Polymeric vesicles in biomedical applications†
René P.
Brinkhuis
,
Floris P. J. T.
Rutjes
and
Jan C. M.
van Hest
*
Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. E-mail: j.vanhest@science.ru.nl; Fax: +31243653393; Tel: +31 243653204
First published on 5th May 2011
Abstract
Polymeric vesicles, or polymersomes, are nano- to micrometre sized polymeric capsules with a bilayered membrane. Applications of these vesicles are foreseen in nanomedicine, in vivo imaging and drug delivery. These applications put many restrictions on the choice of polymer, the size and the surface of the vesicle. In this respect much can be learned and translated to polymersome science from lines of research with a longer history of practical knowledge such as liposomal formulation and polymer drug conjugation. The dimensions of a vesicle, such as size and shape can be controlled for polymersomes and will influence the in vivo circulation time. The surface can be adjusted to induce stealth character, or chemically modified to introduce targeting moieties. And last but not least the choice of block copolymers—the building blocks of a polymersome—can introduce features like biocompatibility, inherent or induced permeability and triggered release. In this review we will discuss the recent advances in polymersome science with regard to biomedical applications and will specifically address the abovementioned features which affect their biological behaviour.
 René P. Brinkhuis | René Brinkhuis (1981) received his M Sc in chemistry in 2008, at the Radboud University Nijmegen (NL). As part of the M Sc study he did research internships in the department of Solid State Chemistry of Prof. Elias Vlieg as well as the department of Bio-Organic Chemistry, both at the Radboud University Nijmegen. In 2008 he started as a PhD student in a combined project in the groups of Prof. Jan van Hest and Prof. Floris Rutjes, where he is currently working on polymersome-mediated smart drug delivery over the blood brain barrier. |
 Floris P. J. T. Rutjes | Prof. Floris Rutjes (1966), PhD at the University of Amsterdam in 1993, postdoc with Prof. K. C. Nicolaou (Scripps, La Jolla, USA), was appointed full professor in synthetic organic chemistry at Radboud University Nijmegen in 1999. His research interests comprise the application of catalysis in the synthesis of bioactive molecules, the development of molecular diagnostic tools for life science applications, and synthesis in microreactors. He has received several awards including the Gold Medal of the Royal Netherlands Chemical Society (KNCV, 2002), the AstraZeneca award for research in organic chemistry (2003) and the title of ‘Most entrepreneurial scientist of the Netherlands’ (2008). |
 Jan C. M. van Hest | Prof. Dr Ir. Jan C.M. van Hest received his M Sc in polymer chemistry in 1991, at the Eindhoven University of Technology, and his doctorate in 1996 under supervision of Prof. Dr E. W. (Bert) Meijer. As a post-doctoral researcher he investigated the incorporation of artificial amino acids in proteins using protein engineering under supervision of Prof. Dr D. A. Tirrell, at the university of Massachusetts in Amherst. He then joined the chemical company DSM, where he worked as research scientist and as group leader on the development of innovative material concepts. In 2000 he was appointed as a full professor of bio-organic chemistry at the Radboud University Nijmegen. His current interests focus on translating natural structural concepts into bio-inspired materials with the aid of state of the art organic and polymeric chemistry techniques and protein engineering. |
1. Introduction
Polymeric vesicles, or polymersomes, are nano- to micrometre-sized polymeric capsules with a bilayered membrane which is comprised of amphiphilic block copolymers. Although the aggregation behaviour of amphiphilic polymers was noticed before,1–4 the first systematic study after the formation of polymersomes was reported by Discher et al.5 In this paper the name polymersomes was coined, in analogy to liposomes. When the bilayer membrane architecture is regarded, the similarity between polymersomes and liposomes is obvious in the sense that both species are composed of a bilayer of amphiphiles enclosing an aqueous compartment. There are however also major differences. The building blocks of liposomes are in most cases naturally occurring phospholipids with a molecular weight well below 1 kDa; a polymersome is constructed of amphiphilic block copolymers with a molecular weight up to 100 kDa. This higher molecular weight of the building blocks manifests itself in a tougher, less permeable and less fluidic membrane and as a result superior physical and chemical stability are obtained. With regard to the use of high molecular weight building blocks and the resulting robustness of the capsule structure one can argue that polymersomes resemble to a certain extent viral capsids6 and therefore can be an interesting candidate for in vivo and cellular delivery.Amphiphilic block copolymers have not only been found to form vesicular structures; in fact, the formation of a bilayered vesicular structure puts quite some restrictions on the overall block copolymer composition. Depending on the ratio between the hydrophobic and hydrophilic part of the polymer, spherical micelles, rods and vesicles have been found to form spontaneously.7 Which morphology is found is dependent on the geometry of the amphiphile, which can vary between cylindrical and conical depending on the ratio of the hydrophobic and hydrophilic segments. For small molecular weight amphiphiles this geometry is captured in the dimensionless packing parameter8,9 as is depicted in Fig. 1, which can to some extent also be translated to polymeric amphiphiles. More intuitively, however, the hydrophilic fraction (f) is better suited to predict the expected morphology.6,7Block copolymers with a hydrophilic fraction of more than 45–50 percent will mostly yield micelles. In the region where f is around 35 ± 10 percent in many cases polymersomes are observed and in case the hydrophilic fraction is less than 25 percent, inverted structures can be expected. Finally, there is a small region where worm-like micelles have been reported, when the hydrophilic fraction is around 50 percent. Although this basic empirical rule holds quite well in most cases, the exact aggregation behaviour can depend strongly on the type of block copolymer and the conditions applied.
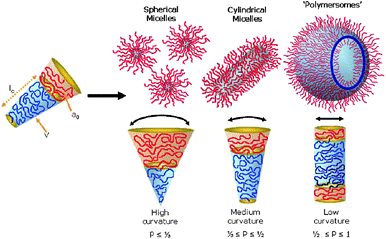 | ||
| Fig. 1 Several structures formed by the self-assembly of amphiphilic block copolymers as determined by the geometry of the amphiphile. The geometry is captured by the dimensionless packing parameter p = v/(a0lc).9 Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
Polymersomes have been prepared in a variety of different ways. Among them two methods seem to be favoured. First of all the “solvent switch” method is broadly applied.10,11 The block copolymer is dissolved in an organic solvent which is a good solvent for both blocks. The organic solvent is diluted with or injected in a non-solvent of one of the blocks, mostly water or a buffered solution, until the desired aggregates are formed. Residual organic solvents are removed by dialysis or are allowed to evaporate. One of the major drawbacks is the need of an organic solvent which is not always compatible with applications in which bioactive compounds have to be encapsulated. Furthermore, traces of the organic solvents tend to stay behind which is not desirable for in vivo applications. The second general method is a technique based on rehydration.11 A polymer film is cast on a substrate and an aqueous solution is used to rehydrate the polymer to form vesicles. Furthermore, methods like electroformation, bulk rehydration and rehydration from pre-treated substrates have been applied. The choice of the method of formation has a big influence on the properties such as the size of the obtained vesicles.
Vesicles are thought to form in a two-step procedure. In an early stage of formation a flat bilayer is formed from micelles or polymer clusters, as depicted in Fig. 2a and b. Thereafter under the influence of a curvature change, and therefore a change in the packing parameter, this sheet will form a closed structure or a vesicle.12,13 This mechanism is depicted in the cartoon presented in Fig. 3. This sequence of events leads to statistical encapsulation of compounds dissolved in the water layer, but it does not provide an explanation for the many fascinating shapes found experimentally.12 More recent simulation studies show the possibility of an alternative, more complex, mechanism in which vesicles do not evolve from the closing of a bilayer membrane, but evolve from micelles that grow and change morphology.13,14 It should be noted that for the second pathway the loading efficiency of hydrophilic compounds would be lower compared to the former one since there is no closure, and hence encapsulation step involved. Experimentally, polymersome formation along path I is supported by Du and Chen15 (Fig. 2). They were able to trap and visualize transition states like micelles and lamellae that eventually formed polymersomes by systematically adjusting the solvent polarity and locking the structure chemically prior to transmission electron microscope (TEM) imaging. Experimental evidence for the second route was found by Adams et al.16 They showed how indeed poor loading efficiencies for hydrophilic compounds were found. These two seemingly contradictive experimental results show that it is possible that more mechanisms of formation do exist, depending on the exact conditions applied.13
 | ||
| Fig. 2 Several stages of polymersome formation captured by Du and coworkers15 as a function of water/organic solvent ratio. First spheres and rods are formed that transform into lamella which close to form polymersomes due to an increase in water content. | ||
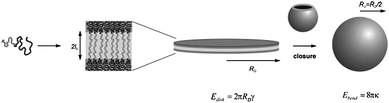 | ||
| Fig. 3 Cartoon of polymersome formation along path I as described by Antonietti et al.12 First spheres that transform into sheets are formed which close in a second step to form polymersomes. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
The field of polymersome research has been reviewed extensively in the past6,9,11,17–24 and often the high potential of polymersomes for biomedical applications such as smart drug delivery systems, in vivo imaging vehicles or artificial organelles is mentioned. Many of these applications involve cell specific interactions and/or require non-toxic and tuneable in vivo behaviour, and these criteria put quite some restrictions on e.g. the choice of polymer and the polymersome dimensions. In this review we address specifically the factors that affect the use of polymersomes in biomedical applications. In the next sections we will focus on which components are suitable for the construction of polymeric vesicles for this specific field of application. We will furthermore address the importance of size, topology and tuneable stability and permeability. Finally, a selection of functionalization methods will be discussed. In addition we will illustrate these topics with recent research and try to extrapolate facts from liposomal research and apply them on polymersomes.
2. Block copolymers
The starting point of formation for any polymersome is the amphiphilic diblock or triblock copolymers that are the building blocks of the membrane. More than a decade of research into polymeric vesicles not only yielded some understanding underlying the formation of block copolymer nanostructures, but also allowed researchers to use their full creativity in the synthesis of new amphiphilic polymers. This resulted in an extensive library of block copolymers known today to form aggregates. For a comprehensive overview of polymers used for polymersome formation, one is referred to some recent reviews on biohybrid amphiphiles23 and synthetic amphiphilic block copolymers.9,25 This section will focus on the most common types of polymers used in biomedical polymersome research.For biomedical applications it is obligatory to use biodegradable or at least biocompatible polymers as building blocks. The field of polymer drug conjugates has already a long history of practical knowledge on suitable polymers that are in clinical use/evaluation nowadays.26,27 These polymers include synthetic polymers such as poly(ethylene glycol) (PEG), poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA), poly(vinyl pyrrolidone) (PVP) and poly(ethylene-imine) (PEI), but also natural polymers like dextran, dextrin and pseudosynthetic polymers such as poly-L-glutamic acid (PGA) and poly(L-lysine) are commonly used. The translation from polymer drug conjugates to polymersome-forming amphiphilic block copolymers is not always straightforward, but many examples can be found in literature in which biocompatible and bio-inspired block copolymers have been employed to form polymeric vesicles.
2.1 Synthetic polymers
The first well-studied polymersome-forming system was based on the biocompatible block copolymer poly(ethylene oxide)-b-poly(ethyl ethylene) (PEO-b-PEE).5 Although the PEE block is highly biocompatible, it is the PEO block that gives these polymersomes useful in vivo characteristics. The water-soluble poly(ethylene oxide) (PEO) or poly(ethylene glycol) (PEG) is a polymer which is generally known for its good biocompatibility and excellent in vivo behaviour. Application of a PEG mantle is used in liposome formulations to strongly reduce in vivo and in vitro non-specific protein adsorption, resulting in stealth behaviour and therefore prolonged circulation times.28,29 PEG has been coupled to many hydrophobic blocks such as the non-degradable polystyrene (PS) and polybutadiene (PBd),30 or biodegradable polymers such as polycaprolactone (PCL) or poly(lactic acid) (PLA).31–33 Each hydrophobic polymer introduces specifically desired characteristics in the polymersome. Polystyrene as an example has a relatively high glass transition temperature (Tg) and will yield after removal of the organic solvent a rigid semi-crystalline membrane. Polybutadiene on the other hand has a Tg well below room temperature which will result in a flexible and fluidic membrane which allows for extrusion and therefore size control after polymersome formation. Polymersomes of polybutadiene-b-polyethylene glycol (PBd-b-PEG) are generally considered as fully biocompatible and are therefore popular in research toward in vivo applications like drug delivery. However, polybutadiene is not biodegradable, at least not on the timescale of days. PCL and PLA on the other hand are biodegradable and biocompatible and are therefore also often encountered as the hydrophobic block.30–33Instead of using the neutral and inert PEG block as a hydrophilic element, also some more functional moieties can be applied. The most straightforward way is to introduce ionic character in a block copolymer. In this way the water-soluble EO block was replaced for polyacrylic acid (PAA) to introduce negatively charged acid residues,34 but also the zwitterionic, phospholipid mimic, poly(2-methacryloyloxy-ethyl phosphorylcholine) (PMPC) has been applied as the hydrophilic block by Armes and co-workers.35–37 They furthermore showed how polymersomes with interesting pH-dependent properties are obtained if both blocks contain zwitter-ionic or ionisable monomers as is the case with vesicles prepared from poly(2-(diisopropylamino)ethyl methacrylate) (PDPA) as the hydrophobic and PMPC35 as the hydrophilic block. The PDPA block is hydrophobic at pH above 6.4 but readily becomes hydrophilic and protonated below this pH. This results in a sharp transition from polymersomes to free dissolved block copolymers upon lowering the pH, which can be used as a drug release mechanism.
The previous example involved an external stimulus (pH change) to release the content from the vesicle, but also amphiphilic block copolymers with inherently leaky properties have been reported. Polymersomes of polystyrene-b-poly3-(isocyano-L-alanyl-aminoethyl) thiophene (PS–PIAT) turned out to be excellent candidates for nanoreactors, since these polymersomes are permeable for small molecules whereas large molecules such as enzymes stay trapped inside.3,38 Although not fully understood, this permeability is probably a result of the helical rodlike secondary structure of the hydrophilic PIAT block, which frustrates the packing of the polymers in a closed bilayer.
These selected examples already show a few of the many possibilities to alter and tune the characteristics of the polymersomes, like robustness, charge and permeability. One crucial property however remains biocompatibility and/or biodegradability. It is therefore a logical step that many researchers have developed amphiphilic block copolymers capable of forming polymersomes which are partly based on naturally occurring building blocks such as amino acids, nucleotides and carbohydrates.
2.2 Bio(hybrid) polymers
Oligo- and polypeptides are highly versatile and biodegradable polymersome building blocks, since their properties can be varied in many different ways. A wide variety of secondary structures can be created based on the primary sequence of amino acids. By selecting the correct amino acid residues stimulus-responsive behaviour is introduced, such as a pH dependent charge and hence solubility.39 One of the first reports on the formation of vesicular structures from peptide hybrids made use of an antibacterial hydrophobic helical peptide, Gramicidin A. By coupling Gramicidin A with PEG an amphiphilic block copolymer was obtained40 which readily formed a variety of aggregated structures in aqueous solution. Among the obtained structures a vesicle-like aggregate was found, which was called a peptosome.2,41 Since then the name peptosome has been more generally used for peptide-containing polymersomes. This early example involved a hydrophobic peptide which was buried under a PEG mantle. However, in most other cases the peptide moiety is the hydrophilic part, forming the periphery of the vesicles. This was for example demonstrated by Dirks et al.42 who coupled a tri-peptide, Gly–Gly–Arg–AMC, via the copper-catalysed azide–alkyne Huisgen [3 + 2] cycloaddition to polystyrene. Even more common than the use of oligopeptides is the application of polypeptides as building blocks in polymersome-forming block copolymers. Peptides consisting of a repetition of the same amino acid, and block copolymers of homopolypeptides can nowadays be obtained in a controlled fashion by the polymerisation of N-carboxyanhydrides (NCA) with a nickel catalyst and an amine initiator.43,44 In this way PBd was used as the amine functional initiator to polymerize for example PBd–poly(L-glutamate)45–48 and PBd–poly(L-lysine)49 which both have been extensively studied with respect to their aggregation behaviour. These examples involve a combination of synthetic polymers and polypeptides, but also block copolymers of which both the hydrophobic and the hydrophilic part are composed of polypeptide hybrids that have been designed to form polymersomes.39,50,51 In two recent examples Kimura et al. showed the usefulness of peptosomes for in vivo cancer imaging.52 They constructed peptosomes out of a block copolymer composed of poly(N-methylglycine) and poly(γ-methyl-L-glutamate) (PMLG) in which a near infrared dye was encapsulated. These peptosomes showed good in vivo circulation times and low recognition by the reticuloendothelial system (RES) (blood clearance mostly by liver and spleen).Another group of naturally occurring building blocks which have recently gained more attention are carbohydrates or polysaccharides.22,53–55 In natural systems saccharides are conjugated to the surface of cellsvia the lipids that make up the cell membrane (glycolipids), and they are also found conjugated to proteins (glycoproteins). These glycoconjugates play an important role in biological processes such as cellular recognition and pathogen infections. In many cases these processes are mediated via interactions between saccharides and receptor proteins, called lectins. In general lectins have only a low affinity for saccharides, so this needs to be compensated by multivalent interactions via the so-called cluster glycoside effect. Application of (poly)saccharides in polymersomes as the hydrophilic part of block copolymers or as the end group of block copolymer amphiphiles allows for the construction of highly functionalised and multivalent vesicle surfaces.
Although quite some research has focused on the synthesis of polysaccharide block copolymers not many examples have been reported that actually form polymersomes.22 An example which also demonstrates specific recognition is a fairly simple rod–coil amphiphile, tetra(p-phenylene)-block-PEG12-α-D-mannopyranoside.56 This molecule formed small polymersomes which specifically interacted with the pili of a specific Escherichia coli bacterial strain as depicted in Fig. 4. When the head group was replaced for galactose, polymersomes were still formed, but without the binding properties. Another example involved a β-cyclodextrin head group coupled to PS which also formed vesicles.57 The hydrophobic interior of β-cyclodextrin was used to attach hydrophobic fluorescent dyes and the adamantane-coupled enzyme horseradish peroxidase (HRP) to the surface.
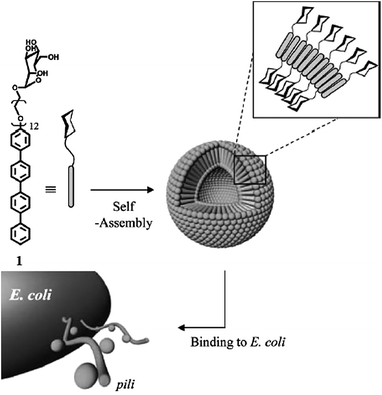 | ||
| Fig. 4 Rod coil amphiphile which forms small vesicles. The mannopyranoside head group induces recognition and binding to the pili of Escherichia coli. Replacing this moiety for galactose had no influence on the aggregate formation, but resulted in loss of recognition.56 | ||
Block copolymers comprising polysaccharides such as dextran-b-poly(benzyl-L-glutamate) (PBLG)53 dextran-b-PS and hyaluronan-b-PBLG58 have also been used for vesicle formation. In these cases polymeric vesicles were obtained via the solvent switch method. Furthermore, dextran-b-PS was found to form both micelles and vesicles, depending on the PS length. The usefulness of these peptide–carbohydrate hybrid polymersomes in drug delivery was nicely demonstrated in a series of recent papers by Lecommandoux et al.59–61 They prepared doxorubicin loaded hyaluronan-b-PBLG polymersomes and showed both in vivo and in vitro (tumor)-targeting by the hyaluronan periphery as well as release and shrinkage of the tumors in animal models. Finally, an elegant first example of a thermoresponsive saccharide-containing block copolymer was reported by Otsuka et al.62 They coupled maltoheptaose (mal7) to poly(N-isopropylacrylamide) (PNIPAM) via click chemistry to obtain polymersomes. They showed how above the lower critical solution temperature (LCST) of PNIPAM polymersomes were spontaneously formed whereas below the LCST the assemblies were dissolved again.
2.3 Polymeric vesicles with non-bilayer structure
There is also a group of vesicle-forming polymers which strictly speaking do not yield a bilayer structure. An interesting example is the self-assembly into vesicles of ABA or ABC triblock copolymers.63 Although the overall composition is similar to a vesicle bilayer: namely two hydrophilic outer blocks separated by a hydrophobic inner domain, the entire structure is a single layered vesicle. By changing the blocks A, B or C one can even direct the orientation of the polymers in the membrane, thereby obtaining asymmetric membranes that display different functional groups on the inside and outside as was elegantly demonstrated by W. Meier and co-workers. Very recently Percec et al.64 reported an in depth investigation into a variety of amphiphilic or Janus dendrimers that formed a variety of aggregates, among them a vesicular structure that was named dendrimersomes. A peptidic-dendron polymersome was also reported to form polymersome-like structures.65 Another interesting system is the so-called polyion polymer complex or PICsome.66–70 They are based on two polymers with opposite charges that self-assemble and phase separate into vesicular structures This is an example of a series of polymers that recognize each other and form recognition-induced aggregates, the so called RIP's.71–73 A final example of vesicles that do not exhibit a bilayered membrane was developed by Caruso et al.74 They showed how enzymes can be entrapped in vesicles that were formed by a layer-by-layer deposition of anionic and cationic polymers.3. Structural factors
For in vivo and biomedical applications of polymersomes it is important to ascertain that a particle circulates for sufficient time to reach the place where it is wanted. This can be achieved by protecting the particle from interactions with the immune system. We have already mentioned examples of polymers that exhibit such stealth-like behaviour which was attributed to the PEG surface of the particles. It should be noted however that although PEGylation is a good start to rule out non-specific interactions, it is by itself not enough to escape clearance by the RES. Although not all factors influencing in vivo circulation times have been fully investigated yet for polymersomes, the analogy between polymersomes and liposomes allows for some extrapolation.From liposome research it is known that factors like stealth-like behaviour, size and shape play an important role.75,76 Therefore this section will review how to control these parameters. Furthermore we will highlight some research where these effects have been observed.
3.1 Stealth-like behaviour
To prolong blood circulation time and prevent recognition by the RES a PEG mantle is applied in liposomal formulations by introducing up to ten percent PEGylated phospholipids. PEG has very useful characteristics in the sense that it blocks (almost all) non-specific protein adsorption and induces no immune response, basically giving a vessel stealth characteristics.28,29 These stealth liposomes have blood circulation half times in the order of 15 hours and will circulate in traceable amounts for many days. Discher and coworkers investigated the in vivo fate of polymersomes formed from PBd–PEG by preparing polymersomes with a fluorescent dye entrapped.77,78 These polymersomes were inherently hundred percent PEGylated. The blood circulation half life was determined by counting fluorescent dots in blood samples derived at different time points. Compared to PEGylated liposomes the circulation half life was found to be even longer and polymersomes could still be found after several days. These results show that PEGylated polymersomes also have stealth-like behaviour. It should be noted that not only PEG has the useful characteristics of reducing interaction with opsonins and preventing clearance by the reticuloendothelial system (RES). Also the zwitterionic polymer PMPC has been recognized to reduce the adsorption of opsonins.79 Other possibilities might be found with pHPMA, and oligosaccharides such as cyclodextrin might be used since these polymers are already under investigation as polymer-conjugated therapeutics.26 Finally in liposomal formulations poly(oxazoline) conjugates also showed prolonged circulation times.80 The non-adsorptive character and hydration shell of the hydrophilic mantle is obligatory to create stealth-like particles, but it is certainly not enough.3.2 Size
Although not fully investigated for polymeric vesicles it is known from liposome science that size has a big influence on blood circulation times, RES recognition, biodistribution and the mechanism of cell uptake.75,81Fig. 5 shows a literature compilation by Harashima et al. on how the circulation time of liposomes is influenced by the size of the liposome. What is not clear from this picture is that there is also a lower limit for prolonged circulation times. The junctions in vascular endothelium of healthy tissue vary depending on the tissue type. In most tissues these openings are below 2–6 nm, so too small for liposomes and polymersomes to enter. Organs and tissues with discontinuous endothelium like the fenestrated endothelium of kidney glomerulus or sinusoidal endothelium of the liver and spleen can have pores of 40–60 and up to 150 nm.82 Particles with a size below these values will therefore be excreted or trapped within the tissue resulting in clearance from the circulation. It is notable that tumors can have discontinuous microvasculature with pore sizes above 100 nm. In fact, this is the basis for non-targeted delivery to tumors since particles that are small enough to enter will accumulate in the tumor. Taking these mechanisms into account the optimum size for circulation in the blood stream is expected to be around 80–150 nm. It might be expected that a similar trend is valid for polymersomes and the polymersome size should be controlled to achieve optimal blood circulation half lives.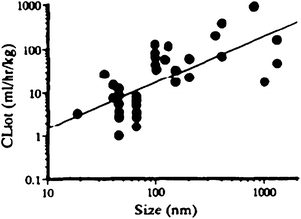 | ||
| Fig. 5 Compilation of literature by Harashima and Kiwada75 on the effect of particle size on the blood clearance of liposomes. | ||
For polymersomes the eventual vesicle size can be influenced at different points in the process of preparation. The first level to exert influence is on the polymer level. For example, it has been shown that there is correlation between amphiphile molecular weight and aggregate size.83 Furthermore, the polydispersity10 of the block copolymer and the ratio between blocks can have an influence on the size distribution.
One stage further, one can influence the size using different methods of vesicle formation.84 Rehydration methods and solvent displacement85 or injection86 methods have been applied and mostly yield 50 to 800 nanometre-sized polymersomes. On the other hand electroformation will yield micrometre-sized giant polymersomes. Howse et al.84 recently showed how they were able to use surface-directed templating with different mesh surfaces to influence the size of PEO–PBO vesicles. They were able to deposit block copolymer domains of predefined size on a grid and showed how rehydration from these grids yielded control over polymersome size as depicted in Fig. 6. Smaller polymer domains yielded smaller polymersomes (5–10 μm) with in all cases small size distributions. The presence of pH-responsive, ionizable groups can also allow control over size during vesicle formation by changing the pH or ionic strength.48 A certain amount of control over polymersome size was shown by Shen and Eisenberg.87 They were able to influence the vesicle size by changing the solvent nature and composition during solvent displacement formation of PS–PAA polymersomes. Luo and Eisenberg85 showed how also binary organic solvent systems can be used to influence the size. Furthermore the presence of additives like dendrimers,88 (homo)polymers10 and surfactants89 have been shown to influence the average polymersome size and polydispersity.10
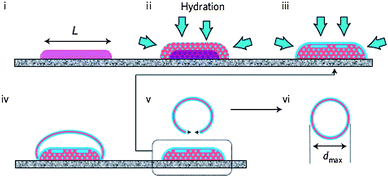 | ||
| Fig. 6 Rehydration of amphiphilic block copolymers from a surface template. The dimensions of the polymer domain (L) dictate the eventual polymersome size (dmax).84 (i) Block copolymer domain of predefined dimensions, (ii) hydration and phase separation, (iii) further hydration and lamellae formation, (iv) expansion, (v) detachment and (vi) energy minimalisation resulting in polymersome formation. | ||
Finally polymersomes can be resized after they have been formed. In this respect the most straightforward way to obtain polymersomes of a predefined size is by extrusion. This basically means that a preformed sample of polymersomes is pressed through a filter with nanopores.78 This procedure has also been proven to be applicable on larger scale by the use of hollow nanofibers.90 The only restriction is that the polymeric membrane should remain fluidic till the end of the procedure, therefore polymers with a low Tg are needed. In the stage after formation it is also possible to exert influence on the size via more sophisticated methods than extrusion. Eisenberg and co-workers reported how membrane fusion and fission driven by a change in dioxane/water content (change of interfacial energy) allowed vesicles to grow and shrink reversibly.85,87
Another example involves the swelling of the membrane by an external stimulus. In this way pH-responsive blocks have been introduced in the corona to trigger size change, but also a change in secondary structure of polypeptides has been employed.45 In a recent example by Yu et al.91 the formation of so-called “breathing vesicles” from PEO–PS–PDEA was demonstrated. By variation of the pH they were able to swell and shrink the polymersome (membrane) with a factor of ∼1.9 changing simultaneously the membrane permeability as depicted in Fig. 7. This approach shows how a well-chosen balance between solubility and hydration will keep the aggregate together but still allows considerable changes. If this balance is not carefully chosen, the polymersome will probably disassemble into its polymeric constituents.
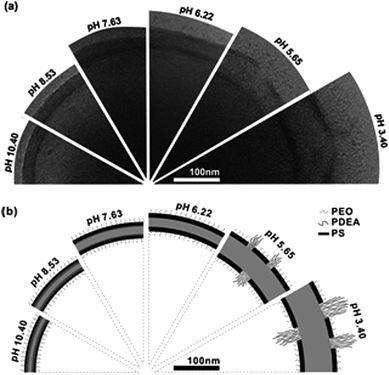 | ||
| Fig. 7 Reversible change of the PEG-b-PS-b-PDEAMA membrane upon pH change. (A) Cryo-TEM images of the vesicle wall structure at several pH values. (B) Schematic illustration of the presumed membrane structure at corresponding pH values.91 | ||
3.3 Shape
The influence of shape and topology on in vivo behaviour is not so well studied for liposomes and polymersomes. In fact, probably the most illustrative example for the influence of shape in amphiphilic assemblies for in vivo behaviour comes from rod like micelles as studied by Geng et al.92 They showed how worm-like micelles, which are nonspherical in nature, have blood circulation times exceeding those of spherical particles like liposomes and polymersomes by far even if the persistence length at the moment of injection is several microns. Micelles or worm-like micelles93–95 do not have an aqueous lumen and therefore do not allow for delivery of hydrophilic compounds, but also worm-like polymersomes have been observed and described96,97 that do enable the encapsulation of hydrophilic compounds, and which should have similar behaviour.These cylindrical shapes are not the only non-spherical shaped vesicles that have been reported. In fact a realm of non-spherical aggregates is possible.12 This has already been demonstrated for giant phospholipid vesicles (10–50 μm) of which it is known that they can transform shape from spherical into a variety of nonspherical shapes upon a change in their environment.98,99 Theoretical studies on triblock copolymers by Liet al.100 showed that this transformation should also be possible for polymersomes as depicted in the phase diagram in Fig. 8.
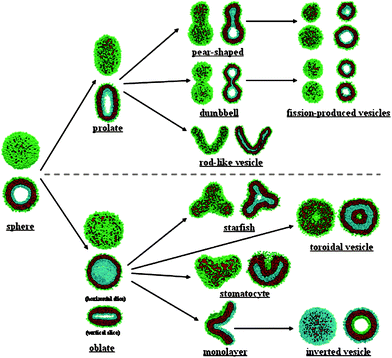 | ||
| Fig. 8 Morphologies possible for triblock copolymers as determined theoretically by Liet al.100 | ||
Recently, experiments have been performed in which non-spherical polymeric assemblies were created chemically15 or via kinetic entrapment.101 It should therefore be possible in the near future to investigate the effect of shapes other than rodlike or spherical on in vivo behaviour.
4. Release and permeabilisation mechanisms
One of the advantages of polymersomes over liposomes is their more robust and therefore less leaky membrane, which is very useful for improved circulation times and the prevention of uncontrolled release of drugs. However, at one point almost each in vivo application involves a disruption step to release the vesicle content or make the content accessible. Despite the fact that polymersomes display improved stability, many successful examples exist of stimuli–responsive vesicles, which release their contents upon action of an external trigger.8,19,23 It is also possible to achieve permeability control, i.e. controlled pore formation, without complete disruption of the vesicular structure. In the next section we will highlight a few recent and illustrative examples.4.1 Triggered disruption
The most straightforward example of a release mechanism is actually degradation of the biodegradable polymers of which the vesicle is composed. Polymersomes of PCL-b-PEG released their cargo due to biodegradation based on (pH driven) hydrolysis of the polymers. During this process a transformation change from polymersomes to rods and eventually micelles was in some cases observed due to the change in hydrophilic fraction (f). By mixing in a stable PBd-b-PEG block copolymer it was possible to tune the speed of degradation.6,30 Besides hydrolysis also disruption based on chemical alteration of the block copolymer by oxidation97 or reduction have been reported,19 pH-triggered release mechanisms which in principle are reversible have also been reported. A nice example of a polymersome that can disassemble upon a pH change was already encountered in Section 2.1. Armes and coworkers used the biocompatible zwitterionic PMPC-b-PDPA block copolymer to form polymersomes at pH above six. By lowering the pH a sharp transition was observed from vesicles to molecularly dissolved polymers due to protonation of the PDPA block. The polymersome content was thereby instantaneously released. The driving force for polymersome formation is in most cases based on phase separation due to the amphiphilic character of the block copolymer. By switching the character of the hydrophobic block to hydrophilic it is possible to dissolve the whole polymersome and release the content. Recently Kim et al.102 reported a system based on this principle. They developed a stimulus-responsive amphiphilic polymer containing boronic acid moieties in the hydrophobic domain that, upon increase of pH and in the presence of monosaccharides such as D-glucose, became soluble in an aqueous environment. Another interesting example was reported by Lecommandoux. They prepared polymersomes of a block copolymer comprising two polypeptides, poly-L-glutamate-b-poly-L-lysine (PGA-b-PLys). By changing the pH they were able to switch the solubility of both blocks, effectively flopping the whole membrane.103 Those examples all involve a change in pH as the trigger, but there are more stimuli explored.Also changes in temperature can be used for altering the hydrophilicity of a block copolymer.19 From literature there are quite some polymers, peptides and proteins which demonstrate LCST behaviour as was mentioned before in section 2.2 with the example by Otsuka et al.62Fig. 9 shows another very versatile example based on P(DEAEMA98-b-NIPAM392), a so-called “schizophrenic” polymer.104,105 This system was shown to actually reversibly dissolve and assemble into polymersomes, as well as transform micelles to polymersomes based on either a pH and/or a temperature trigger.104 Based on a pH-induced conformational change Bellomo et al.39 reported a release mechanism for polymersomes composed of a dipeptide block copolymer which contained a stable hydrophilic α-helix of PEGylated poly-L-lysine and a hydrophobic α-helix of poly(L-leucine0.3-co-L-lysine0.7). Upon lowering the pH the α helix of the hydrophobic block transformed into a random coil due to protonation of the lysine residues. This conformational change was found to disrupt the vesicle, allowing the transport of calcium ions and dyes in and out of the polymersome.
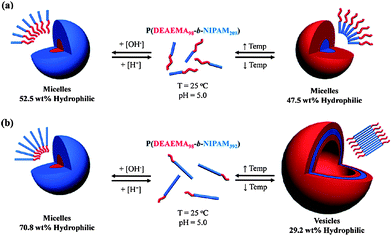 | ||
| Fig. 9 Mechanism of aggregation and dissolution of schizophrenic vesicles as described by Smith et al.104 | ||
Researchers also have developed methods of disruption, based on external stimuli such as ultrasound and light. An interesting system was reported by Mabrouk et al.106 as depicted in Fig. 10. They described how the incorporation of azobenzene moieties allowed for a conformational change in the membrane upon illumination with UV light. Their polymersomes were disrupted within a second. Fig. 11 shows another example of a light-driven vesicle shape transformation and release. Robbins et al.107 reported how polymersomes formed from PBd-b-PEG with porphyrins entrapped in the membrane and proteins in the lumen disrupted in response to light. When either the protein or the porphyrin was left out this effect was negligible, indicating a synergy between the fluorophore and the encapsulated enzyme.
 | ||
| Fig. 10 Polymersome disruption by illuminating with UV light. A fraction of the polymeric building blocks is functionalized with azobenzene moieties. Illumination with light induced a conformational change, disrupting the polymersome and releasing its content.106 | ||
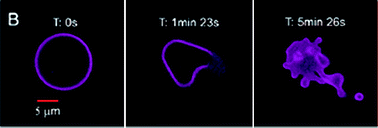 | ||
| Fig. 11 Polymersomes loaded with enzymes in the lumen and porphyrins in the membrane respond to light by a morphology change and eventually release of content.107 | ||
4.2 Controlled permeability
For some applications a complete disruption of the vesicle is not desired. Rather, the polymersome should be semi-permeable. This is for example the case in enzyme therapeutics or artificial organelles, in which enzymes are encapsulated in the vesicle. The polymersome functions as a protective cage, i.e. keeping the enzymes in and allowing substrate and products to diffuse in and out of the polymersome. Different strategies have been reported on how to create in a controlled fashion these semi-permeable or leaky polymersomes.An example of an inherently leaky polymersome is based on the earlier mentioned PS-b-PIAT block copolymer system. These polymersomes have been used as biocatalytic reactors, by encapsulating enzymes in the lumen. It was shown that substrate diffused through pores in the membrane, and the encapsulated enzyme was not able to escape, basically making it a nanoreactor with a semi-permeable membrane.38 This line of research was developed further by creating a nanoreactor108 capable of performing a three-enzyme cascade reaction. The cascade scheme is depicted in Fig. 12 and shows how one enzyme is entrapped in the lumen, a second is captured in the hydrophobic membrane and finally a third enzyme is coupled covalently to the surface via clickable anchors. Another system that has been reported by Kataoka to assemble in a semi-permeable membrane is based on oppositely charged block copolypeptides which yield PICsomes.70 These PICsomes have also been used as biocatalytic nanoreactors. Although these two examples show the usefulness of semi-permeable vesicles, in both cases the permeability was not tuneable.
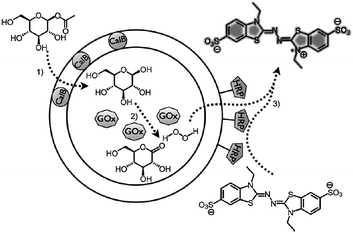 | ||
| Fig. 12 Polymersome nano-reactor performing an three enzyme cascade reaction. All three enzymes are associated with the PS–PIAT vesicle and leaving out one will stop the whole cascade.108 Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
Meier and co-workers followed a bio-inspired approach for the construction of semi-permeable vesicles.109 They nicely showed that bacterial transmembrane channel proteins OmpF and aquaporin110,111 could be successfully reconstituted into the membrane of polymersomes composed of the triblock copolymer poly(2-methyl oxazoline)-b-poly(dimethyl siloxane)-b-poly(2-methyl oxazoline) (PMOXA–PDMS–PMOXA), which made these vesicles selectively porous. In nature many transmembrane pores have a directed orientation, but upon reconstitution into liposomes or polymersomes they tend to adopt a random orientation, basically blocking out fifty percent of the channels. By using an asymmetric ABC type of triblock copolymer they were able to form vesicles with either the A or the C block on the outside,111 effectively constructing an asymmetric membrane as depicted in Fig. 13. They reconstituted aquaporin 0 in these membranes and were able to tune the direction insertion. An elegant approach towards the construction of semi-permeable polymersomes112 was the reconstitution of LamB protein in PDMS-b-PMOXA polymersomes. This protein was recognized by bacteriophage lambda and used to dock the phage, after which it injected its DNA into the polymersome, as nicely captured by TEM imaging in Fig. 14.
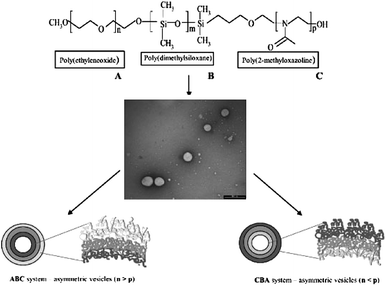 | ||
| Fig. 13 Asymmetric polymersome membranes obtained from ABC triblock copolymers allow for the directed reconstitution of transmembrane channels.111 By changing the molecular weight of the A and C block either the A or the C block is directed outwards. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
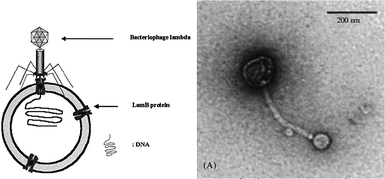 | ||
| Fig. 14 Schematic and TEM image of a polymersome in which membrane protein LambB is reconstituted.112 This membrane protein is recognized by bacteriophage lambda which is shown to dock on the polymersome to inject its RNA. | ||
In another example Choi and Montemagno113 showed that it is also possible to reconstitute two different proteins, bacteriorhodopsin and F1-ATP-ase, in a polymeric vesicle membrane, as depicted in Fig. 15. ATP-ase can be regarded as a proton gradient driven motor that, while rotating, converts ADP into ATP. As noted ATP-ase needs a proton gradient to work. This gradient is supplied by bacteriorhodopsin, which is activated by a light source. Choi and coworkers nicely showed how the enzymes worked together. If no light was applied there was no production of ATP. This research is one of the first systems reported where a complex biocatalytic process is performed in a polymersome nanoreactor.
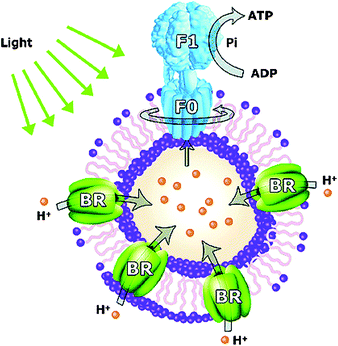 | ||
| Fig. 15 Light driven nanoreactor. Under the influence of light bacteriorhodopsin (BR) establishes a proton gradient, which is used by ATP-ase to produce ATP from ADP.113 | ||
Only few examples of synthetic approaches towards controlled semi-permeable polymersomes have been reported. One of the first of these approaches was reported by Tsourkas and co-workers.114 They designed polymersomes based on PBd-b-PEG and mixed in phospholipids, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-choline (POPC). After they crosslinked the PBd membrane POPC was extracted leaving a porous vesicle. The same method was applied by exchanging POPC for the biodegradable block copolymer poly(caprolactone)–PEG. After crosslinking and hydrolysis a highly permeable polymersome was obtained.115 Recently, another fully synthetic approach to controlled permeable polymersomes was reported by Kim et al.116 They used the boronic acid based block copolymers described in Section 4.1 and mixed it with PS–PEG, which forms a semi-crystalline membrane. Upon washing away the boronic acid polymer by applying sugar and base they were able to form pores in the membrane of which the size was tuneable by the ratio of boronic acid block copolymer and PS–PEG. This procedure could be used in combination with the encapsulation of CalB, allowing the substrate to pass the membrane while keeping CalB enclosed. They also showed how bigger pores allowed faster diffusion of substrate, as determined by the reaction rate.
5. Surface functionality
PEGylated liposomes are neutral and are considered to be long circulating vesicles, with circulation times up to days. When compared to natural systems there is however room for improvement. Red blood cells for example circulate up till 100 days. Their cell surface has an overall negative charge due to the external glycocalyx moieties. For liposomes the effect of surface charge has therefore been studied, but the general trend is not so obvious and seems to be influenced by the size of the liposome, especially in rat and mice studies.75 In rabbits a clear trend was observed which was in favour of neutral PEGylated liposomes (t1/2 = 19.3 h), whereas negative and PEGylated negatively charged liposomes showed circulation half times of only 9.6 and 16.5 hours respectively.117 Charge is only one of the possible functionalities which affect the interaction of polymersomes with biological systems. Another aspect is surface topology. Research on both will be discussed in the next sections.5.1 Surface topology
Christian et al.118 recently reported on the synthesis and in vivo evaluation of polymersomes with a red blood cell like surface (i.e. negatively charged surface). They prepared polymersomes of PBd-b-PEG with a near infrared dye entrapped, and compared them with polymersomes where a fraction of PBd-b-PAA was introduced to obtain a negative surface charge. After 24 hours they showed how polymersomes with negative surfaces predominantly accumulated in the liver. Liposomes often are covered with up to ten percent of PEG chains that induce stealth character. In general it is thought that the conformation of the surface PEG chains has an influence on the stealth effect. PEG chains can either adopt a mushroom conformation to cover the full surface or a fully stretched conformation depending on the available space. Smart et al.119 studied these conformations for polymersomes both theoretically and experimentally. For polymersomes it is not so straightforward to reduce the number of PEG chains in the corona. Often PEG is a part of the block copolymer, covalently linked to the hydrophobic block and it is needed to form and stabilize the polymersome in solution. By the introduction of a hydrazone bond between PS and PEG (PS–Hz–PEG) He et al.120 showed how polymersomes constructed of this polymer were able to shed off their PEG mantle by adjusting the pH. Brinkhuis et al.121 used this concept on PBd–Hz–PEG vesicles and systematically mixed in a non-hydrolysable analogue. They showed that after vesicle formation the number of PEG chains could be reduced to five percent without losing the colloidal stability. This is already an example where the surface structure is adapted, but more complex surface topologies have also been reported, involving domain formation.In liposomes it is possible to create surface domains via micro-phase separation either assisted or not assisted by the addition of ions.122 This domain formation was recently also described in polymersomes.123 The effect of more complex surface topologies on cell interactions was shown by Battaglia et al.124 They were able to create domains within the polymersome surface by mixing different ratios of two triblock copolymers based on PMPC. They nicely showed how the ratio of polymers influenced the size and number of domains present in their polymersomes as depicted in Fig. 16. Furthermore, this figure indicates how the cellular uptake is influenced by the surface topology and size of these patchy polymersomes, pointing out the importance of surface topology.
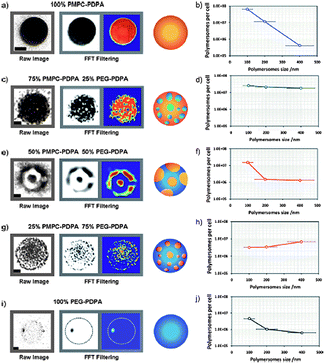 | ||
| Fig. 16 (a) Effect of the ratio of PMPC–PDPA and PEG–PDPA on the domain size of patchy polymersomes. (b) The number and size of domains influence the cell binding and uptake of patchy polymersomes.124 Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
5.2 Surface functionalisation
In many biomedical applications reported thus far, such as in vivo imaging, polymersomes have been used which contained a functional content, e.g. near infrared fluorescent dyes52,118,125 and MRI contrast agents,114,115 but which did not have any surface functionality. In fact, they were mostly designed for prolonged in vivo circulation times, which means that they were predominantly PEGylated. However, for imaging and drug delivery it might be desirable to functionalize the periphery with ligands or moieties that allow (specific) recognition. In principle two approaches can be followed for this purpose. In the first method the functional group is attached to the polymer prior to vesicle formation. One of the first examples of this approach was the preparation of a functionalized polymersome for cellular delivery via the conjugation of the cell penetrating peptide Tat to PBd–PEG. After the polymersomes were formed they showed good cellular uptake and were able to deliver a near infrared dye loaded polymersome to dendritic cells.125 However this approach is not always trivial. If the functionality that needs to be introduced is too hydrophobic, the moiety might fold back in the membrane upon polymersome formation, reducing its availability. Furthermore, if bigger moieties need to be introduced, this will put quite some restrictions on the preparation methods and also the expected behaviour of the amphiphiles. Therefore researchers have also developed a second approach which entails post-functionalization of polymersomes either via covalent or via strong non-covalent interactions, as recently reviewed by Meier and coworkers.24In many cases non-covalent interactions are reversible and this can be useful, but also makes the system less robust. A non-covalent interaction that is almost as strong as a chemical bond is the interaction between biotin and streptavidin.126 Hammer and co-workers127 used polymersomes of PBD–PEG end-functionalized with biotin (actually biocytin) to study the adhesiveness of these polymersomes to streptavidin, both surface immobilized and free in solution. They showed that when biotin was attached to a longer PEG chain than the non-functionalized membrane polymers there was a difference in adhesive properties. When biotin sticks out of the membrane the adhesion to immobilized streptavidin increased whereas in solution no difference was found. The group of Meier published a series of papers where they used biotin-functional polymers to adhere polymersomes to streptavidin.128,129 In one example130 they used a double biotinylated triblock copolymer PMOXA–PDMS–PMOXA to adhere biotinylated ligands to the surface. In this case streptavidin with four binding sites fulfilled the function of effectively connecting the polymersome and targeting ligand. The fact that streptavidin has four binding sites also puts some restrictions on its applicability. If the concentration of streptavidin becomes too high crosslinking of polymersomes will become dominant and bigger aggregates will form. The interaction of cyclodextrin and adamantane is another example of a host–guest pair that strongly interacts and of which one of the binding partners can be easily immobilized on the surface of a polymeric membrane.57,131
An interesting, strong, metal ion complexation that is often used in protein science to purify proteins from a cell lysate is the nickel–NTA interaction with histidine-tags. The group of Meier showed two132,133 examples where they immobilized the NTA ligand on the surface of PBD–PEG polymersomes. They demonstrated to be able to complex both green fluorescent protein (GFP) and bone morphogenetic protein (BMP) via their His-tags on the polymersome surface.
Functional moieties have also been attached covalently to premade polymersomes. However, there are some restrictions to the chemistry involved. In most cases polymersomes are formed and used in an aqueous environment, so the chemistry should be applicable in water and be selective. A recent report134 on the immobilisation of polymersomes on a substrate exploited peripheral aldehydes to form an imine bond, which is in principle reversible. An irreversible bond that can be formed and is common in bio-conjugation techniques is the amide bond. Carboxylic acids present on the periphery of polymersomes were activated as NHS esters32 which allowed the attachment of several proteinsvia available amine residues. Another coupling strategy adopted from bioconjugation is the use of the maleimide functionality. Maleimides readily react with thiols which can be made available in a controlled fashion in both peptides and proteins by the introduction of a cysteine residue. In this way Pang et al.135 showed how they were able to couple mouse-anti-rat monoclonal antibodies for brain delivery in rats to the surface of polymersomes.
As depicted in Fig. 17 Opsteen et al.136 reported the use of the copper-catalysed [2 + 3] Huisgen cycloaddition to immobilise fluorescent dansyl, GFP and biotin on the surface of azide end-functional PS–PAA vesicles. PS–PAA was easily synthesised viaATRP and the introduction of the azide moiety was achieved by substitution of the bromine end group present after ATRP. Not all block copolymers however allow for easy modification. Therefore, van Dongen et al.137 adopted a strategy in which they mixed in up to ten percent of an acetylene-functional PS–PEG in their PS–PIAT polymersomes. These so-called anchors allowed for the immobilisation of enzymes on the vesicle surface. In order to steer away from the toxic Cu catalyst, in recent years also strain-promoted copper-free click reactions have been developed.138 Other click strategies have also been adopted. Very recently Petersen et al.139 introduced the vinyl sulfone moiety in polymersomes. This moiety also reacts fast and selective with thiols. They showed how via this method RGD peptides could be coupled on the periphery of their poly(methyl caprolactone)-b-PEG polymersomes.
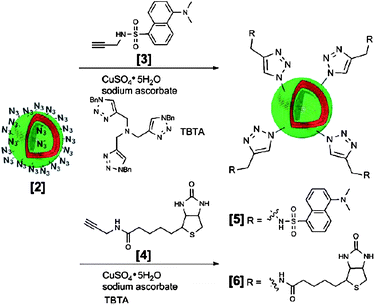 | ||
| Fig. 17 Surface functionalization of polymersomes assembled from PS–PAA–N3 with molecular probesvia click chemistry. The same methodology has been applied to immobilize proteins, e.g. green fluorescent protein.136 | ||
Beside the use of surface functionalisation to improve drug delivery or in vivo imaging, it has also been employed to improve on the cellular uptake of the polymersome nanoreactors as discussed in section 4.2.
A first report of such an artificial organelle inside a cell was provided by Ben-Haim et al.140 They showed how PMOXA–PDMS–PMOXA polymersomes functionalized with oligonucleotide polyguanylic acid (polyG) are efficiently recognized by macrophages and engulfed. A selection of other cell types did not show significant cell uptake, underlining the specificity for macrophages. After assessing the fate and distribution of the polymersomes inside the macrophages they loaded the newly constructed organelle with trypsin, an intestinal protease, and delivered it to the cell. As a traceable substrate they chose the hydrophobic tripeptide serine protease specific substrate BZiPAR (bis-(CBZ-Ile-Pro-Arg)-R110), which can diffuse passively through both cell membranes and the polymeric membrane. First they blocked the cell's own protease to prevent false positives. After the substrate reached the polymersome loaded trypsin in the cell, the substrate was converted to release the coupled Rhodamine effectively making the cells fluorescent.
In a more recent example van Dongen et al.138 used the inherently semi-permeable polymersome based on PS-PIAT to construct an artificial organelle. They ligated the cell penetrating peptide Tat to the surface of their polymersomes to induce cellular uptake by HeLa cells. The enzyme horseradish peroxidase (HRP), which was encapsulated in the polymersome, subsequently catalysed the oxidation of compound 3,3′,5,5′tetramethylbenzidine (TMB) by hydrogen-peroxide. The oxidation product of TMB was easily detected and showed good conversion inside the cell. This model reaction was used to demonstrate that the artificial organelle was capable of neutralizing oxidative species in a cellular environment.
Conclusion
Polymersomes are new versatile carriers that have high potential for in vivo imaging and nanotherapeutics. Although quite different from liposomes general aspects known from liposomal science can be used to design polymersomes with specific functions. Control can be exerted over size, shape, surface function and topology, which will influence the in vivo circulation time and therefore the applicability of these nanocarriers. A large variety of examples can nowadays be found in literature that show how polymersome research has caught up with liposomal science in the past decade and in many cases adds extra dimensions to what is possible.References
- J. C. M. Vanhest, D. A. P. Delnoye, M. W. P. L. Baars, M. H. P. Vangenderen and E. W. Meijer, Science, 1995, 268, 1592–1595 CrossRef CAS.
- K. Fujita, S. Kimura and Y. Imanishi, Langmuir, 1999, 15, 4377–4379 CrossRef CAS.
- J. J. L. M. Cornelissen, M. Fischer, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427–1430 CrossRef CAS.
- L. F. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777–1779 CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- J. Z. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef CAS.
- M. Antonietti and S. Forster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- T. Uneyama, J. Chem. Phys., 2007, 126, 114902 CrossRef.
- X. H. He and F. Schmid, Macromolecules, 2006, 39, 2654–2662 CrossRef CAS.
- J. Z. Du and Y. M. Chen, Macromolecules, 2004, 37, 5710–5716 CrossRef CAS.
- D. J. Adams, S. Adams, D. Atkins, M. F. Butler and S. Furzeland, J. Controlled Release, 2008, 128, 165–170 CrossRef CAS.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS.
- K. T. Kim, S. A. Meeuwissen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2010, 2, 844–858 RSC.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, C. A. David, S. S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS.
- C. Schatz and S. Lecommandoux, Macromol. Rapid Commun., 2010, 31, 1664–1684 CrossRef CAS.
- S. F. M. van Dongen, H. P. M. de Hoog, R. J. R. W. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS.
- S. Egli, H. Schlaad, N. Briuns and W. Meier, Polymers, 2011, 3, 28 Search PubMed.
- J. A. Opsteen, J. J. L. M. Cornelissen and J. C. M. van Hest, Pure Appl. Chem., 2004, 76, 1309–1319 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, Polymer Therapeutics II: Polymers as Drugs, Conjugates and Gene Delivery Systems, 2006, vol. 193, pp. 1–65 Search PubMed.
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235–237 CrossRef CAS.
- M. C. Woodle, Chem. Phys. Lipids, 1993, 64, 249–262 CrossRef CAS.
- F. Ahmed, A. Hategan, D. E. Discher and B. M. Discher, Langmuir, 2003, 19, 6505–6511 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, G. Srinivas, A. Brannan, F. Bates, M. L. Klein, T. Minko and D. E. Discher, Mol. Pharmaceutics, 2006, 3, 340–350 CrossRef CAS.
- F. H. Meng, G. H. M. Engbers and J. Feijen, J. Controlled Release, 2005, 101, 187–198 CrossRef CAS.
- F. H. Meng, C. Hiemstra, G. H. M. Engbers and J. Feijen, Macromolecules, 2003, 36, 3004–3006 CrossRef CAS.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894–3900 CrossRef CAS.
- J. Z. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef CAS.
- H. Lomas, J. Z. Du, I. Canton, J. Madsen, N. Warren, S. P. Armes, A. L. Lewis and G. Battaglia, Macromol. Biosci., 2010, 10, 513–530 CrossRef CAS.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. M. Christianen, J. C. Maan, A. E. Rowan and R. J. M. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- S. Kimura, D. H. Kim, J. Sugiyama and Y. Imanishi, Langmuir, 1999, 15, 4461–4463 CrossRef CAS.
- S. Kimura, Y. Muraji, J. Sugiyama, K. Fujita and Y. Imanishi, J. Colloid Interface Sci., 2000, 222, 265–267 CrossRef CAS.
- A. J. T. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. J. L. M. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. P. J. T. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172–4174 RSC.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- T. J. Deming, Adv. Polym. Sci., 2006, 202, 1–18 CAS.
- F. Checot, S. Lecommandoux, Y. Gnanou and H. A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef CAS.
- H. Kukula, H. Schlaad, M. Antonietti and S. Forster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef CAS.
- F. Checot, S. Lecommandoux, H. A. Klok and Y. Gnanou, Eur. Phys. J. E, 2003, 10, 25–35 CrossRef CAS.
- F. Checot, A. Brulet, J. Oberdisse, Y. Gnanou, O. Mondain-Monval and S. Lecommandoux, Langmuir, 2005, 21, 4308–4315 CrossRef CAS.
- K. E. Gebhardt, S. Ahn, G. Venkatachalam and D. A. Savin, J. Colloid Interface Sci., 2008, 317, 70–76 CrossRef CAS.
- E. P. Holowka, D. J. Pochan and T. J. Deming, J. Am. Chem. Soc., 2005, 127, 12423–12428 CrossRef CAS.
- J. Gaspard, J. A. Silas, D. F. Shantz and J. S. Jan, Supramol. Chem., 2010, 22, 178–185 CrossRef CAS.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2008, 19, 109–117 CrossRef CAS.
- C. Schatz, S. Louguet, J. F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS.
- B. S. Kim, W. Y. Yang, J. H. Ryu, Y. S. Yoo and M. Lee, Chem. Commun., 2005, 2035–2037 RSC.
- M. Felici, M. Marza-Perez, N. S. Hatzakis, R. J. M. Nolte and M. C. Feiters, Chem.–Eur. J., 2008, 14, 9914–9920 CrossRef CAS.
- K. K. Upadhyay, J. F. Le Meins, A. Misra, P. Voisin, V. Bouchaud, E. Ibarboure, C. Schatz and S. Lecommandoux, Biomacromolecules, 2009, 10, 2802–2808 CrossRef CAS.
- A. Misra, K. K. Upadhyay, A. N. Bhatt, A. K. Mishra, B. S. Dwarakanath, S. Jain, C. Schatz, J. F. Le Meins, A. Farooque, G. Chandraiah, A. K. Jain and S. Lecommandoux, Biomaterials, 2010, 31, 2882–2892 CrossRef CAS.
- A. K. Mishra, K. K. Upadhyay, A. N. Bhatt, E. Castro, K. Chuttani, B. S. Dwarakanath, C. Schatz, J. F. Le Meins, A. Misra and S. Lecommandoux, Macromol. Biosci., 2010, 10, 503–512.
- S. Lecommandoux, K. K. Upadhyay, J. F. Le Meins, A. Misra, P. Voisin, V. Bouchaud, E. Ibarboure and C. Schatz, Biomacromolecules, 2009, 10, 2802–2808 CrossRef CAS.
- I. Otsuka, K. Fuchise, S. Halila, S. Fort, K. Aissou, I. Pignot-Paintrand, Y. G. Chen, A. Narumi, T. Kakuchi and R. Borsali, Langmuir, 2010, 26, 2325–2332 CrossRef CAS.
- H. Iatrou, H. Frielinghaus, S. Hanski, N. Ferderigos, J. Ruokolainen, O. Ikkala, D. Richter, J. Mays and N. Hadjichristidis, Biomacromolecules, 2007, 8, 2173–2181 CrossRef CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS.
- A. Harada, K. Nakanishi, S. Ichimura, C. Kojima and K. Kono, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 1217–1223 CAS.
- B. J. McKenna, H. Birkedal, M. H. Bartl, T. J. Deming and G. D. Stucky, Angew. Chem., Int. Ed., 2004, 43, 5652–5655 CrossRef CAS.
- A. Koide, A. Kishimura, K. Osada, W. D. Jang, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2006, 128, 5988–5989 CrossRef CAS.
- B. Guo, Z. Q. Shi, Y. Yao, Y. F. Zhou and D. Y. Yan, Langmuir, 2009, 25, 6622–6626 CrossRef CAS.
- A. Kishimura, S. Liamsuwan, H. Matsuda, W. F. Dong, K. Osada, Y. Yamasaki and K. Kataoka, Soft Matter, 2009, 5, 529–532 RSC.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085–6088 CrossRef CAS.
- R. J. Thibault, O. Uzun, R. Hong and V. M. Rotello, Adv. Mater., 2006, 18, 2179 CrossRef CAS.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS.
- R. J. Thibault, T. H. Galow, E. J. Turnberg, M. Gray, P. J. Hotchkiss and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 15249–15254 CrossRef.
- F. Caruso, D. Trau, H. Mohwald and R. Renneberg, Langmuir, 2000, 16, 1485–1488 CrossRef CAS.
- H. Harashima and H. Kiwada, Adv. Drug Delivery Rev., 1996, 19, 425–444 CrossRef CAS.
- A. Nagayasu, K. Uchiyama and H. Kiwada, Adv. Drug Delivery Rev., 1999, 40, 75–87 CrossRef CAS.
- P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, J. Controlled Release, 2003, 90, 323–334 CrossRef.
- J. C. M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y. Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef CAS.
- J. Lam, Y. Ma, S. Armes, A. Lewis and S. Stolnik, J. Pharm. Pharmacol., 2004, 56, S70–S70.
- M. C. Woodle, C. M. Engbers and S. Zalipsky, Bioconjugate Chem., 1994, 5, 493–496 CrossRef CAS.
- D. C. Litzinger, A. M. J. Buiting, N. Vanrooijen and L. Huang, Biochim. Biophys. Acta, Biomembr., 1994, 1190, 99–107 CrossRef CAS.
- D. C. Drummond, O. Meyer, K. L. Hong, D. B. Kirpotin and D. Papahadjopoulos, Pharmacol. Rev., 1999, 51, 691–743 CAS.
- W. Wang, A. M. McConaghy, L. Tetley and I. F. Uchegbu, Langmuir, 2001, 17, 631–636 CrossRef CAS.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS.
- L. B. Luo and A. Eisenberg, Langmuir, 2001, 17, 6804–6811 CrossRef CAS.
- C. Sanson, C. Schatz, J. F. Le Meins, A. Brulet, A. Soum and S. Lecommandoux, Langmuir, 2010, 26, 2751–2760 CrossRef CAS.
- H. W. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473–9487 CrossRef CAS.
- X. F. Li, A. Kroeger, T. Azzam and A. Eisenberg, Langmuir, 2008, 24, 2705–2711 CrossRef CAS.
- J. B. Huang, Y. Zhu, B. Y. Zhu, R. K. Li and H. I. Fu, J. Colloid Interface Sci., 2001, 236, 201–207 CrossRef CAS.
- S. Rameez, I. Bamba and A. F. Palmer, Langmuir, 2010, 26, 5279–5285 CrossRef CAS.
- S. Y. Yu, T. Azzam, I. Rouiller and A. Eisenberg, J. Am. Chem. Soc., 2009, 131, 10557–10566 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- J. T. Zhu and W. Jiang, Macromolecules, 2005, 38, 9315–9323 CrossRef CAS.
- Y. Geng and D. E. Discher, J. Am. Chem. Soc., 2005, 127, 12780–12781 CrossRef CAS.
- Y. Kim, P. Dalhaimer, D. A. Christian and D. E. Discher, Nanotechnology, 2005, 16, S484–S491 CrossRef.
- X. Q. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, D. A. Steeber and S. Q. Gong, Biomaterials, 2010, 31, 9065–9073 CrossRef CAS.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS.
- R. Lipowsky, Nature, 1991, 349, 475–481 CrossRef CAS.
- M. Yanagisawa, M. Imai and T. Taniguchi, Phys. Rev. Lett., 2008, 100, 148102 CrossRef.
- X. J. Li, I. V. Pivkin, H. J. Liang and G. E. Karniadakis, Macromolecules, 2009, 42, 3195–3200 CrossRef CAS.
- K. T. Kim, J. H. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908 CrossRef CAS.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef CAS.
- A. E. Smith, X. W. Xu, S. E. Kirkland-York, D. A. Savin and C. L. McCormick, Macromolecules, 2010, 43, 1210–1217 CrossRef CAS.
- V. Butun, N. C. Billingham and S. P. Armes, J. Am. Chem. Soc., 1998, 120, 11818–11819 CrossRef.
- E. Mabrouk, D. Cuvelier, F. Brochard-Wyart, P. Nassoy and M. H. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7294–7298 CrossRef CAS.
- G. P. Robbins, M. Jimbo, J. Swift, M. J. Therien, D. A. Hammer and I. J. Dmochowski, J. Am. Chem. Soc., 2009, 131, 3872 CrossRef CAS.
- S. F. M. van Dongen, M. Nallani, J. L. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem.–Eur. J., 2009, 15, 1107–1114 CAS.
- A. Ranquin, W. Versees, W. Meier, J. Steyaert and P. Van Gelder, Nano Lett., 2005, 5, 2220–2224 CrossRef CAS.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS.
- R. Stoenescu, A. Graff and W. Meier, Macromol. Biosci., 2004, 4, 930–935 CrossRef CAS.
- A. Graff, M. Sauer, P. Van Gelder and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5064–5068 CrossRef CAS.
- H. J. Choi and C. D. Montemagno, Nano Lett., 2005, 5, 2538–2542 CrossRef CAS.
- Z. L. Cheng and A. Tsourkas, Langmuir, 2008, 24, 8169–8173 CrossRef CAS.
- Z. L. Cheng, D. L. J. Thorek and A. Tsourkas, Adv. Funct. Mater., 2009, 19, 3753–3759 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787 CrossRef CAS.
- V. D. Awasthi, D. Garcia, R. Klipper, B. A. Goins and W. T. Phillips, J. Pharmacol. Exp. Ther., 2004, 309, 241–248 CrossRef CAS.
- D. A. Christian, O. B. Garbuzenko, T. Minko and D. E. Discher, Macromol. Rapid Commun., 2010, 31, 135–141 CAS.
- T. P. Smart, O. O. Mykhaylyk, A. J. Ryan and G. Battaglia, Soft Matter, 2009, 5, 3607–3610 RSC.
- L. He, Y. Jiang, C. L. Tu, G. L. Li, B. S. Zhu, C. Y. Jin, Q. Zhu, D. Y. Yan and X. Y. Zhu, Chem. Commun., 2010, 46, 7569–7571 RSC.
- R. P. Brinkhuis, T. R. Visser, F. P. J. T. Rutjes and J. C. M. Van Hest, Polym. Chem., 2011, 2, 550–552 RSC.
- T. Baumgart, S. T. Hess and W. W. Webb, Nature, 2003, 425, 821–824 CrossRef CAS.
- D. A. Christian, A. W. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843–849 CrossRef CAS.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS.
- N. A. Christian, M. C. Milone, S. S. Ranka, G. Z. Li, P. R. Frail, K. P. Davis, F. S. Bates, M. J. Therien, P. P. Ghoroghchian, C. H. June and D. A. Hammer, Bioconjugate Chem., 2007, 18, 31–40 CrossRef CAS.
- P. C. Weber, D. H. Ohlendorf, J. J. Wendoloski and F. R. Salemme, Science, 1989, 243, 85–88 CrossRef CAS.
- J. J. Lin, J. A. Silas, H. Bermudez, V. T. Milam, F. S. Bates and D. A. Hammer, Langmuir, 2004, 20, 5493–5500 CrossRef CAS.
- M. Grzelakowski, O. Onaca, P. Rigler, M. Kumar and W. Meier, Small, 2009, 5, 2545–2548 CrossRef CAS.
- P. Rigler and W. Meier, J. Am. Chem. Soc., 2006, 128, 367–373 CrossRef CAS.
- P. Broz, S. M. Benito, C. Saw, P. Burger, H. Heider, M. Pfisterer, S. Marsch, W. Meier and P. Hunziker, J. Controlled Release, 2005, 102, 475–488 CrossRef CAS.
- M. Y. Guo, M. Jiang and G. Z. Zhang, Langmuir, 2008, 24, 10583–10586 CrossRef CAS.
- R. Nehring, C. G. Palivan, O. Casse, P. Tanner, J. Tuxen and W. Meier, Langmuir, 2009, 25, 1122–1130 CrossRef CAS.
- R. Nehring, C. G. Palivan, S. Moreno-Flores, A. Mantion, P. Tanner, J. L. Toca-Herrera, A. Thunemann and W. Meier, Soft Matter, 2010, 6, 2815–2824 RSC.
- S. Domes, V. Filiz, J. Nitsche, A. Fromsdorf and S. Forster, Langmuir, 2010, 26, 6927–6931 CrossRef CAS.
- Z. Q. Pang, W. Lu, H. L. Gao, K. L. Hu, J. Chen, C. L. Zhang, X. L. Gao, X. G. Jiang and C. Q. Zhu, J. Controlled Release, 2008, 128, 120–127 CrossRef CAS.
- J. A. Opsteen, R. P. Brinkhuis, R. L. M. Teeuwen, D. W. P. M. Lowik and J. C. M. van Hest, Chem. Commun., 2007, 3136–3138 RSC.
- S. F. M. van Dongen, M. Nallani, S. Schoffelen, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Macromol. Rapid Commun., 2008, 29, 321–325 CrossRef CAS.
- S. F. M. van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef.
- M. A. Petersen, L. G. Yin, E. Kokkoli and M. A. Hillmyer, Polym. Chem., 2010, 1, 1281–1290 RSC.
- N. Ben-Haim, P. Broz, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2008, 8, 1368–1373 CrossRef CAS.
Footnote |
| † This work was performed within the framework of the Dutch Top Institute Pharma project # T5-105. |
| This journal is © The Royal Society of Chemistry 2011 |