Synthetic methodology effect on the microstructure and thermal properties of poly(n-butyl acrylate-co-methyl methacrylate) synthesized by nitroxide mediated polymerization†
Nabila
Cherifi
abc,
Adeline
Issoulie
c,
Abdel
Khoukh
c,
Ahmed
Benaboura
b,
Maud
Save
c,
Christophe
Derail
c and
Laurent
Billon
*c
aCentre de recherches Scientifiques et Techniques en Analyse Physico-chimiques (C.R.A.P.C), BP 248 Alger RP, 16004, Alger, Algérie
bUniversité des Sciences et Technique Houari Boumedienne, Faculté de Chimie, Laboratoire de Synthèse Macromoléculaire et Thio-Organique Macromoléculaire, B.P. 32 El-Alia, 16111, Alger, Algérie
cUniversité de Pau et des Pays de l'Adour, CNRS, IPREM - Equipe de Physique et Chimie des Polymères, UMR 5254 Hélioparc, 2 avenue du président Angot, 64053, Pau, France. E-mail: laurent.billon@univ-pau.fr
First published on 26th April 2011
Abstract
In this work, we report the synthesis of poly(n-butyl acrylate-co-methyl methacrylate) copolymers by the nitroxide mediated polymerization (NMP) technique, using N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl)nitroxide (SG1) as a control agent and 2-methylaminoxypropionic-SG1 alkoxyamine (BlocBuilder®) as the initiator. The copolymers are synthesized either by batch or semi-batch processes and the gradient profile is examined via the determination of the instantaneous fraction of monomer incorporated in the copolymer. A control of the molar mass together with low molar mass distribution (Mw/Mn < 1.4) is observed. The dependence of the copolymer glass transition temperature with conversion was followed by differential scanning calorimetry. The copolymers are investigated by carbon nuclear magnetic resonance and heteronuclear multiple bond correlation (HMBC) NMR sequences to study the effect of the monomer addition mode on the microstructure of copolymers. The thermomechanical properties of gradient copolymers are finally reported to establish the effect of the composition on the mechanical behaviour of the copolymers.
Introduction
Free radical polymerization (FRP) is a versatile technique, largely used for industrial manufacturing procedures, which allows polymerization of a wide range of monomers with various functional groups. Nevertheless, due to the spontaneous transfer reactions and irreversible bimolecular terminations existing during FRP, the lifetime of propagating radicals is very short. This induces generally poor control of polymer microstructures with broad molar mass distribution. With the aim of avoiding the disadvantages of free radical synthesis, living controlled radical polymerizations (CRP) were developed in the mid 1990s. They are based on the principle of dynamic equilibration between dormant and active species with decrease of the instantaneous radical concentration. This induces formation of materials with predictable molecular weight and low distribution. Atom Transfer Radical Polymerization (ATRP),1–4 Radical Addition Fragmentation and Transfer (RAFT)5–7 and Nitroxide Mediated Polymerization (NMP)8,9 are the three most successful radical living techniques frequently used to polymerize large families of monomers. These techniques allow the synthesis of a variety of graft, star, block and gradient (co)polymer complex polymeric architectures. Gradient copolymers are a class of materials re-visited by Matyjaszewski et al. in 1996.10–12 They are characterized by a gradual change of the copolymer composition along the chains. Depending on the nature of the monomers used, two types of gradient copolymers can be differently synthesized: (a) spontaneous gradients obtained by batch process, with monomers exhibiting different reactivity ratios. The more active monomer will be rapidly consumed in the beginning of polymerization, contrary to less one, inducing spontaneous changes in composition of growing macromolecules. (b) Forced gradient copolymers synthesized by so called semi batch process, when monomers with close reactivity ratios are used. In this process, one monomer is continuously added to the other during the course of the polymerization, using a chosen addition rate. ATRP,13–19 RAFT,20–22 and NMP23–31 techniques were heavily investigated to synthesize this new class of copolymers. Indeed, the ATRP technique was mainly applied by Matyjaszewski et al.13–17 in synthesize of numerous spontaneous gradients using acrylate/methacrylate or styrene/acrylate pairs of monomers. Syntheses of forced gradients were also reported using poly(methyl methacrylate-grad-2(trimethyl silyl oxy)ethyl methacrylate18 or styrene/acrylonitrile copolymers.19On the other hand, the gradient copolymer of methyl methacrylate (MMA) and methacrylate-terminated poly(dimethylsiloxane),20styrene/butyl acrylate21 were synthesized by RAFT technique. This same process was applied in the synthesis of gradient copolymers based on complex-radical terpolymerization of styrene, maleic anhydride and N-vinylpyrrolidone initiated by γ-rays.22
By using the NMP technique, Mignard et al.23 have reported the synthesis of gradient copolymers of styrene and n-butyl acrylate comonomers. In addition, styrene/4-acetoxystyrene and styrene/4-hydroxystyrene gradients were reported by Torkelson et al.24,25 Trang et al.26 reported the synthesize of poly(methyl methacrylate-co-N,N-dimethyl acrylamide) gradient copolymers and Karaky et al. reported the successful synthesis of spontaneous and forced gradient copolymers: poly(octadecyl acrylate-grad-methyl acrylate),27 poly(N,N-Dimethylacrylamide-grad-butyl acrylate),28,29 poly(styrene-grad-methyl acrylate)30 and poly(styrene-grad-butyl acrylate).31
The poly(n-butyl acrylate-co-methyl methacrylate) polymers were largely studied by different techniques, conventional radical polymerization,32,33 RAFT34 and ATRP.35–39 These two monomers n-BA, MMA present large difference in the monomer reactivity ratios, rMMA = 2.19 and rBA = 0.39,40 and it has been demonstrated viaATRP37,38 that this difference leads to the formation of spontaneous gradient copolymers. However, to the best of our knowledge, the NMP technique was never applied in the synthesis of this couple of comonomers. In this contribution, we have been interested in the use of NMP to synthesise n-butyl acrylate/methyl methacrylate gradient copolymers. Batch and semi batch processes were applied using two addition rates. We focused our attention on the control of the polymerization with a study of kinetic and macromolecular features. The instantaneous composition was concomitantly followed by 1H-NMR.
The recovered copolymers were thoroughly characterized by 13C-NMR, HMBC and DSC experiments in order to confirm the gradient profile. Finally, the effect of the composition on the thermorheological properties is proposed.
Experimental section
Materials
n-Butyl acrylate (BA, Aldrich, 99%), methyl methacrylate (MMA, Aldrich, 99%) were used as received. The alkoxyamine BlocBuilder® (2-methylaminoxypropionic-SG1) used as the initiator and the nitroxide SG1 (88%) were provided by Arkema Chemicals. All solvents were used without further purification.Synthesis of P(BA-co-MMA) copolymers
For all the polymerizations, samples were withdrawn during the polymerization for further NMR, SEC and DSC analyses. The final recovered copolymers were precipitated twice into a methanol/water (90/10) mixture. The final products were filtered and dried in a vacuum oven until constant weights were reached. Table 1 summarizes the experimental conditions of the copolymerizations.
| Experiment | Final nBA (mol) | Final nMMA (mol) |
|
|
MMA addition rate (mL h−1) |
|---|---|---|---|---|---|
| Batch 50/50 | 0.242 | 0.243 | 505 | 0.05 | — |
| Batch 75/25 | 0.48 | 0.16 | 500 | 0.05 | — |
| Semibatch-2.2 | 0.192 | 0.192 | 500 | 0.05 | 2.2 |
| Semibatch-4.3 | 0.192 | 0.192 | 500 | 0.05 | 4.3 |


Analytical techniques
The crude polymer solutions were analyzed by size exclusion chromatography (SEC) at 30 °C with THF as eluent at a flow rate of 1 mL min−1. The SEC system was equipped with three Waters Styragel columns HR 0.5, 2, 4 working in series (separation range 1 × 102 – 3 × 106 g mol−1) and a Viscotek ERC 7515-A refractive index detector. Number-average molar mass (Mn) and dispersity (Mw/Mn) were calculated with a calibration derived from PS standards using OmniSEC® software. All polymers samples were prepared at 1 to 5 g L−1 concentrations and filtered through PVDF 0.45 μm filters.The proton nuclear magnetic resonance (1H NMR) spectra of the crude polymer solutions were recorded at 25 °C using a Bruker Advanced AM400 spectrometer (400 MHz), in CDCl3 as solvent, in order to determine the monomer conversions and the copolymer compositions. The copolymer compositions were determined from the relative area of the proton of OCH3 at 3.5–3.65 ppm corresponding to MMA units and of OCH2 of BA units at 3.8–4.00 ppm. The copolymer microstructures were also analyzed by carbon nuclear magnetic resonance (13C NMR, 100 MHz) and heteronuclear multiple bond correlation (HMBC) NMR sequences in CDCl3.
Differential scanning calorimetry technique (DSC) was carried out on Q100 from TA instruments, to measure the copolymer glass transition temperature (Tg). The samples taken during the polymerization were precipitated into a methanol/water (90/10) mixture. For each measurement, a quantity of 8–10 mg of copolymer was weighed and scanned at 20 °C min−1, from −90 °C to 140 °C, under dry nitrogen (50 mL min−1).
Thermomechanical analysis has been performed by using a constant strain rheometer (RDA II, Rheometrics) where the real part (G′, storage modulus) and the imaginary part (G′′, loss modulus) of the complex shear modulus G* was measured, in the linear domain, at a fixed circular frequency (1 rad s−1) as a function of temperature with a speed of 1 °C min−1. We have also reported tanδ which is the ratio between G′′ and G′. The measurement of G*(T) allows us to determine the different transitions which can influence the mechanical properties, particularly the glassy transition temperature, Tα, defined by the temperature corresponding to the maximum of tanδ. To minimize the compliance of the apparatus according to the behavior of the polymers at the experimental temperature, plate-plate geometry was used with different plate diameters from 5 mm to 15 mm.
Results and discussion
In the present study, the copolymerization of n-butyl acrylate BA and methyl methacrylate MMA was performed by NMP at 115 °C using Blocbuilder alkoxyamine and free SG1 nitroxide. The polymerization was carried out either by batch or in semi-batch processes to target spontaneous and forced gradient copolymers.Nitroxide mediated polymerization of methacrylic esters has presented a great challenge for a long time since they have a large activation–deactivation equilibrium constant. In 2005, Charleux et al.41 presented a theoretical approach to describe the copolymerization kinetics in controlled living copolymerization operating via reversible termination. They theoretically determined the variation of the average activation–deactivation equilibrium constant <K> of the copolymerization and showed that less than 10 mol% of styrene was enough to control the NMP of methyl methacrylate at 90 °C by sufficiently decreasing the value of <K> (<K> ≈ 2 × 10−9 mol L−1).41 As reported in Fig. S1 (ESI†), a molar fraction of 75% of n-butyl acrylate is required to reach a similar <K> value of 2 × 10−9 mol L−1 for its copolymerization with MMA carried out at 115 °C. For the purpose of this work aiming at exploring the properties of poly(BA-co-MMA) copolymers with a minimum content of 25% of MMA, we performed two batch copolymerizations starting with two different initial compositions of the comonomer mixture (BA/MMA: 50/50 and BA/MMA: 75/25) and two semi-batch copolymerizations targeting a final composition of 50/50 in BA/MMA. The <K> value for BA/MMA copolymerization performed at 115 °C starting with a molar fraction of BA in the comonomer mixture of 0.5 is equal to 7 × 10−9 mol L−1 (see Fig. S1†). The experimental conditions of the four polymerizations are gathered in Table 1.
Fig. 1 exhibits the variation of the logarithmic monomer concentration versus time and the evolution of Mn and the dispersity (Mw/Mn) as a function of conversion for both batch copolymerizations. Theoretical Mn of the copolymer, calculated from eqn (1), are also depicted in Fig. 1.
 | (1) |
![Batch copolymerization of BA and MAMA. (a) Variation of ln[M]0/[M] versus time. (b) Evolution of the average molar mass (Mn) and dispersity (Mw/Mn) versus conversion. The black symbols (diamonds) and lines correspond to the Batch 50/50 experiment and the grey symbols (triangles) and lines correspond to the Batch 75/25 experiment. The plain and the dotted lines correspond to the fit of theoretical Mn and the experimental Mn respectively.](/image/article/2011/PY/c1py00066g/c1py00066g-f1.gif) | ||
| Fig. 1 Batch copolymerization of BA and MAMA. (a) Variation of ln[M]0/[M] versus time. (b) Evolution of the average molar mass (Mn) and dispersity (Mw/Mn) versus conversion. The black symbols (diamonds) and lines correspond to the Batch 50/50 experiment and the grey symbols (triangles) and lines correspond to the Batch 75/25 experiment. The plain and the dotted lines correspond to the fit of theoretical Mn and the experimental Mn respectively. | ||
The variation of the logarithmic monomer concentration versus time shows a fast initiation followed by a linear evolution (Fig. 1a). The experimental Mn increases linearly versus conversion for both Batch 50/50 and Batch 75/25 experiments, which is characteristic of a controlled polymerization. We can notice that the experimental Mn values match the theoretical ones for the Batch 75/25 whereas Mn values were slightly above Mn,theo for the Batch 50/50 experiment. This reveals the presence of early irreversible termination reactions with an initiating efficiency of approximately 80%, which was expected from the highest <K> value observed for an initial 50/50 comonomer mixture (see Fig. S1 in the ESI†). Nevertheless, the two copolymers exhibit features of controlled systems since no shoulder was observed in the SEC chromatograms of the final samples (see Fig. S2 in ESI†) together with low dispersity (1.2 < Mw/Mn < 1.4).
Evolutions of the kinetics and macromolecular features of the semi-batch copolymerizations of BA and MMA are displayed in Fig. 2. An increase of Mn with conversion and narrow molar mass distributions with Mw/Mn values below 1.5 are observed. The SEC traces of the copolymers are reported in Fig. S2 (ESI†) and shows symmetrical peaks. On the other hand, the logarithmic monomer concentrations gradually increase with time and should follow eqn (2).
 | (2) |
![Semi-batch copolymerization of BA and MMA. (a) Variation of ln[M]0/[M] versus time. (b) Evolution of molar mass (Mn) and dispersity (Mw/Mn) versus conversion. The squares correspond to the Semibatch-2.2 experiment and the triangles correspond to the Semibatch-4.3 experiment.](/image/article/2011/PY/c1py00066g/c1py00066g-f2.gif) | ||
| Fig. 2 Semi-batch copolymerization of BA and MMA. (a) Variation of ln[M]0/[M] versus time. (b) Evolution of molar mass (Mn) and dispersity (Mw/Mn) versus conversion. The squares correspond to the Semibatch-2.2 experiment and the triangles correspond to the Semibatch-4.3 experiment. | ||
Indeed, the non-linearity of the curve can be explained by the semi-batch process as the continuous addition of MMA into the comonomer mixture induces a change of the MMA comonomer fraction (fMMA) which impacts directly the <kp><K> product, with <kp> the average rate constant of propagation of the copolymerization (see eqn (3) from ref. 41). Fig. 3 depicts the theoretical evolution of the <kp><K> product with the MMA fraction in the comonomer mixture, hence predicting the non linear variation of ln[M]0/[M] versus time (see also Fig. S3 in the ESI†). The numerical values used for plotting eqn (3) are reported in ref. 42–44 and note 45.
 | (3) |
 | ||
| Fig. 3 Predicted values of the product of the average activation-deactivation equilibrium constant with the average propagation rate constant <kp> <K>versus the molar fraction of methyl methacrylate in the comonomer mixture, for nitroxide-mediated copolymerization of BA and MMA at 115 °C. | ||
The variation of instantaneous fraction of monomer (Finst) as a function of the apparent normalized chain length has been largely used to assess the gradient profile of synthetic copolymers.27,46–48 The instantaneous fraction of BA in the copolymers was calculated from eqn (4), where Δ(%conv) denotes the monomer conversion difference in a time interval.
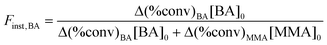 | (4) |
The apparent normalized chain length is calculated by dividing the Mn value of copolymer, recovered at given time and to the final Mn value of the recovered final copolymer. Fig. 4 illustrates the results obtained for the four BA/MMA copolymerizations experiments. In the case of both batch experiments (Batch 50/50 and Batch 75/25), the BA instantaneous fraction increases with the apparent normalized chain length, which is in accordance with the values of reactivity ratios (rMMA = 2.19 and rBA = 0.39).40 Since MMA is more reactive than BA, thus it is firstly incorporated and preferentially consumed. This induces the enrichment of the copolymer in BA at the end of the copolymerization, and consequently the formation of instantaneous gradient copolymers.
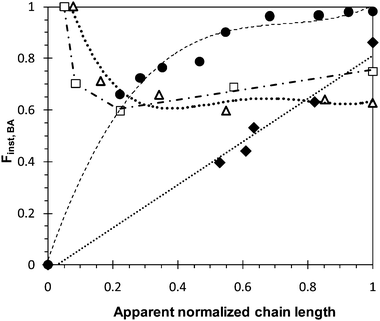 | ||
| Fig. 4 Evolution of the instantaneous fraction of BA (Finst) for synthesized P(BA-co-MMA) copolymers as a function of the apparent normalized chain length: (◆) Batch 50/50, (●) Batch 75/25, (□) Semibatch-2.2 and (△) Semibatch-4.3. | ||
For Semibatch-2.2 and Semibatch-4.3 experiments, BA monomer was first polymerized in bulk for one hour up to 10% conversion (DP ≈ 50) before applying a semi-batch process by incorporating MMA at two different addition rates. As shown by Fig. 4, from the moment the addition of MMA started, we first observed a decrease in the instantaneous fraction of BA in the copolymer with an increase of the apparent normalized chain length until reaching a plateau of Finst,BA. Hence, gradient copolymers by semi-batch synthesis were also obtained, but with gradient shape different to that obtained by batch synthesis.
Several research groups used the nuclear magnetic resonance (NMR) technique for determining the copolymer microstructure, particularly the sequence distribution and tacticity.36,49–53 In the present study, we have been interested in investigating the microstructure of the different synthesized copolymers by NMR. The aim was to compare the copolymer microstructure according to the gradient profile (Fig. 4) which depends on the polymerization process (batch or semi-batch). In 2001, According to Aerdts’ method,49 Madruga et al.36 reported the study of stereochemical arrangement of monomers in poly(BA-co-MMA) copolymers synthesized by ATRP and FRP techniques. They reported seven distinguishable signals in the carbon carbonyl region of the 13C NMR spectra. Their relative intensities changed with the composition of monomer in the copolymer and depended on both the diad tacticity (racemic (r) or meso (m)) and the proportion of the different diads and triads sequences. Four of these signals were assigned to sequences and configurations of MMA (M)-centered triads. The three others corresponded to BA (B)-centered triads.36
The carbonyl area of the copolymers synthesized in the present work was analyzed by carbon NMR. Fig. 5 shows the corresponding NMR spectra in the carbonyl regions of both Batch 75/25 and Semibatch-2.2 and Fig. S4 in the ESI† displays the NMR spectra of Semibatch-4.3 copolymer. The signals of the MMA (M) centered triads are assigned to (A) MMM (rr), (B) MMM (mr + rm), BMM (mr + rr), (C) MMM (mm), BMM (rm + mm), MMB (mr + mm) and BMB (mm + rm + mr), and finally (D) BMB (rr) triads. The signals of the BA (B) centered triads are assigned to (E) MBM, (F) MBB and BBB triads (G).
 | ||
| Fig. 5 13C NMR spectra showing the carbonyl region of (a) Batch 75/25 and (b) Semibatch-2.2 copolymers as function of time and apparent normalized chain length. | ||
The comparison between the intermediate 13C NMR spectra recorded at different polymerization time intervals for the Batch 75/25 experiment, as shown in Fig. 5, reveals an increase in the relative intensity of BBB triad signal versus time, accompanied with a decrease in the relative intensities of (A), (B), (C) and (E) signals. This confirms the enrichment of the copolymer chain in BA, as observed in Fig. 4. In contrast, the series of 13C NMR spectra for the Semibatch-2.2 copolymer present a decrease in the relative intensity of BBB triad signal versus time, together with a gradual increase in the relative intensities of (A), (B), (C) and (E) signals (see Fig. 5). These results confirm the gradual incorporation of MMA units in the copolymer chains. Similar results are observed for the Semibatch-4.3 copolymer (see Fig. S4 in the ESI†).
Two dimensional NMR (2D NMR) experiments are very useful for the investigation of polymer microstructures50–53 considering different techniques especially Heteronuclear Single Quantum Coherence (HSQC), Total Correlated Spectroscopy (TOCSY) and Heteronuclear Multiple Bond correlation (HMBC).
Brar et al.53 have extensively studied the microstructure of spontaneous poly(MMA-g-BA) copolymers produced by ATRP with a complete assignment of the peaks by 2D HSQC, 2D TOCSY and 2D HMBC experiments. In this work, we have been interested in the characterization of the synthesized copolymers by 2D-HMBC NMR spectroscopy in the area of the α-methyl, methylene and methyne protons coupled with carbonyl carbons, in order to illustrate the formation of MMMM or BBBB tetrads in copolymers, not observed by 13C NMR. We focused our attention on the crosspeaks observed at: (1) 177.24/0.94 ppm assigned to the coupling of carbonyl carbons with α-methyl proton in the triad MrMmM. The three crosspeaks (2) 177.24/1.91 ppm, (3) 177.42/1.86 ppm and (4) 177.00/1.37 ppm, assigned to the coupling of the carbonyl carbon with the methylene protons of the MrMmMrM tetrads. Also, the crosspeaks at (5) 177.05/0.81 ppm, characteristic of the coupling of α-methyl protons with carbon carbonyle in MrMB triad, (6) 177.05/1.81 ppm assigned to the correlation of methine proton of triad MMB with MrMB of the carbonyl carbon. (7) 174.90/2.19 ppm, assigned to BBM triads of CH. The three crosspeaks (8) 174.64/1.33, (9) 174.42/1.85 and (10) 174.42/1.36 correspond to BBmBB tetrads. As depicted in Fig. 6, by comparing between HMBC spectra of Batch 75/25, at 45 min, 300 and 570 min, we notice the appearance of crosspeaks (8), (9) and (10), reflecting the enrichments of chains in BBmBB tetrads. Fig. 7 shows results obtained for Semibatch-2.2. After 60 min, only crosspeaks characteristic of polybutyl acrylate are observed, since it was firstly homopolymerized. At 300 min, we notice the formation of MrMB and MrMM triads. At 720 min, the cross-peaks (2), (3) and (4), characteristics of the tetrads MrMmMrM are observed, confirming the enrichment of polymeric chains in MMA units.
 | ||
| Fig. 6 2D HMBC NMR spectra for Batch75/25 in CDCl3 (as function of time, apparent normalized chain length). | ||
 | ||
| Fig. 7 2D HMBC NMR spectra for Semibatch-2.2 in CDCl3 (as function of time, apparent normalized chain length). | ||
The four poly(BA-co-MMA) copolymers were analyzed by differential scanning calorimetry to follow the glass transition temperature (Tg) of the copolymer as a function of the apparent normalized chain length (see Fig. 8) in order to compare with the gradient profile. Indeed, many studies have reported that depending on the nature of monomers and synthesis conditions, gradient copolymers may present different thermal behaviours.25,28,38,54–56 Torkelson et al.25 reported the presence of one broad Tg in poly(styrene-grad-4-hydroxystyrene), the temperature transition width being as large as 65–85 °C. A microphase separation was highlighted in other gradient copolymers by the presence of two glass transition temperatures, as reported by Karaky et al.28,29 in the poly(N,N-dimethylacrylamide-grad- butyl acrylate) gradient copolymer.
 | ||
| Fig. 8 Evolution of the glass transition temperatures (Tg) as a function of apparent normalized chain length in synthesized poly(BA-co-MMA) copolymers: (◆) Batch 50/50, (●) Batch 75/25, (□) Semibatch-2.2 and (△) Semibatch-4.3. | ||
As depicted in Fig. 8, we noticed that the two batch copolymers exhibit a unique Tg intermediate between those of two PBA and PMMA homopolymers (Tg, PBA ≈ −54 °C; Tg,PMMA ≈ 100 °C for DP > 100). The characteristics of the final copolymers are reported in Table 2. For both Batch 75/25 and Batch 50/50 experiments, the value of the Tg remains constant with the increase of the apparent normalized chain length despite the enrichment in BA highlighted in Fig. 4 and 5. Similar results were reported by Madruga et al. when studying poly(BA-co-MMA) spontaneous gradient copolymers obtained by ATRP.38 It has been reported that Tg values remained constant with the increase of monomer conversion. This was explained by the presence of two opposite factors which can simultaneously affect the glass transition temperature values. The first one is the continuous increase in molar mass with conversion inducing an increase in Tg of copolymers, while the second one is a composition variation with enrichment in BA causing a decrease in Tg. These two effects compensate each other, and therefore the Tg values appear constant. The same explanation was also reported for poly(allyl methacrylate-g-butyl acrylate) copolymers56 and remains valid in the case of our synthesized Batch 50/50 and Batch 75/25 copolymers. In contrast, the Semibatch-2.2 copolymer exhibits a large Tg equal to −3.2 °C and a second weak transition at 82 °C (see Fig. S5 in ESI†). In the case of the Semibatch-4.3 copolymer, a first Tg equal to −0.6 °C was observed together with a second very weak transition at 56 °C (see Fig. S5 in the ESI†).
| Experiment | Overall monomer conversion (%) | BA molar fraction in the copolymer (%) | M n (g mol−1) | M w/Mn | T g a(°C) | T g th b(°C) | T α c(°C) |
|---|---|---|---|---|---|---|---|
| a Experimental Tg measured by DSC. b Theoretical Tg calculated from eqn (5). c Experimental Tg measured by rheology. | |||||||
| Batch50/50 | 70 | 37 | 48600 | 1.24 | 22 | 20 | 31.6 |
| Batch75/25 | 90 | 73 | 55800 | 1.33 | −22 | −25 | n.a. |
| Semibatch-2.2 | 91 | 56 | 61900 | 1.32 | −3.2 (82) | −6.2 | 1.6 |
| Semibatch-4.3 | 88 | 46 | 55100 | 1.29 | −0.6 (56) | 6.8 | 24.5 |
In addition, the increase of Tg values with the apparent normalized chain length confirms the gradient profile of the both Semibatch copolymers (see Fig. 8). The increase of Tg values confirmed the incorporation of MMA along the copolymer chains and the production of forced poly(BA-grad-MMA) gradient copolymers. The two weak glass transition temperatures at 82 °C and 56 °C only observed in final products are not reported in this figure. Table 2 gathers the experimental Tg values obtained for the final copolymers and measured by DSC and rheology. The difference between the two Tg values is a classical feature.57 In the same table, we collected the theoretical Tg values determined by the Fox equation (see eqn (5) which is commonly applied to predict glass transition temperatures, especially in statistical copolymers.
 | (5) |
We have reported the thermomechanical analysis of Batch 50/50, Semibatch-2.2 and Semibatch-4.3 in the Fig. 9. The three gradient copolymers exhibit similar global behaviour particularly at higher temperatures. The glass-transition temperature is clearly affected by the composition as reported in Table 1. Each sample exhibits a narrow rubbery zone in the intermediate temperatures domain (tanδ < 1) just after the transition region. This particularity is certainly due to the molar mass of PBA domains which present a limit for entanglements (MwPBA part ≈ 2 × Me).58 Finally, all samples exhibit a terminal domain at relatively low temperatures (from 55 °C to 75 °C lower than Tg of PMMA) in accordance with non-segregation observed by AFM investigation. This last point is still underway and will have to be evaluated by a spectromechanical analysis which is relevant to explore the segregation of block copolymer.59
 | ||
| Fig. 9 Temperature dependence of the storage modulus (G′ – left axis) and tan δ (right axis) for Batch 50/50 (37% mol BA with Mn = 48 600 g mol−1, Ip = 1.24, tan δ = 32 °C), Semibatch-2.2 (56% mol BA with Mn = 61 900 g mol−1, Ip = 1.32, tan δ = 12.3 °C) and Semibatch-4.3 (46% mol BA with Mn = 55 100 g mol−1, Ip = 1.29, tan δ = 24.5 °C). | ||
Conclusion
In this work, copolymers of poly(butyl acrylate-grad-methyl methacrylate) were synthesized by nitroxide mediated polymerization, applying both the batch and the semi batch processes. The obtained materials were characterized by H-NMR and SEC analysis. It has been found that criteria for controlled radical polymerization were verified, with controlled molecular weight and narrow dispersities. The gradient profiles of synthesized materials were verified by the study of variation of instantaneous fractions of butyl acrylateversus apparent normalized chain length in copolymers. On the other hand, 13C-NMR spectroscopy and hetero nuclear multiple bond correlation experiments (HMBC) were also applied to study the microstructure of synthesized copolymers and the distribution of BA, MMA units along polymeric chains. The evolution of glass transition temperatures with total conversion of monomers during polymerization were determined using the DSC technique, and permitted us to confirm the gradient profile of copolymers. In addition, thermomechanical analysis of these materials demonstrates that the physical properties depend on the synthesis method which has an effect on the final composition of gradient copolymer and corresponding molecular weight.Acknowledgements
NC thanks the Algerian Ministry of high education and scientific research (MESRS-PNE program) for funding. The authors are grateful to Arkema for providing the SG1 nitroxide and BlocBuilder® alkoxyamine. The authors would like to thank G. Clisson for his help with SEC.References and notes
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- J. Qiu, B. Charleux and K. Matyjaszeski, Prog. Polym. Sci., 2001, 26, 2083 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszeski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- J. C. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- B. Charleux and J. Nicolas, Polymer, 2007, 48, 5813 CrossRef CAS.
- D. Greszta and K. Matyjaszewski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1996, 36, 569 Search PubMed.
- T. Pakula and K. Matyjaszewski, Macromol. Theory Simul., 1996, 5, 987 CrossRef CAS.
- D. Greszta and K. Matyjaszewski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1996, 37(2), 569 CAS.
- M. J. Ziegler and K. Matyjaszewski, Macromolecules, 2001, 34, 415 CrossRef CAS.
- S. C. Hong, J. F. Lutz, Y. Inoue, C. Strissel, O. Nuyken and K. Matyjaszewski, Macromolecules, 2003, 36, 1075 CrossRef CAS.
- S. Qin, J. Saget, J. Pyun, S. Jia, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2003, 36, 8969 CrossRef.
- H. Lee, K. Matyjaszewski, S. Yu and S. S. Sheiko, Macromolecules, 2005, 38, 8264 Search PubMed.
- D. Neugebauer, M. Theis, T. Pakula, G. Wegner and K. Matyjaszewski, Macromolecules, 2006, 39, 584 Search PubMed.
- H. G. Börner, D. Duran, K. Matyjaszewski, M. Silva and S. S. Sheiko, Macromolecules, 2002, 35, 3387 CrossRef.
- D. Greszta, K. Matyjaszewski and T. Pakula, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1997, 38, 709 CAS.
- H. Shinoda, K. Matyjaszewski, L. Okrasa, M. Mierzwa and T. Pakula, Macromolecules, 2003, 36, 4772 CrossRef CAS.
- X. Sun, Y. Luo, R. Wang, B. Li, B. Liu and S. Zhu, Macromolecules, 2007, 40, 849 Search PubMed.
- Z. Hu and Z. Zhang, Macromolecules, 2006, 39, 1384 Search PubMed.
- E. Mignard, T. Leblanc, D. Bertin, O. Guerret and W. F. Reed, Macromolecules, 2004, 37, 966 CrossRef CAS.
- M. K. Gray, H. Zhou, S. T. Nguyen and J. M. Torkelson, Macromolecules, 2004, 37, 5586 CrossRef CAS.
- J. Kim, M. M. Mok, R. W. Sandoval, D. J. Woo and J. M. Torkelson, Macromolecules, 2006, 39, 6152 Search PubMed.
- N. Trang, T. Phan, S. Maiez-Tribut, J. Pascault, A. Bonnet, P. Gerard, O. Guerret and D. Bertin, Macromolecules, 2007, 40, 4516 Search PubMed.
- K. Karaky, G. Clisson, G. Reiter and L. Billon, Macromol. Chem. Phys., 2008, 209, 715 Search PubMed.
- K. Karaky, C. Derail, G. Reiter and L. Billon, Macromol. Symp., 2008, 267, 31 Search PubMed.
- K. Karaky, C. Pouchan, J. Desbrières and L. Billon, Macromolecules, 2007, 40, 458 CrossRef CAS.
- K. Karaky, E. Pere, C. Pouchan, H. Garay, A. Khoukh, J. Desbrières, J. Francois and L. Billon, New J. Chem., 2006, 30, 698 RSC.
- K. Karaky, E. Pere, C. Pouchan, J. Desbrières, C. Derail and L. Billon, Soft Matter, 2006, 2, 770 RSC.
- P. Bujak, N. Henzel and M. Matlengiewicz, Int. J. Polym. Anal. Charact., 2008, 13, 149 Search PubMed.
- E. L. Madruga and M. Fernández-García, Macromol. Chem. Phys., 1996, 197, 3743 Search PubMed.
- X. Zhang, J. Gao, Y. Luo and B. Li, Acta Polym. Sin., 2008, 1, 83 Search PubMed.
- S. V. Arehart and K. Matyjaszewski, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1999, 40, 458 Search PubMed.
- J. L. de la Fuente, M. Fernández-García, M. Fernández-Sanz and E. L. Madruga, Macromolecules, 2001, 34, 5833 Search PubMed.
- M. J. Ziegler and K. Matyjaszewski, Macromolecules, 2001, 34, 415 CrossRef CAS.
- M. Fernández-García, J. L. De La Fuente, M. Fernández-Sanz and E. L. Madruga, Macromol. Rapid Commun., 2001, 22, 1046 Search PubMed.
- A. Nese, J. Mosnáček, A. Juhari, J. A. Yoon, K. Koynov, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2010, 43, 1227 Search PubMed.
- G. Sebastian Roos, Axel H. E. Müller and K. Matyjaszewski, Macromolecules, 1999, 32, 8331 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191 CrossRef CAS.
- G. S. Ananchenko, M. Souaille, H. Fischer, C. Le Mercier and P. Tordo, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3264 CrossRef CAS.
- P. Lacroix-Desmazes, J. F. Lutz, F. Chauvin, R. Severac and B. Boutevin, Macromolecules, 2001, 34, 8866 CrossRef CAS.
- The numerical values used for plotting eqn (3) are as follows: rMMA = 2.19, rBA = 0.39 (ref. 40), kp, MMA (115 °C) = 2575 L mol−1s−1 (ref. 42), kp, BA (115 °C) = 82 245 L mol−1s−1 (ref. 42), KMMA (100 °C) = 1.30 × 10−7 mol−1 L−1 (ref. 43), KBA (112 °C) = 4.34 × 10−11 mol−1 L−1 (ref. 44).
- K. Matyjaszewski, M. J. Ziegler, S. V. Arehart, D. Greszta and T. Pakula, J. Phys. Org. Chem., 2000, 13, 775 CrossRef.
- M. Yu. Zaremski, D. I. Kalugin and V. B. Golubev, J. Polym. Sci., 2009, 51, 103 Search PubMed.
- M. Kryszewski, Polym. Adv. Technol., 1998, 9, 244 Search PubMed.
- A. M. Aerdts, A. L. German and G. P. M. van der Velden, Magn. Reson. Chem., 1994, 32, S80 Search PubMed.
- A. S. Brar, G. Singh and R. Shankar, J. Mol. Struct., 2004, 703, 69 Search PubMed.
- A. S. Brar and Puneeta, J. Appl. Polym. Sci., 2006, 99, 2694 Search PubMed.
- A. S. Brar, S. Hooda and R. Kumar, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 313 Search PubMed.
- A. S. Brar and S. Kaur, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1100 Search PubMed.
- C. L. H. Wong, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2842 CrossRef CAS.
- M. M. Mok, J. Kim and J. M. Torkelson, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 48 CrossRef CAS.
- R. Paris and J.l. de la Fuente, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1845 Search PubMed.
- G. Marin, Ph. Vandermaesen and J. Momornicki, J. Adhes., 1991, 35, 23 Search PubMed.
- N. Jullian, F. Léonardi, B. Grassl, J. Peyrelasse and C. Derail, Appl. Rheol., 2010, 20(3), 33685 Search PubMed.
- N. Jullian, L. Rubatat, J. Peyrelasse, P. Gerard and C. Derail, J. Rheol., 2011, 55(2), 379 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00066g |
| This journal is © The Royal Society of Chemistry 2011 |