (5) with diiodobenzene (6) afforded polymer P3 in 95% isolated yield with a number-average molecular weight (Mn) of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 (Scheme 3).22 The obtained polymer P3 had good solubility in common organic solvents such as THF, CHCl3, CH2Cl2, and toluene because of their alkyl side chains, and could be characterized using 1H and 13C NMR spectra. In addition, thin films were readily prepared by casting and spin-coating techniques from a toluene solution. Mizoroki–Heck coupling23 and Suzuki–Miyaura coupling24 were also employed for the synthesis of [2.2]paracyclophane-containing PAV- and PA-type conjugated polymers P49c and P5,9k respectively (Schemes 4 and 5).
500 (Scheme 3).22 The obtained polymer P3 had good solubility in common organic solvents such as THF, CHCl3, CH2Cl2, and toluene because of their alkyl side chains, and could be characterized using 1H and 13C NMR spectra. In addition, thin films were readily prepared by casting and spin-coating techniques from a toluene solution. Mizoroki–Heck coupling23 and Suzuki–Miyaura coupling24 were also employed for the synthesis of [2.2]paracyclophane-containing PAV- and PA-type conjugated polymers P49c and P5,9k respectively (Schemes 4 and 5).
![Synthesis of PAE-type polymer P3 using pseudo-para-dibromo[2.2]paracyclophane (3).](/image/article/2011/PY/c0py00421a/c0py00421a-s2.gif) | ||
| Scheme 2 Synthesis of PAE-type polymer P3 using pseudo-para-dibromo[2.2]paracyclophane (3). | ||
![Synthesis of PAE-type polymer P3 using pseudo-para-diethynyl[2.2]paracyclophane (5).](/image/article/2011/PY/c0py00421a/c0py00421a-s3.gif) | ||
| Scheme 3 Synthesis of PAE-type polymer P3 using pseudo-para-diethynyl[2.2]paracyclophane (5). | ||
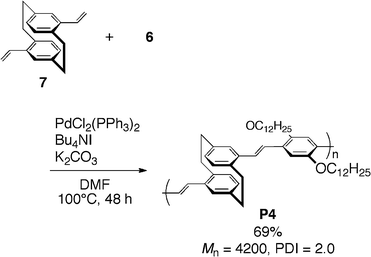 | ||
| Scheme 4 Synthesis of PAV-type polymer P4. | ||
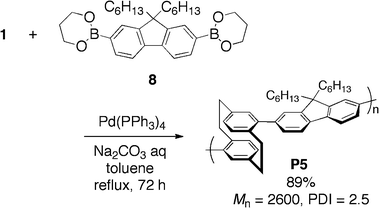 | ||
| Scheme 5 Synthesis of PA-type polymer P5. | ||
[2.2]Paracyclophane leads to the construction of π-stacked structures of π-electron systems. As shown in Fig. 1, the incorporation of [2.2]paracyclophane into the conjugated polymer backbone resulted in the formation of a π-stacked structure in the single polymer chain. That is, [2.2]paracyclophane-containing conjugated polymer P3 comprises the stacked π-electron system of compound 9. Thus, [2.2]paracyclophane acts not as the [2.2]paracyclophane unit in the conjugated polymer backbone, but as the bridge unit that combines the π-electron systems in close proximity. Therefore, the physical properties of the [2.2]paracyclophane-containing conjugated polymer strongly depend on the stacked π-electron system, i.e., compound 9.
![Schematic figure of [2.2]paracyclophane-containing through-space conjugated polymer and stacked π-electron system, compound 9.](/image/article/2011/PY/c0py00421a/c0py00421a-f1.gif) | ||
| Fig. 1 Schematic figure of [2.2]paracyclophane-containing through-space conjugated polymer and stacked π-electron system, compound 9. | ||
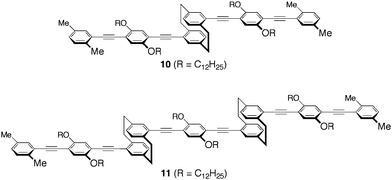
Fig. 2A shows the normalized UV-vis absorption spectra of compounds 9–11 and polymer P3 in diluted CHCl3 solutions (1.0 × 10−5 M). Compounds 10 and 11 are the [2.2]paracyclophane-containing oligomers, in which two and three π-electron systems are stacked, respectively. There was an apparent red-shift of the spectra as the number of the stacked π-electron systems increased; the λmax of 9–11 and P3 appeared at 371, 378, 381, and 387 nm, respectively. The partially overlapped arrangement of the π-electron systems leads to a through-space interaction and thus to an extension of the through-space π-conjugation length of the polymer.
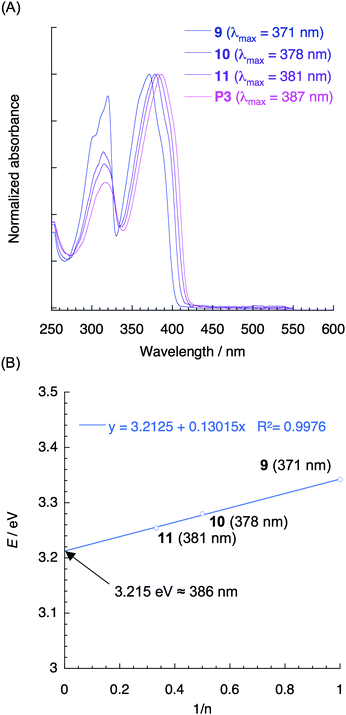 | ||
| Fig. 2 (A) UV-vis absorption spectra of compounds 9–11 and polymer P3 in CHCl3 (1.0 × 10−5 M). (B) Plot of E (eV) versus 1/n (n = the number of repeating units) for compounds 9–11. | ||
It is well established that the relationship between the number of repeating units and the absorption maximum λmax for the conjugated oligomers provides estimates of λmax for polymers with a sufficiently high Mn. This relationship is applied to the through-space conjugated system;25 a plot of the HOMO–LUMO band-gap energy (E) estimated from the λmax against the reciprocal of the number of stacked π-electron systems (1/n) is shown in Fig. 2B. The linear correlation appears to be reasonably good, and the coefficient of determination (R2) was calculated to be 0.998. A linear extrapolation to 1/n = 0 yielded the HOMO–LUMO band-gap energy of 3.2 eV for the polymer, which corresponded to the λmax of 386 nm. This result was consistent with the experimental value of λmax = 387 nm for polymer P3, which possesses approximately 21 stacked π-electron systems on average. According to the empirical power-law relationship (E = 1/λmax ≈ n−x), the effective conjugation repeating unit number (n) was estimated to be approximately 9. This implies that through-space conjugated polymers consisting of one-dimensionally stacked-π-electron systems can be potentially applied as single molecular wires.
PAV-type through-space conjugated polymers can be accessed through the Knoevenagel reaction in addition to Heck coupling.23 The synthetic routes for the cyano-substituted PAVs3b are shown in Schemes 6 and 7.26 The reaction of pseudo-para-diformyl[2.2]paracyclophane (12) with bis(cyanomethyl)benzene derivative (13) in the presence of tert-BuOK was carried out to afford the corresponding cyano-substituted through-space PAV P6 in 94% isolated yield with the Mn of 6600 and the PDI of 3.0 (Scheme 6). The through-space PAV P7 was also obtained by the Knoevenagel reaction of monomers 14 and 15 in 96% isolated yield with the Mn of 8000 and the PDI of 2.3, as shown in Scheme 7. The difference between PAVs P6 and P7 was the substitution position of the cyano groups in the vinylene units; they were substituted at the α- and β-positions from the dialkoxyphenylene units in P6 and P7, respectively.
![Synthesis of cyano-substituted PAV-type polymer P6 using pseudo-para-diformyl[2.2]paracyclophane (12).](/image/article/2011/PY/c0py00421a/c0py00421a-s6.gif) | ||
| Scheme 6 Synthesis of cyano-substituted PAV-type polymer P6 using pseudo-para-diformyl[2.2]paracyclophane (12). | ||
![Synthesis of cyano-substituted PAV-type polymer P7 using pseudo-para-bis(cyanomethyl)[2.2]paracyclophane (14).](/image/article/2011/PY/c0py00421a/c0py00421a-s7.gif) | ||
| Scheme 7 Synthesis of cyano-substituted PAV-type polymer P7 using pseudo-para-bis(cyanomethyl)[2.2]paracyclophane (14). | ||
The optical properties of PAVs P6 and P7, as well as of model compounds 16 and 17, are summarized in Table 1. The λmax of P6 and P7 were observed at 380 and 436 nm, respectively, which were bathochromically shifted in relation to those of 16 (λmax = 370 nm) and 17 (λmax = 413 nm), respectively, because of the through-space conjugation.
| Compound | λ Abs,max a/nm | λ PL,max b/nm | Φ PL c |
|---|---|---|---|
| a λ Abs,max of UV-vis absorption spectra in CHCl3 (3.0 × 10−5 to 2.0 × 10−4 M). b λ PL,max of photoluminescence spectra in CHCl3 (2.6 × 10−6 to 3.0 × 10−6 M). c Relative photoluminescence quantum efficiencies calculated by using 9-anthracenecarboxylic acid in CH2Cl2 as a standard. | |||
| P6 | 327, 380 | 477 | <0.01 |
| P7 | 360, 436 | 510 | 0.32 |
| 16 | 315, 370 | 484 | <0.01 |
| 17 | 330, 413 | 491 | 0.30 |
PAVs P6 and P7 exhibited PL peaks in sufficiently diluted CHCl3 solution with maxima at around 477 and 510 nm, respectively. It is noted that their PL quantum efficiencies (ΦPL) were quite different; they were estimated to be less than 0.01 and 0.32, respectively. The model compounds 16 and 17 had the same tendency, and their ΦPL were less than 0.01 and 0.30, respectively. The remarkably weak emissions of P6 and 16 are derived from the substitution position of the cyano groups. As shown in Fig. 3, π-conjugation between the dialkoxyphenylene unit and the cyano groups at the α-positions of the vinylene units in P6 and 16 is broken, whereas the π-conjugation lengths in P6 and 17 are well extended. Density functional theory (DFT) calculations and steric effects by X-ray crystallography27 (Fig. 3) also support this optical behavior. These results indicate that the properties of the through-space conjugated polymer reflect those of the stacked π-electron system.
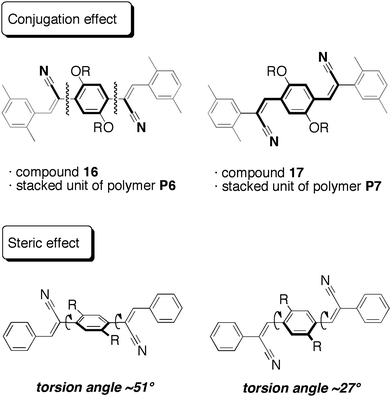 | ||
| Fig. 3 Schematic figures for conjugation and steric effects of cyano-substituted through-space conjugated polymers P6 and P7. | ||
A new type of donor–acceptor through-space conjugated polymer was synthesized.28 The polymer comprised donor and acceptor π-electron systems that are alternately π-stacked in short distance and held by covalent bonds. It is noted that the construction of π-stacked donor–acceptor systems has been achieved by a supramolecular approach.29 The synthetic route for the donor–acceptor-stacked through-space conjugated polymer is outlined in Scheme 8. Fluorene and benzothiadiazole were selected as donor and acceptor π-conjugated units, respectively. The treatment of monomers 18 and 19 in the presence of a catalytic amount of palladium complex and bulky phosphine30 afforded the corresponding PA-type through-space conjugated polymer P8 in 68% isolated yield with the Mn of 17000 and the PDI of 2.9. In the obtained polymer P8, the donor and acceptor π-conjugated systems are linked alternately via the through-space interactions in the single polymer main chain (Scheme 8); compounds 20 and 21 represent the donor and acceptor units of P8, respectively.
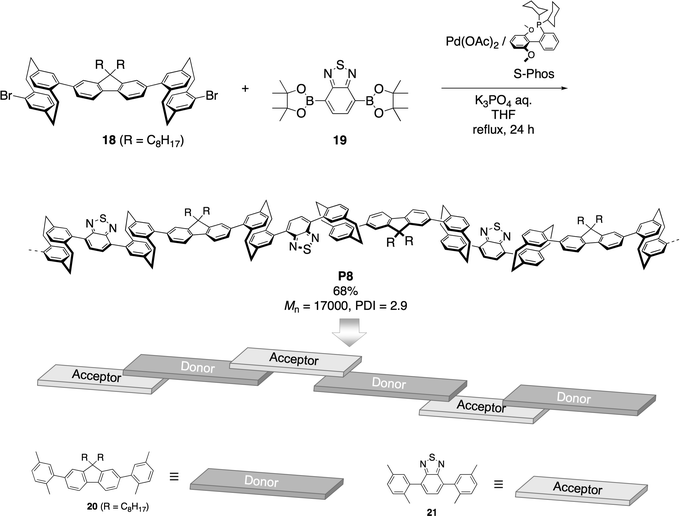 | ||
| Scheme 8 Synthesis of donor–acceptor-stacked polymer P8 and its schematic figure. | ||
The UV-vis absorption spectra (1.0 × 10−5 M in CHCl3) and photoluminescence spectra (1.0 × 10−6 M in CHCl3) of 20, 21, and P8 are shown in Fig. 4A–C, respectively. In Fig. 4A, the typical π–π* transition band of the fluorene moiety in 20 was observed at around 300 nm, and the maximum of the emission peak was at 364 nm with a vibrational structure excited at 260 nm. Compound 21 exhibited absorption bands at around 310 and 360 nm (Fig. 4B), which were attributed to the π–π* transition band of the xylyl—phenylene–xylyl backbone and the charge-transfer (CT) band from the benzene to the thiadiazole moieties, respectively. When the CT band of 21 at 360 nm was excited, it emitted green light at 467 nm.
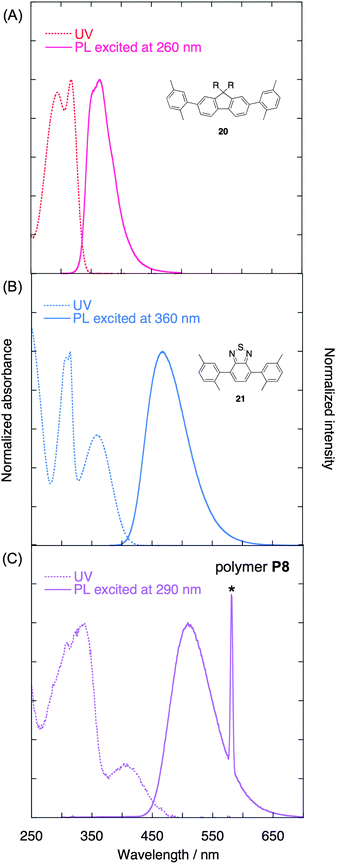 | ||
| Fig. 4 (A) UV-vis absorption spectrum in CHCl3 (1.0 × 10−5 M) and photoluminescence spectrum in CHCl3 (1.0 × 10−6 M) of compound 20. (B) UV-vis absorption spectrum in CHCl3 (1.0 × 10−5 M) and photoluminescence spectrum in CHCl3 (1.0 × 10−6 M) of compound 21. (C) UV-vis absorption spectrum in CHCl3 (1.0 × 10−5 M) and photoluminescence spectrum in CHCl3 (1.0 × 10−6 M) of polymer P8. *Scattered light was observed at around 580 nm. | ||
The absorption spectrum of P8 exhibited a peak at around 325 nm and a broad peak at around 400 nm, as shown in Fig. 4C. Compared to the absorption spectra of 20 and 21 (Fig. 4A and B), the spectrum of P8 was characteristic of the overlapping spectra of donor and acceptor π-conjugated systems. Fig. 4C includes the photoluminescence spectrum of P8 excited at 290 nm in diluted CHCl3 solution; at this excitation wavelength, the donor fluorene moieties were effectively excited, and the emission peak of the acceptor benzodithiadiazole moieties was observed at around 500 nm. The photoluminescence spectrum of the donor 20 (red solid line in Fig. 4A) substantially overlapped with the absorption spectrum of the CT band of the acceptor 21 (blue dotted line in Fig. 4B), which resulted in the effective Förster-type31 intramolecular energy transfer from the donor π-conjugated units to the acceptor π-conjugated units in the polymer backbone.
3. Pseudo-ortho-disubstituted [2.2]paracyclophane-containing through-space conjugated polymers
Disubstituted [2.2]paracyclophane has several stereoisomers such as ortho-, meta-, para-, pseudo-ortho-, pseudo-meta-, pseudo-para-, and pseudo-geminal-disubstituted [2.2]paracyclophanes. Pseudo-para-dibromo[2.2]paracyclophane (3), which was discussed above, can be readily obtained from the dibromination of commercially available [2.2]paracyclophane. Given the low solubility of pseudo-para-dibromo[2.2]paracyclophane (3), it can be obtained by recrystallization.32 After thermal isomerization of 3,32 separation by recrystallization yields pure pseudo-ortho-dibromo[2.2]paracyclophane (22), as shown in Scheme 9.![Synthesis of pseudo-ortho-disubstituted [2.2]paracyclophane-containing polymer P9.](/image/article/2011/PY/c0py00421a/c0py00421a-s9.gif) | ||
| Scheme 9 Synthesis of pseudo-ortho-disubstituted [2.2]paracyclophane-containing polymer P9. | ||
The monomer, pseudo-ortho-diethynyl[2.2]paracyclophane (24), was obtained as shown in Scheme 9. Due to the low reactivity of 22 for Sonogashira–Hagihara coupling, the diethynyl monomer 24 was prepared by diformylation of 22 and successive elimination of 1,1-dihaloalkene.33 The polymerization of 24 with 4 was carried out by the Sonogashira–Hagihara reaction to afford the corresponding PAE type through-space conjugated polymer P9, consisting of the pseudo-ortho-linked [2.2]paracyclophane repeating unit in 23% isolated yield with the Mn of 3800 and the PDI of 1.3, as shown in Scheme 9.34
The optical profiles of pseudo-ortho-linked polymer P9 were similar to those of pseudo-para-linked polymer P3 as well as compound 9 (Table 2). This is because both P3 and P9 comprise the same π-electron system, i.e., model compound 9 is stacked straight (for P3) or zigzag (for P9). As shown in Fig. 5A, the absorption spectrum of P9 was largely consistent with that of P3, and the maximum of the absorption peak of P9 appeared at 377 nm. This λmax value was smaller than that of P3 (λmax = 387 nm) because of the shorter chain length of P9 relative to that of P3. Fig. 5B shows the photoluminescence spectra of P3 and P9 in diluted CHCl3 solution (1.0 × 10−7 M) excited at each absorption maximum. Their PL spectra, which were similar to the spectrum of 9 despite their π-stacked structures, exhibited a clear vibrational structure with peak maxima at around 400 nm. In addition, the photoluminescence quantum efficiencies of P3, P9, and 9 were found to be 0.82, 0.86, and 0.86, respectively (Table 2). This result indicates that the through-space conjugated polymers emit bright blue light from the chromophore state rather than the phane state (an excimer-like state).35
| Compound | λ Abs,max a/nm | λ PL,max b/nm | Φ PL c |
|---|---|---|---|
| a λ Abs,max of UV-vis absorption spectra in CHCl3 (1.0 × 10−5 M). b λ PL,max of photoluminescence spectra in CHCl3 (1.0 × 10−7 M). c Relative photoluminescence quantum efficiencies calculated by using 9-anthracenecarboxylic acid in CH2Cl2 as a standard. | |||
| P3 | 319, 386 | 411 | 0.82 |
| P9 | 319, 377 | 407 | 0.86 |
| 9 | 320, 369 | 398 | 0.86 |
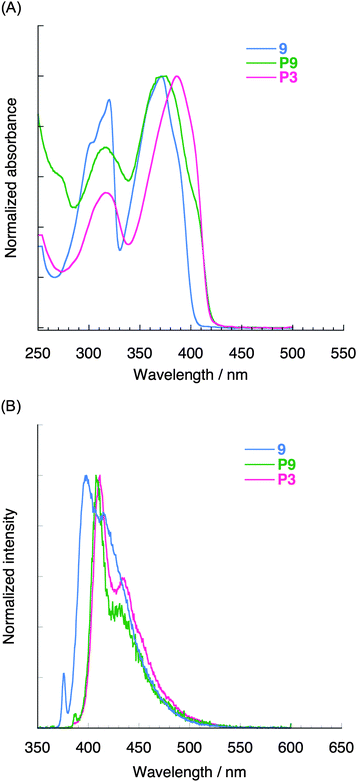 | ||
| Fig. 5 (A) UV-vis absorption spectra in CHCl3 (1.0 × 10−5 M) of 9, P9, and P3. (B) Photoluminescence spectra in CHCl3 (1.0 × 10−7 M) of 9, P9, and P3. | ||
4. Pseudo-geminal-disubstituted [2.2]paracyclophane-containing through-space conjugated polymers
The through-space conjugated polymer P10, composed of pseudo-geminal-disubstituted [2.2]paracyclophane, was recently synthesized by Jagtap and Collard.36Pseudo-geminal-diethynyl[2.2]paracyclophane 28 was prepared viamethyl [2.2]paracyclophane-4-carboxylate 25,37 as outlined in Scheme 10. Selective formylation of 25 was achieved using TiCl4 to yield the corresponding pseudo-geminal-disubstituted [2.2]paracyclophane 26,38 which was converted to the key monomer 28 by Hopf's procedures.39 Employing the Sonogashira–Hagihara coupling by the PdCl2(PPh3)2/CuI catalytic system afforded PAE type through-space conjugated polymer P10 in 60% yield with the Mn of 5000 and the PDI of 1.7 (Scheme 10).![Synthesis of pseudo-geminal-disubstituted [2.2]paracyclophane-containing polymer P10.](/image/article/2011/PY/c0py00421a/c0py00421a-s10.gif) | ||
| Scheme 10 Synthesis of pseudo-geminal-disubstituted [2.2]paracyclophane-containing polymer P10. | ||
A π-stacked structure, in which PAE π-electron systems 29 fully overlapped in close proximity, was constructed in a single polymer chain. The optical profiles supported the π-stacked structure, as summarized in Table 3. The absorption edge of pseudo-geminal-linked P10 was observed at around 470 nm, whereas those of pseudo-para- and pseudo-ortho-linked P3 and P9 appeared at around 430 nm. Furthermore, the photoluminescence spectrum of P10 was broad and featureless. The maximum of the peak appeared at around 530 nm with a Stokes shift of 171 nm, which was much larger than those of pseudo-para- and pseudo-ortho-linked P3 and P9 (Table 3). These results strongly suggest that the PAE π-electron systems form π-stacked structures in the ground state and the excited state.
| Compound | λ Abs,max/nm | λ Abs,edge/nm | λ PL,max/nm | Stokes shift/nm |
|---|---|---|---|---|
| a The UV-vis absorption and photoluminescence spectra was shown in ref. 36. | ||||
| P3 | 319, 386 | 430 | 411 (vibrational) | 25 |
| P9 | 319, 377 | 430 | 407 (vibrational) | 30 |
| P10 a | 320, 359 | 470 | 530 (broad) | 171 |
| 29 | 321, 374 | 410 | 402 (vibrational) | 28 |
5. Conclusion and outlook
This review summarizes recent reports on the synthesis of through-space conjugated polymers consisting of the [2.2]paracyclophane repeating units in the conjugated polymer backbone. Polymers can be easily obtained by common polymerization methods such as the palladium-catalyzed coupling reaction, Knoevenagel reaction, etc. Introduction of the [2.2]paracyclophane skeletons into the conjugated polymer leads to the formation of new π-electron systems in the polymer, which induces intramolecular π-stacking in a primary polymer structure. [2.2]Paracyclophane does not exist solely as a [2.2]paracyclophanylene unit in the polymer; it acts as a bridge unit binding the π-electron systems at short distances. Therefore, this class of through-space conjugated polymers would be regarded as those comprised of stacked π-electron systems rather than [2.2]paracyclophane repeating units. This means that each π-electron system possesses each HOMO–LUMO energy band-gap in a [2.2]paracyclophane-containing through-space conjugated polymer. That is, each π-electron system consists of more than one band gap. In contrast, a common π-conjugated polymer basically consists of a conduction band and a valence band. Incorporation of various aromatic groups as comonomers into the through-space conjugated polymers is possible, resulting in easy control of each energy band-gap in stacked π-electron systems. Through-space conjugated polymers are emerging as one of the most promising single molecular wires in which energy and charges are transferred via the through-space interactions among the stacked π-electron systems.Notes and references
- (a) Science and Applications of Conducting Polymers, ed. W. R. Salaneck, D. T. Clark and E. J. Samuelsen, Adam Hilger, Bristol, 1991 Search PubMed; (b) Handbook of Organic Conductive Molecules, ed. H. S. Nalwa, Wiley, Chichester, 1997 Search PubMed; (c) Handbook of Conducting Polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker, New York, 3rd edn, 2006 Search PubMed.
- A. C. Grimsdale and K. Müllen, Adv. Polym. Sci., 2006, 199, 1–82 CAS.
- (a) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. Friend, H. P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–554 CrossRef CAS; (b) N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628–630 CrossRef; (c) U. Scherf, Top. Curr. Chem., 1999, 201, 163–222 CAS.
- (a) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS; (b) Poly(arylene ethynylene)s, in Adv. Polym. Sci., ed. C. Weder, 2005, vol. 177 Search PubMed.
- (a) Y. Morisaki and Y. Chujo, in Conjugated Polymer Synthesis, ed. Y. Chujo, Wiley-VCH, Weinheim, 2010, ch. 5 Search PubMed; (b) T. Nakano, Polym. J., 2010, 42, 103–123 CrossRef CAS.
- (a) B. O'Regan and M. Graetzel, Nature, 1991, 353, 737–740 CrossRef CAS; (b) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef; (c) Organic Photovoltaics: Materials, Device Physics, and Manufacturing Technologies, ed. C. Brabec, V. Dyakonov and U. Scherf, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (d) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- (a) Organic Light Emitting Devices: Synthesis, Properties and Application, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- (a) Cyclophane Chemistry: Synthesis, Structures and Reactions, ed. F. Vögtle, John Wiley & Sons, Chichester, 1993 Search PubMed; (b) Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (c) H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 9808–9812 CrossRef CAS.
- (a) Y. Morisaki and Y. Chujo, Macromolecules, 2002, 35, 587–589 CrossRef CAS; (b) Y. Morisaki and Y. Chujo, Chem. Lett., 2002, 194–195 CrossRef CAS; (c) Y. Morisaki, T. Ishida and Y. Chujo, Macromolecules, 2002, 35, 7872–7877 CrossRef CAS; (d) Y. Morisaki and Y. Chujo, Polym. Bull., 2002, 49, 209–215 CrossRef CAS; (e) Y. Morisaki, F. Futoshi and Y. Chujo, Organometallics, 2003, 22, 3553–3557 CrossRef CAS; (f) Y. Morisaki and Y. Chujo, Macromolecules, 2003, 36, 9319–9324 CrossRef CAS; (g) Y. Morisaki and Y. Chujo, Macromolecules, 2004, 37, 4099–4103 CrossRef CAS; (h) Y. Morisaki, T. Ishida, H. Tanaka and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5891–5899 CrossRef CAS; (i) Y. Morisaki, N. Wada and Y. Chujo, Polym. Bull., 2005, 53, 73–80 CrossRef CAS; (j) Y. Morisaki, N. Wada and Y. Chujo, Polymer, 2005, 46, 5884–5889 CrossRef CAS; (k) Y. Morisaki and Y. Chujo, Bull. Chem. Soc. Jpn., 2005, 78, 288–293 CrossRef CAS; (l) Y. Morisaki and Y. Chujo, Tetrahedron Lett., 2005, 46, 2533–2537 CrossRef CAS; (m) Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2006, 45, 6430–6437 CrossRef CAS; (n) Y. Morisaki and Y. Chujo, Prog. Polym. Sci., 2008, 33, 346–364 CrossRef CAS; (o) Y. Morisaki, T. Murakami and Y. Chujo, Macromolecules, 2008, 46, 5960–5963 CrossRef; (p) Y. Morisaki, T. Murakami, T. Sawamura and Y. Chujo, Macromolecules, 2009, 42, 3656–3660 CrossRef CAS; (q) Y. Morisaki, T. Murakami and Y. Chujo, J. Inorg. Organomet. Polym. Mater., 2009, 19, 104–112 CAS; (r) Y. Morisaki, L. Lin and Y. Chujo, Polym. Bull., 2009, 62, 737–747 CrossRef CAS; (s) L. Lin, Y. Morisaki and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7003–7011 CrossRef CAS.
- (a) Y. Morisaki, T. Ishida and Y. Chujo, Polym. J., 2003, 35, 501–506 CrossRef CAS; (b) Y. Morisaki, T. Ishida and Y. Chujo, Org. Lett., 2006, 8, 1029–1032 CrossRef CAS; (c) Y. Morisaki, T. Ishida and Y. Chujo, Polym. Bull., 2006, 57, 623–630 CrossRef CAS; (d) Y. Morisaki, T. Ishida and Y. Chujo, C. R. Chim., 2009, 12, 332–340 CrossRef CAS.
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915–916 CrossRef CAS.
- R. A. Meyers, J. W. Hamersma and H. A. Green, J. Polym. Sci., Part B: Polym. Lett., 1972, 10, 685–689 CrossRef CAS.
- (a) K. P. Sivaramakrishnan, C. Samyn, I. J. Westerman, D. T. Wong and C. S. Marvel, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1083–1094 CrossRef CAS; (b) D. M. Chang and C. S. Marvel, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2507–2515 CrossRef CAS; (c) S. Lin and C. S. Marvel, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1151–1157 CrossRef CAS.
- (a) S. Mizogami and S. Yoshimura, J. Chem. Soc., Chem. Commun., 1985, 427–428 RSC; (b) S. Mizogami and S. Yoshimura, J. Chem. Soc., Chem. Commun., 1985, 1736–1738 RSC.
- (a) D. M. Collard and B. Lee, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2000, 41, 241 CAS; (b) F. Salhi, B. Lee, C. Metz, L. A. Bottomley and D. M. Collard, Org. Lett., 2002, 4, 3195–3198 CrossRef CAS; (c) F. Salhi and D. M. Collard, Adv. Mater., 2003, 15, 81–85 CAS.
- (a) L. Guyard and P. Audebert, Electrochem. Commun., 2001, 3, 164–167 CrossRef CAS; (b) L. Guyard, P. Audebert, W. R. Dolbier, Jr and J.-X. Duan, J. Electroanal. Chem., 2002, 537, 189–193 CrossRef CAS.
- (a) J. Nishimura and S. Yamashita, in Cyclopolymerization and Polymers with Chain-ring Structures, ed. G. B. Butler and J. E. Kresta, American Chemical Society, Washington, DC, 1982, pp. 177–195 Search PubMed; (b) J. Furukawa and J. Nishimura, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 85–90 CrossRef CAS; (c) J. Furukawa and J. Nishimura, J. Polym. Sci., Polym. Symp., 1976, 56, 437–446 Search PubMed; (d) J. Nishimura and S. Yamashita, Polym. J., 1979, 11, 619–627 CrossRef CAS; (e) J. Nishimura, M. Mimura, N. Nakazawa and S. Yamashita, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 2071–2084 CrossRef CAS; (f) J. Nishimura, M. Furukawa, S. Yamashita, T. Inazu and T. Yoshino, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 3257–3268 CrossRef CAS.
- (a) D. T. Glatzhofer and D. T. Longone, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 947–954 CrossRef CAS; (b) D. T. Longone and D. T. Glatzhofer, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1725–1733 CrossRef CAS; (c) J. Ulan'ski, J. Kubacki, I. Glowacki, M. Kryszewski and D. T. Glatzhofer, J. Appl. Polym. Sci., 1992, 44, 2103–2106 CrossRef CAS; (d) D. J. Guerrero and D. T. Glatzhofer, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 457–464 CrossRef CAS; (e) J. Ulan'ski, J. Sielski, D. T. Glatzhofer and M. Kryszewski, J. Phys. D: Appl. Phys., 1990, 23, 75–78 CrossRef CAS.
- S. Iwatsuki, T. Itoh, M. Kubo and H. Okuno, Polym. Bull., 1994, 32, 27–34 CrossRef CAS.
- N. Wada, Y. Morisaki and Y. Chujo, Macromolecules, 2009, 42, 1439–1442 CrossRef CAS.
- (a) Y. Tohda, K. Sonogashira and N. Hagihara, Synthesis, 1977, 777–778 CrossRef CAS; (b) K. Sonogashira, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-VCH, New York, 2002, pp. 493–529 Search PubMed.
- Unpublished result.
- (a) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CAS; (b) R. F. Heck and J. P. Nolley, Jr, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- (a) N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) R. Rathore, S. H. Abdelwahed and L. A. Guzei, J. Am. Chem. Soc., 2003, 125, 8712–8713 CrossRef CAS; (b) T. Nakano and T. Yade, J. Am. Chem. Soc., 2003, 125, 15474–15484 CrossRef CAS; (c) N. Caraballo Martínez, M. del Rosario Colorado Heras, M. Mba Blázquez, J. Osío Barcina, A. García Martínez and M. del Rosario Torres Salvador, Org. Lett., 2007, 9, 2943–2946 CrossRef CAS.
- (a) Y. Morisaki, L. Lin and Y. Chujo, Chem. Lett., 2009, 38, 734–735 CrossRef CAS; (b) Y. Morisaki, L. Lin and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5979–5988 CrossRef CAS.
- (a) F. Lange, D. Hohnholz, M. Lerze, H. Ryu, M. Mohloch and R. F. Hanack, Synth. Met., 1999, 101, 652–653 CrossRef CAS; (b) M. Hohloch, C. Maichle-Moössmer and M. Hanack, Chem. Mater., 1998, 10, 1327–1332 CrossRef CAS.
- L. Lin, Y. Morisaki and Y. Chujo, Int. J. Polym. Sci., 2010, 908128 Search PubMed.
- (a) I. Yamaguchi and T. Yamamoto, Chem. Lett., 2002, 938–939 CrossRef CAS; (b) R. S. Lokey and B. L. Iverson, Nature, 1995, 375, 303–305 CrossRef CAS; (c) X. Zhao, M.-X. Jia, X.-K. Jiang, L.-Z. Wu, Z.-T. Li and G.-J. Chen, J. Org. Chem., 2004, 69, 270–279 CrossRef CAS; (d) S. Ghosh and S. Ramakrishnan, Macromolecules, 2005, 38, 676–686 CrossRef CAS; (e) W. Zhang, W. R. Dichtel, A. Z. Stieg, D. Benitez, J. K. Gimzewski, J. R. Heath and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6514–6519 CrossRef CAS.
- S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 CrossRef CAS.
- T. Förster, Naturwissenschaften, 1946, 33, 166–175 CrossRef CAS.
- H. J. Reich and D. J. Cram, J. Am. Chem. Soc., 1969, 91, 3517–3526 CrossRef CAS.
- L. Bondarenko, I. Dix, H. Hinrichs and H. Hopf, Synthesis, 2004, 2751–2759 CAS.
- Y. Morisaki, N. Wada, M. Arita and Y. Chujo, Polym. Bull., 2009, 62, 305–314 CrossRef CAS.
- These phenomena were established by Bazan and co-workers using [2.2]paracyclphane-containing stilbenoid oligomers: see: (a) W. J. Oldham, Jr, Y. J. Miao, R. J. Lachicotte and G. C. Bazan, J. Am. Chem. Soc., 1998, 120, 419–420 CrossRef; (b) G. C. Bazan, W. J. Oldham, Jr, R. J. Lachicotte, S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 1998, 120, 9188–9204 CrossRef CAS; (c) S. Wang, G. C. Bazan, S. Tretiak and S. Mukamel, J. Am. Chem. Soc., 2000, 122, 1289–1297 CrossRef CAS.
- S. P. Jagtap and D. M. Collard, J. Am. Chem. Soc., 2010, 132, 12208–12209 CrossRef CAS.
- M. Psiorz and R. Schmid, Chem. Ber., 1987, 120, 1825–1828 CrossRef CAS.
- H. Zitt, I. Dix, H. Hopf and P. G. Jones, Eur. J. Org. Chem., 2001, 2298–2307.
- (a) E. V. Sergeeva, V. I. Rozenberg, D. Y. Antonov, E. V. Vorontsov, Z. A. Starikova, I. V. Fedyanin and H. Hopf, Chem.–Eur. J., 2005, 11, 6944–6961 CrossRef CAS; (b) L. Bondarenko, S. Hentschel, H. Greiving, J. Grunenberg, H. Hopf, I. Dix, P. G. Jones and L. Ernst, Chem.–Eur. J., 2007, 13, 3950–3963 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |