RAFT polymerization mediated bioconjugation strategies
Volga
Bulmus
*ab
aDepartment of Chemical Engineering, Izmir Institute of Technology, Gulbahce, Izmir 35430, Turkey
bSchool of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: volgabulmus@iyte.edu.tr; vbulmus@unsw.edu.au; Fax: +90 232 750 6645; Tel: +90 232 750 6660
First published on 31st May 2011
Abstract
This review aims to highlight the use of RAFT polymerization in the synthesis of polymer bioconjugates. It covers two main bioconjugation strategies using the RAFT process: (i) post-polymerization bioconjugations using pre-synthesized reactive polymers, and (ii) bioconjugations via in situpolymerization using biomolecule-modified monomers or chain transfer agents.
 Volga Bulmus | Assoc. Prof. Volga Bulmus received her B.E. degree in Chemical Engineering and Ph.D. degree in Bioengineering from Hacettepe University (Turkey). Between 2001 and 2003, she worked as a postdoctoral research fellow in the Bioengineering Department at University of Washington (WA, USA). In 2004, she joined the Centre for Advanced Macromolecular Design at the University of New South Wales (UNSW) (NSW, Australia) as a Vice Chancellor's Research Fellow. In 2008, she was appointed as a tenured academic by the School of Biotechnology and Biomolecular Sciences (BABS) at UNSW. In 2010, she joined the Department of Chemical Engineering at Izmir Institute of Technology (Iztech) (Turkey) as an Associate Professor. Bulmus' research interests include design, synthesis, and evaluation of well-defined, nanoscale-tailored polymeric systems and polymer bioconjugates for nanomedicine and biotechnology applications. She has published 60+ journal articles in the relevant fields. |
1 Introduction
1.1 Polymer bioconjugates
Polymer bioconjugates are formed by coupling synthetic or biological polymer chains to biological molecules via covalent bonds or bioaffinity interactions. Structural and functional properties, such as stability, solubility, biocompatibility and bioactivity, of biomolecules are usually altered upon coupling of polymer chains.1–4 New features and functionality can also be imparted to biomolecules by polymer conjugation, inducing novel behaviors such as stimuli-responsive phase-separation,5,6self-assembly,7,8 and patterning behaviors.9,10Polymer bioconjugates find applications in different fields of (nano)biotechnology, biomedicine and pharmaceutical technologies. For example, polyethylene glycol conjugates of several therapeutic proteins have been used for treatment of diseases in humans. Readers are referred to several excellent reviews on different aspects of the biomolecule-polymer conjugates.11–25Initial studies in the field of polymer bioconjugates commenced in the mid 1970s. Reactive, linear/soluble polymers were conjugated to enzymes to improve heat stability.26,27 In 1977, Davis and Abuchowski discovered the non-immunogenic properties of polyethylene glycol (PEG) and developed a method of attaching PEG to proteins (PEGylation) which prevented the recognition of proteins by the immune system and slowed their breakdown in the body.28 Following studies in later years explored in more detail the conjugation of varying enzymes and other proteins with stimuli-responsive polymers29–31 and other water-soluble polymers.32,33 These early studies revealed the potential of enyzme/protein-polymer conjugates in bioseparations, bioreactions, diagnostics and drug delivery. The approval of bovine adenosine deaminase – PEG conjugates by FDA in 1990 followed by intereferon-alpha, l-asparaginase and granulocyte colony-stimulating factor conjugates of PEG, to enter the market as pharmaceuticals for human use, has further proved the enormous potential of biomolecule-polymer conjugates in biomedicine and pharmaceutical technologies.
In accord with the increasing utility of polymer bioconjugates in different areas of biomedicine1,2,34,35 and (nano)biotechnology5,7–10,36 research has been driven into generating homogeneous and well-defined conjugates manifesting uniformity in biohybrid properties and reproducible biological activity. The properties of biomolecule-polymer conjugates usually need to be tailored at molecular level to generate conjugates for a given application. Molecular weight and its distribution, conjugation site, molecular architecture, solubility, chemical and biological functionality which appear to be the most important properties for bioconjugate designs, need to be well-controlled to establish a solid correlation between the performance and bioconjugate design.1,2,12,34
1.2 Reversible addition fragmentation chain transfer (RAFT) polymerization
Reversible addition fragmentation chain transfer (RAFT) polymerization, first reported in 1998 by Moad, Rizzardo and Thang at CSIRO,37 is a living radical polymerization (LRP) technique, a free radical polymerization technique which is not subject to termination or transfer reactions and yields polymer chains that are able to re-propogate by addition of free radicals and monomers.
RAFT polymerization requires the use of thiocarbonylthio moiety containing chain transfer agents (RAFT agents) (Scheme 1). The RAFT mechanism and the appropriate RAFT agent structures have been recently detailed in a number of review articles.16,25,38,39 Briefly, during a RAFT process (Fig. 1), the oligomeric radicals formed at the initiation stage of polymerization add to the highly reactive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond of the RAFT agents. Fragmentation of these radical intermediates results in the formation of oligomeric RAFT agents and R group radicals. The R-radicals should initiate the growth of polymer chains. The growing polymeric radicals add to the polymeric RAFT agents forming stabilized radical intermediates, following by the fragmentation to the polymeric RAFT agents and polymeric radicals. At the end of polymerization, dormant polymeric RAFT agents together with terminated polymeric radicals are obtained.
S bond of the RAFT agents. Fragmentation of these radical intermediates results in the formation of oligomeric RAFT agents and R group radicals. The R-radicals should initiate the growth of polymer chains. The growing polymeric radicals add to the polymeric RAFT agents forming stabilized radical intermediates, following by the fragmentation to the polymeric RAFT agents and polymeric radicals. At the end of polymerization, dormant polymeric RAFT agents together with terminated polymeric radicals are obtained.
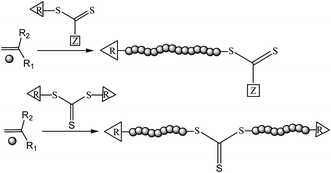 | ||
| Scheme 1 Schematic of the polymer synthesis by RAFT process using a thiocarbonylthio or a symmetric trithiocarbonate RAFT agent. | ||
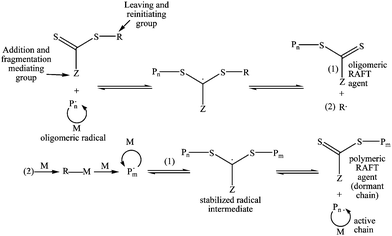 | ||
| Fig. 1 Reversible addition fragmentation chain transfer mechanism. | ||
RAFT polymerization benefits and suffers from all characteristics of free radical polymerization process: It takes place under facile reaction conditions. It is compatible with a wide range of monomers, and tolerates varying functional chemical groups. While RAFT polymerization is subject to undesirable termination reactions, such reactions are greatly minimized with respect to conventional free radical polymerization. This feature provides control over the molecular weight and narrows the molecular weight distribution of the polymer chains produced by the RAFT technique. In addition, RAFT polymerization yields polymers with defined chain end functionality as the alpha- and omega-termini of living polymer chains are capped with R- and Z- groups, respectively, of the RAFT agents, with the exception of symmetric trithiocarbonate and Z-connected multi-RAFT agents (Scheme 1).38,39 In cases where symmetrical trithiocarbonates or Z-connected multi-RAFT agents are used, the Z-group is located in the core and connects the arms of the RAFT-synthesized polymer.40,41 It is also possible to create various polymer architectures such as block and graft copolymers, stars, and nanostructures using RAFT polymerization.42,43,44–51
RAFT polymerization offers a highly versatile platform for controlled synthesis and molecular engineering of polymer bioconjugates.13–16,25 The strength of the RAFT approach for generation of polymer bioconjugates lies in its ability to control the polymerization of a wide range of monomers in varying solvents including water, at moderate temperatures, using only chain transfer agents and common free radical initiators (without the need for any additional polymerization component such as metal catalysts and sacrificial initiators). Moreover, it enables the synthesis of well-defined polymers with defined and spatially-controlled chemical functionalities such as pendant-, mid-junction, alpha- and omega-end-group functionalities, suitable for performing bioconjugations. In addition, the ability of RAFT polymerization to synthesize designed architectures especially block copolymers, gradient copolymers, stars and branched structures potentially makes the generation of bioconjugates with varying architectures possible, envisaging new or improved applications of polymer bioconjugates.
1.3 Scope of the review
This article aims to highlight the use of RAFT polymerization in synthesis of polymer bioconjugates by reviewing some highlights in the recent literature. It focuses only on the synthetic aspects of bioconjugation strategies utilizing RAFT polymerization. It does not intend to comprehensively summarize all of the work to date in the field. Readers are also referred to several excellent reviews covering comprehensively the use of living radical polymerizations in bioconjugations.13–16,25Within this review, the term “biomolecule” has been used to refer to the molecules originating from a living organism and the synthetic analogs of such molecules. Thus, the conjugates of synthetic peptides and nucleic acids have been included in the review.
The review covers two main bioconjugation strategies:
(i) Post-polymerization bioconjugations (Scheme 2): Preparation of bioconjugates using pre-synthesized reactive polymers, either functionalized by modification of their RAFT end-group or synthesized directly by using functionalized RAFT agents, has been reviewed. Bioconjugations have been classified according to the reactivity of the polymers used.
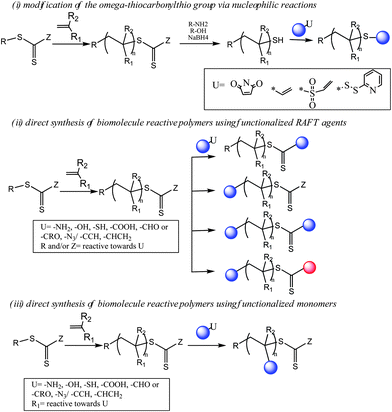 | ||
| Scheme 2 Post-polymerization bioconjugation strategies using RAFT technique. | ||
(ii) Bioconjugations via in situ polymerization (Scheme 3): Bioconjugations via in situRAFT polymerization using biomolecule-modified monomers or RAFT agents have been reviewed.
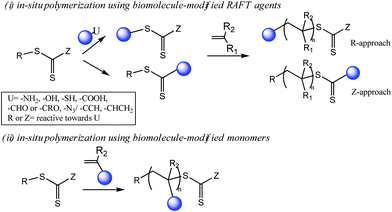 | ||
| Scheme 3 Bioconjugation strategies via in situRAFT polymerization. | ||
2 RAFT polymerization approach to polymer bioconjugates
2.1 Post-polymerization bioconjugation strategy
In general, the conjugation of pre-synthesized, end-group functionalized polymers to biomolecules has been the method of choice for preparation of bioconjugates using the RAFT polymerization approach. This method usually requires multiple steps including the synthesis and purification of polymers, conjugation to biomolecules and the purification of the final conjugates, which significantly reduces the overall yield. Despite this, the post-polymerization bioconjugation strategy has been commonly used, mainly because of its compatibility with fragile biomolecules. As it minimizes the number of steps involving the biomolecules, the risk of altering the biomolecule's conformation and activity is reduced.There are, in general, two common methods for generating end-group functionalized RAFT-polymers for bioconjugations (Scheme 2): (i) post-polymerization modification of the thiocarbonylthio end-group of the synthesized polymers41,52,53 and (ii) employing functionalized RAFT agents for direct synthesis of polymers with α- and/or ω- functionalities reactive towards biomolecule’s functional groups.
With the former approach, ω-thiol-terminated polymers have been successfully generated by reaction of nucleophiles such as amines,54–57hydroxides55,58–60 and reducing agents such as boronhydrides61–65 with the thiocarbonylthio group of the RAFT-synthesized polymers. The thiol-functional polymers could be further modified with thiol-reactive reagents, such as maleic anhydride,66 for creating highly reactive sites for conjugation with biomolecules. Alternatively, thiol-functional polymers can be used directly for bioconjugations without any further modification.62,67–70 The removal of thiocarbonylthio group from the bioconjugate structure may be an advantage of this approach as the reactive thiocarbonylthio group may cause concentration-dependent cytotoxicity if the bioconjugates are intended to be used in living organisms.71,72 α- and/or ω-functionality can be also incorporated to a polymer chain during RAFT polymerization by using a RAFT agent with functionalized R- and/or Z-groups, respectively. When designing R- and Z- groups, amines, thiols, carboxylic acids and ketones/aldehydes, which are the reactive groups of biological molecules, commonly used for bioconjugations, have been considered.56,66,69,70,73–107 Bioconjugation strategies combining click reactions such as azide-alkyne cycloadditions and thiol-ene additions with end-group functionalized RAFT polymers have also attracted attention because of high efficiency of such reactions (vide infra).
Among the post-polymerization bioconjugation strategies, conjugation of relatively small molecular weight biomolecules such as sugar residues, oligopeptides and vitamins such as biotin and folic acid, to the pendant-group functionalized polymers have been also reported (Scheme 2).73,74,77,78,80,81
The examples of post-polymerization bioconjugations of reactive RAFT polymers have been presented in the following sections.
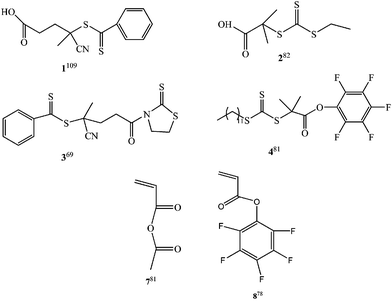
The bioconjugation reactions between carboxylic acid and amine or hydroxyl groups usually require the use of carboxylic acid-activating agents to increase the conjugation yields. Thus, carboxylic acid-terminated polymers synthesized by RAFT polymerization have been activated using N-hydroxysuccinimide (NHS),112,113pentafluorophenyl (PFP)82 or 2-mercaptothiozaline (3, Fig. 2).69 For example, the carboxyl end-group of semi-telechelic poly(N-isopropylacrylamide) (pNIPAAm) synthesized by RAFT polymerization using 2-ethylsulfanylthiocarbonylsulfany1-2-methyl propionic acid (2, Fig. 2) as a carboxylic acid-bearing RAFT agent was modified with tetrafluorophenol to yield amine-reactive ester groups for conjugation to amine groups of anti-streptavidin and anti-Plasmodium falciparum histidine-rich protein 2 (PfHRP2) antibodies. These conjugates were used to capture and detect a model streptavidin antigen and subsequently to clinical ranges of the malaria antigen Plasmodium falciparum histidine-rich protein 2 (PfHRP2) from human plasma for a fluidic system.82
Following a more direct approach, RAFT agents can be first functionalized with carboxylic acid active esters, and used for conjugations with amine or hydroxyl groups.75,81,83,104 For example, (2-oxopropyl)acrylate (5, Fig. 2) was efficiently polymerized in the presence of a pentafluorophenyl (PFP)-ester functionalized trithiocarbonate RAFT agent (4, Fig. 2) to yield polymers with low polydispersity index (PDI < 1.15) and pendant ketone groups for the attachment of aminooxy glycans, as well as alpha-terminal PFP ester and trithiocarbonate groups.81
Active ester approach has been also used for conjugation of biomolecules to pendant groups of polymers.73,74,77,78 For example, pentafluorophenyl acrylate (FP-A, 6, Fig. 2) was polymerized in the presence of 3-(benzylsulfanylthiocarbonylsulfanyl)-propionic acid as a RAFT agent.78Amine-functional sugars, D-glucosamine and D-galactosamine, were conjugated via nucleophilic addition to P(FP-A) in the presence of triethylamine. The slow addition of reagents was noted to be critical to ensure a full conversion of the activated ester.
A thiazolidine-2-thione mid-functionalized RAFT agent (Fig. 3) was used for RAFT polymerization of N-(2-hydroxypropyl)methacrylamide (HPMA) to generate polymers with mid-chain thiazolidine-2-thione functionality.88 These mid-functionalized polymers yield branched biomolecule conjugates with an “umbrella-like” polymeric structure (Fig. 3). The branched polymer-lysozyme conjugates showed enhanced stability and biological activity with respect to similar molecular weight linear polyHPMA-lysozyme bioconjugates.88
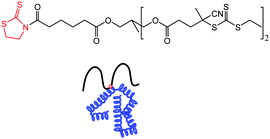 | ||
| Fig. 3 Thiazolidine-2-thione mid-functionalized RAFT agent and schematic of umbrella-like (mid-functional) polymer attachment to a protein.88 | ||
Aldehyde- and hydrazide functional polymers were also used in conjugations with biomolecules through amine or hydroxyl groups. For example, Godula and Bertozzi80 have reported a relatively easier generic strategy for the synthesis of glycopolymers with a broad scope of glycan structures. The proposed strategy was based on the ligation of free reducing sugars to an acryloyl hydrazide polymer with a biotin-end group (Fig. 4). A variety of reducing glycans ranging in structure from simple mono- and disaccharides to considerably more complex oligosaccharides were conjugated to the hydrazide pendant groups of the polymer under acidic conditions in the presence of an aniline catalyst. Glycopolymers were obtained in good to excellent yields (37–85%) except sialyl N-acetyllactosamine and sialyl Lewis glycans that were incorporated with 20 and 17% yield, respectively.
Utilizing amine reactive polymers for preparation of protein- or peptide-conjugates may result in the formation of heterogenous conjugates due to the presence of a high number of amine containing amino acid residues in proteins and peptides. One route to site-selective conjugation of one polymer chain per protein using amine-reactive polymers is to target only N-terminus of proteins/peptides by precisely adjusting the pH of the conjugation reaction to deprotonate only the α-amino group of the terminal residue.2
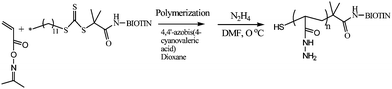 | ||
| Fig. 4 RAFT polymerization of acetoxime acrylate in the presence of a biotinylated chain transfer agent and subsequent treatment with hydrazine to generate α-biotin,ω-thiol poly(hydrazide acrylate).80 | ||
RAFT agents bearing α-allyl or α,ω-bis-allyl groups have been described.116,117 Allyl-pendant functionalized monomers have also been polymerized by RAFT process.118 Allyl-functionality is of particular interest as it can be exploited for modification via orthogonal thiol-ene reactions.118–121Maleimide-terminated or -pendant functionalized polymers have also been synthesized using the RAFT process.86,91,92,98,122Poly(N-isopropylacrylamide) (PNIPAM) prepared by RAFT polymerization was aminolyzed to yield thiol-terminated chains that were subsequently reacted with excess 1,8-bis-maleimidodiethyleneglycol. The resulting maleimide-terminated polymer was reacted with BSA and ovalbumin to yield heterotelecehlic polymer protein conjugates by two consecutive Michael addition thiol-ene reactions.98Vinyl sulfone is another group which is highly reactive toward thiols. The reduction of the RAFT dithiobenzoate group and simultaneous trapping with divinyl sulfone to produce Michael acceptor polymers was recently reported.123 The semitelechelic vinyl sulfone polymers were conjugated via a highly efficient reaction to the free cysteine residue of bovine serum albumin (BSA). Importantly, after polymer attachment, the activity of the BSA was 92% of the unmodified biomolecule.124
Pyridyl disulfide (PDS) is an effective active group towards the selective-exchange reaction with thiols under mild conditions.125 The formation of reversible disulfide linkages upon thiol-PDS reaction makes pyridyl disulfide a useful group for preparation of reversible bioconjugates.126–128 The combination of PDS group with RAFT polymerization was first reported with a PDS-functionalized trithiocarbonate RAFT agent.129 Given its versatility, the PDS group has been widely used in RAFT-mediated bioconjugations.40,56,67–70,93,94,105,106,114,127 Various PDS functionalized RAFT-agents including a mid-chain PDS functional chain transfer agent87 have been reported.40,89,114,130–134 For example, Duvall et al.90 synthesized a new PDS functional RAFT agent, trithiocarbonic acid 1-cyano-1-methyl-3-[2-(pyridin-2-yldisulfanyl)ethylcarbamoyl]propyl ester ethyl ester to generate PDS-functionalized block copolymers. The PDS functionality of the block copolymer provided a reversible peptide conjugation site. A cell-internalized proapoptotic peptide was conjugated via a disulfide-thiol exchange reaction with a conjugation yield of 75%. In another study,114 well-defined α-PDS functionalized poly(oligoethylene glycol acrylate)s that were synthesized using a PDS-modified trithiocarbonate RAFT agent was conjugated to 5′-thiol-modified small interfering RNA (siRNA) to increase the nuclease stability of siRNA.135
It is also possible to incorporate ω-PDS functionality by in situaminolysis of the thiocarbonylthio end group of RAFT-polymers in the presence of 2,2′-dithiodipyridine.56,67,68–70,136 This approach produces PDS-functional polymers with high yields (close to 90%) without accompanying side-reactions such as disulfide or thiolactone formation. The “in situaminolysis” approach can also be performed in the presence of ene-bearing molecules to generate polymers with different ω-functionality with high yields.68,136
Pyridyldisulfide ethylmethacrylate was (co)polymerizedvia RAFT process using a dithiobenzoate chain transfer agent to yield well-defined (co)polymers with PDS pendant groups.43,93,106 The PDS pendant groups were utilized to conjugate anticancer drugs95 and fluorescent probes.105 Following a different approach, a well-defined N-(2-hydroxypropyl)methacrylamide-s-N-(3-aminopropyl)methacrylamide (HPMA-S-APMA) copolymer, synthesized viaRAFT polymerization, was modified with N-succinimidyl 3-(2-pyridyldithio)-propionate (SPDP) to convert to the pendant amine groups to PDS functionality. 5′-sense strand thiolated RNAs were then coupled to the polymer through a disulfide exchange with pendant PDS moieties, giving a conjugation yield of 89 ± 4%.84 Here it is worth noting that the authors used single-strand RNA for conjugations to obtain relatively high conjugation yields. The complementary strand RNAs were then base-paired with RNA-polymer conjugates. The conjugation of macromolecules to pendant functionalized polymers usually results in low conjugation yields due to steric hindrance effects.
Except a few recent studies,84,95,100 the primary and secondary amine functionalities, have not been directly accessible viaRAFT polymerization mainly due to the degradation of RAFT agents during polymerization. This limitation has been overcome by several indirect routes including the protection of amine groups by tert-butyloxycarbonate (t-BOC) during polymerization.107
Recently, following a direct approach, Deng et al.96 synthesized diblock copolymers with 2-lactobionamidoethyl methacrylamide and 3-aminopropyl methacrylamide hydrochloride blocks via chain extension RAFT polymerization in water using azobis(4-cyanovaleric acid) (ACVA) as an initiator and 4-cyanopentanoic acid dithiobenzoate as a RAFT agent. The primary amine pendant groups of the copolymer were modified with biotinyl-N-hydroxysuccinimide ester to prepare glyconanoparticles for biomolecular recognition processes against avidin and a lectin. Similarly, Henry et al.97 used N-(2-aminoethyl)maleimide trifluoroacetate to introduce a single primary amine group to the omega-terminus of poly(dimethylaminoethyl methacrylate) and poly(N-isopropyl acrylamide) and also to a specialized block copolymer. Addition of a single maleimide monomer to the polymer allowed the functional group to be located at the junction of two blocks. The polymers were functionalized with an amine-reactive fluorescent dye or folic acid with a conjugation efficiency of 86 and 94%, respectively. It was noted that the triflate salt of N-(2-aminoethyl)maleimide (AM) prevented reduction of the RAFT agent during polymerization.
In studies by York et al.84,85 and Alidedeoglu et al.100 homo- and copolymers of a primary amine containing monomer, N-(3-aminopropyl)methacrylamide (APMA) were prepared in an aqueous acetic buffer (pH 5.2 to prevent degradation of dithiobenzoate functionality) using 4-cyanopentanoic acid dithiobenzoate as a RAFT agent. APMA provided amine functionality, allowing conjugation of cell-targeting folate derivatives and o-glucuronic acid.
These recent studies focussing on the synthesis of amine-functionalized RAFT polymers and their use for bioconjugations are significant as the degradation of RAFT agents in the presence of amines has been one of the main barriers to the use of RAFT technique for bioconjugations. Especially since carboxylic acid-amine reactions have been among the most widely used chemistries for bioconjugations, and various amine-reactive reagents (such as fluorescent probes) and biomolecules (such as proteins and peptides containing acidic residues) are widely available, the ability to synthesize amine-functionalized polymersvia the RAFT technique clearly makes the technique more practical for bioconjugation applications.
A copper-catalyzed azide-alkyne click reaction was used to couple a self-assembling, azide functionalized, cyclic octapeptide with an alkyne functionalized RAFT polymer.102 Following the same methodology, immunogenic peptides (a tetrapeptide and an eicosapeptide) from the cancer-associated glycoprotein MUC1 were conjugated with poly(NIPAAm).103 In this case, 3-(trimethylsilyl)prop-2-ynyl-2-(butylthiocarbonothioylthio) propanoate was used as a functional RAFT agent to directly synthesize poly(NIPAAm) with α-trimethylsilyl protected alkyne group.
While copper catalyzed click reactions are highly efficient, the use of metal catalyst may cause concerns for conjugations of proteins, peptides and DNA/RNA which contain moieties complexing with metals.141 In this context, thiol-ene additions present a better alternative path to bioconjugates.99,104,137 The thiocarbonylthio functionality of RAFT polymers has been in situ aminolyzed to thiol in the presence of ene-containing biomolecules such as maleimide-biotin and sugar (meth)acrylates which have led to highly efficient bioconjugations.56,67,69,79In situaminolysis approach avoids the formation of disulfide interchain couplings, usually observed with two-step aminolysis and conjugation strategy. It is worth noting that the use of tri-n-butyl phosphine as a reductant in the two-step strategy can also avoid the formation of disulfides.142
Aminooxy functionalized polymers can be conjugated to ketone or aldehyde engineered biomolecules via a click mechanism. A well-defined PNIPAAm (Mn = 4200 Da by NMR; PDI = 1.14 by GPC) was synthesized in the presence of a Boc-protected aminooxy trithiocarbonate chain transfer agent.100 Following the removal of the Boc group, the polymer’s aminooxy terminal was conjugated with N′-levulinyllysine-modified BSA in solution or aldehyde-modified heparin on a gold surface, forming oxime bonds. While the conjugationsvia this strategy are highly chemoselective and occur without side reactions with functional groups on proteins, they require proteins engineered with levulinic acid or aminooxyacetic acid143 to create ketone or aldehyde functionality on the protein surface.
Another example of protein–ligand interactions in RAFT-mediated bioconjugations was demonstrated by Chang et al.90 Reduced glutathione-modified PNIPAAm, synthezied using a RAFT agent modified with pyridyl disulfide and subsequently conjugated with glutathioneviathiol-disulfide exchange reaction, was used to capture glutathione-S-transferase (GST) via affinity binding. The polymer demonstrated specificity only for GST among other proteins such as BSA and lysozyme.
In an interesting study, Tominey et al.145 proposed the development of RAFT-synthesized polymeric artificial receptors for proteins. A number of functionalized monomers were copolymerized using a water-soluble symmetric trithiocarbonate RAFT agent. Cytochrome C, hemoglobin, BSA, histone, lysozyme, proteinase K were tested for selective binding to RAFT polymers. The pair with the highest affinity was found to be a bisphosphonate-containing RAFT polymer and histidine-rich histone (Kd = 16 nM). While RAFT polymerization appears to be the most amenable technique for the generation of biomimetic structures for such precise applications, the polymerization conditions need to be optimized very carefully to minimize the polydispersity of the polymers produced, as polydispersity, in this case, is extremely critical for the binding events due to the different affinities and binding stoichiometries of polymers of different chain length.
Other than the (strept)avidin–biotin pair, there are only a few other protein–ligand pairs, as detailed above, that have been investigated in RAFT-mediated bioconjugation strategies. Cofactor reconstitutions, metal-protein ligand coordinations, dye-affinity interactions remain to be investigated for bioconjugation of RAFT polymers using non-covalent interactions.
2.2 Bioconjugations via in situpolymerization strategy
An alternative route to well-defined bioconjugates, first proposed by Maynard and co-workers for ATRP-mediated bioconjugations,127,128 is the in situ generation of conjugates by polymerization of biomolecule-modified RAFT agents or biomolecule-modified monomers. The in situpolymerization strategy has a number of advantages over the post-polymerization conjugation strategy: (i) the overall number of synthetic steps are reduced, (ii) it is easier to control the number and conjugation site of the attached polymer, (iii) purification steps are simplified, (iv) the conjugation yields are higher, (v) better-defined bioconjugates with greatly reduced heterogeneity are obtained. Together with these advantages, an important issue with the in situpolymerization approach is that although the number of overall synthetic steps is less, there are more steps involving the biomolecule in comparison with the post-polymerization conjugation strategy. Thus the treatment conditions (for example: solvent, temperature, radical concentration, presence of salts) during the steps involving fragile biomolecules such as proteins, large peptides and DNA/RNA, need to be carefully optimized to preserve the conformation and activity of the biomolecule. The same concern is also valid for the preservation of thiocarbonylthio structure in the presence of certain biomolecules such as amine deprotected peptides.The advantages of RAFT polymerization, important for bioconjugations, over the other living radical polymerization techniques can be listed as follow: The variety of the monomers that can be polymerized by RAFT is large. The use of metal catalysts and additional agents such as sacrifical initiators is not needed. The synthesis of well-defined biomolecule-polymer conjugates in easily detectable quantities using only RAFT agent-modified biomolecules and common free radical initiators is possible.
Peptide,146–149protein134,150,151 or DNA-modified152 RAFT agents have been used for in situ bioconjugate formation viaRAFT polymerization. The in situRAFT polymerization approach using peptide-RAFT agents has been performed only with simple and protected peptides, mainly due to the degradation problem of the RAFT agents in the presence of deprotected peptides. However, recently, a few studies reported the successful RAFT polymerization of monomers functionalized with deprotected, longer chain peptides (vide infra).153,154
The synthesis of protein-macroRAFT agents (Fig. 5) was performed using a selective thiol-disulfide exchange reaction of Z-group pyridyl disulfide-functionalized RAFT agents with the single non-oxidized cysteine (cys-34) residue of BSA.134,151 Using BSA-RAFT agents, BSA-PNIPAAm, BSA-POEG-A, BSA-PHEA conjugates were in situ generated at ambient temperatures using azo-initiators or γ-radiation. De et al.150 used the R-approach for modification of the RAFT agent with BSA (Fig. 5) viathiol-maleimide addition reaction. Disulfide bonds present in native BSA were reduced to increase the number of free thiols per protein, providing multiple attachment sites. The resultant BSA-macroRAFT agent was used to control the polymerization of NIPAAm at room temperature using similar conditions to those reported previously.151 The R-group-protein-modified RAFT agent design reduces steric hindrance during polymerization while the Z-group- modified RAFT agent design is affected by steric hindrance and may reduce the polymerization efficiency. Also, conjugating via Z-approach results in hydrolyzable conjugates, due to the presence of the labile C–S bond between the biomolecule and the polymer chain, which might be favorable for certain applications.
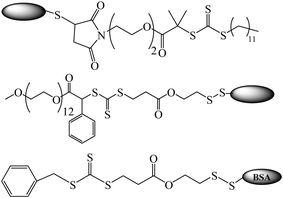 | ||
| Fig. 5 Bovine serum albumin (BSA)-macro RAFT agents.134,150,151 | ||
Peng and Lin152 reported the synthesis of surface-anchored DNA-RAFT agent to prepare a DNA-b-polymer-grafted gold surface. An N-hydroxysuccinimide activated trithiocarbonate RAFT agent (2-(1-carboxy-1-methylethylsulfanylthiocarbonylsulfanyl)-2-methyl propionic acid) was attached to 5′-amino, 3′-disulfide-functionalized single stranded DNA through its 5′-amino end. The attachment of the DNA-RAFT agent to the gold surface was performed after the reduction of the 3′-disulfide group of DNA by DTT addition. Importantly, this step did not degrade the RAFT agent structure or caused any unwanted interaction between the RAFT agent and the gold surface. It was previously shown131 that a PDS-functionalized trithiocarbonate RAFT agent can bind to gold surface viadisulfide reduction and gold-thiol binding preserving the trithiocarbonate group intact. The OEG-MA monomer was polymerized from the surface-grafted DNA-RAFT agent under unconventional polymerization conditions (using AIBN as an initiator in water at 30 °C for 5 h).152Free radical polymerization in solution, as the side reaction, was found to increase with raised temperature, consequently no polymer growth was observed on the surface at higher polymerization temperatures. An important observation was that the grafting kinetics in the presence of DNA molecules, even at a relatively low grafting density, was faster, compared to the surface modified with a small molecule coupled-RAFT agent.
RAFT polymerization of biomolecule-modified monomers has also been investigated to prepare a number of biomolecule-polymer pendant conjugates, usually using saccharide residues82,96,97,155–157 and peptides153,154,158 as a biomolecule. In a study aimed to develop a gene carrier,154methacrylamide monomers of a DNA condensing peptide (K-12) and an endosomal escape peptide (K6H5) were RAFT-copolymerized with N-(2-hydroxypropyl(methacrylamide) (HPMA) under aqueous conditions using ethyl cyanovaleric trithiocarbonate as a RAFT agent and VA-044 as an initiator in acetate buffer (pH 5.1) at 44 °C for 48 h. An important note is that the peptides used in polymerizations were deprotected. An acetic acid buffer at pH 5.1 with a molar strength of 1 M was used to ensure that ε-amines of L-lysine were fully protonated, thereby protecting the trithiocarbonate from nucleophilic attack. Statistical copolymers with highly controlled molecular weight and composition were obtained. The copolymers efficiently condensed DNA into small particles which were stable even in a physiologically-relevant salt solution. With increasing peptide content, the peptide-based polymers demonstrated gene delivery efficiencies to HeLa cells that were comparable to branched polyethylenimine.
Conclusions
The strength of the RAFT approach for generation of polymer bioconjugates lies in its ability to control the polymerization of a wide range of monomers in varying solvents including water, at moderate temperatures, using only chain transfer agents and common free radical initiators. The simple set up of RAFT polymerization and the commercial availability of RAFT agents make the RAFT technique accessible to researchers from different fields including biotechnology and biomedicine and most amenable to the generation of bioconjugates with improved properties. Accordingly, RAFT-mediated strategies have been increasingly used to prepare biomolecule-polymer conjugates with well-defined composition, size, molecular architecture and conjugation site. Especially with the combination of orthogonal chemistries with RAFT polymerization, access to well-defined conjugates with designed architectures at high yields has been possible. These synthetic abilities envisage new or improved applications of biomolecule-polymer conjugates. The impact of the RAFT technique on preparation of bioconjugates should be significant, particularly for drug delivery, diagnostics/biosensors and biopurifications where the physicochemical features of bioconjugates need to be tuned very precisely to have desirable and consistent performance.References
- W. P. Gao, W. G. Liu, T. Christensen, Z. M. R. Zalutsky and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16432 CrossRef CAS.
- W. P. Gao, W. G. Liu, J. A. Mackay, M. R. Zalutsky, E. J. Toone and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15231 CrossRef CAS.
- J. M. Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539 CrossRef CAS.
- S. M. Ryan, X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51 CrossRef CAS.
- Z. L. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. H. Chen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472 CrossRef CAS.
- S. F. M. Van Dongen, M. Nallani, J. Cornelissen, R. J. M. Nolte and J. C. M. Van Hest, Chem.–Eur. J., 2009, 15, 1107 CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263 CrossRef CAS.
- K. L. Christman, E. Schopf, R. M. Broyer, R. C. Li, Y. Chen and H. D. Maynard, J. Am. Chem. Soc., 2009, 131, 521 CrossRef CAS.
- K. L. Christman, R. M. Broyer, Z. P. Tolstyka and H. D. Maynard, J. Mater. Chem., 2007, 17, 2021 RSC.
- G. Pasut and F. M. Veronese, Adv. Drug Delivery Rev., 2009, 61, 1177 CrossRef CAS.
- A. Monzur and S. Brocchini, Adv. Drug Delivery Rev., 2006, 58, 1671 CrossRef.
- G. N. Grover and H. D. Maynard, Curr. Opin. Chem. Biol., 2010, 14, 818 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- K. Velonia, Polym. Chem., 2010, 1, 944 RSC.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563 RSC.
- A. K. Shakya, H. Sami, A. Srivastava and A. Kumar, Prog. Polym. Sci., 2010, 35, 459 CrossRef CAS.
- L. A. Canalle, D. W. P. M. Lowik and J. C. M. Van Hest, Chem. Soc. Rev., 2010, 39, 329 RSC.
- F. H. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 218.
- J. H. Jeong, H. Mok, Y. K. Oh and T. G. Park, Bioconjugate Chem., 2009, 20, 5 CrossRef CAS.
- J. F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958 CrossRef CAS.
- J. F. Lutz and H. G. Borner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS.
- A. S. Hoffman and P. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. B. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS.
- R. Epton, G. Marr and G. J. Morgan, Polymer, 1977, 18, 319 CrossRef CAS.
- R. Epton, M. E. Hobson and G. Marr, Polymer, 1977, 18, 1203 CrossRef CAS.
- A. Abuchowski, T. Vanes, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CAS.
- G. H. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509 CrossRef CAS.
- Z. L. Ding, G. H. Chen and A. S. Hoffman, Bioconjugate Chem., 1996, 7, 121 CrossRef CAS.
- T. Shimoboji, Z. L. Ding, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 2002, 13, 915 CrossRef CAS.
- W. N. E. Van Dijk-Wolthuis, P. van de Wetering, W. L. J. Hinrichs, L. J. F. Hofmeyer, R. M. J. Liskamp, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 1999, 10, 687 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS.
- S. Shaunak, A. Godwin, J.-W. Choi, S. Balan, E. Pedone, D. Vijayarangam, S. Heidelberger, I. Teo, M. Zloh and S. Brocchini, Nat. Chem. Biol., 2006, 2, 161 219, 221.
- C. Fleming, A. Maldjian, D. Da Costa, A. K. Rullay, D. M. Haddleton, J. St. John, P. Penny, R. C. Noble, N. R. Cameron and B. G. Davis, Nat. Chem. Biol., 2005, 1, 270 CrossRef CAS.
- A. J. Dirks, R. J. M. Nolte and J. Cornelissen, Adv. Mater., 2008, 20, 3953 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- G. Moad, M. Chen, M. Haussler, A. Postma, E. Rizzardo and S. H. Thang, Polym. Chem., 2011, 2, 492 RSC.
- J. Q. Liu, H. Y. Liu, V. Bulmus, L. Tao, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1399 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551 CrossRef CAS.
- L. J. Wong, S. Sevimli, H. M. Zareie, T. P. Davis and V. Bulmus, Macromolecules, 2010, 43, 5365 CrossRef CAS.
- S. Kirkland-York, K. Gallow, J. Ray, Y.-L. Loo and C. Mccormick, Soft Matter, 2009, 5, 2179 RSC.
- M. G. Kellum, A. E. Smith, S. K. York and C. L. Mccormick, Macromolecules, 2010, 43, 7033 CrossRef CAS.
- A. E. Smith, X. W. Xu, D. A. Savin and C. L. Mccormick, Polym. Chem., 2010, 1, 628 RSC.
- A. E. Smith, X. W. Xu, S. E. Kirkland-York, D. A. Savin and C. L. Mccormick, Macromolecules, 2010, 43, 1210 CrossRef CAS.
- E. Schopf, R. Broyer, L. Tao, Y. Chen and H. D. Maynard, Chem. Commun., 2009,(32), 4818 RSC.
- J. Q. Liu, L. Tao, W. R. Yang, D. Li, C. Boyer, R. Wuhrer, F. Braet and T. P. Davis, Langmuir, 2010, 26, 10068 CrossRef CAS.
- M. Glassner, J. P. Blinco and C. Barner-Kowollik, Polym. Chem., 2011, 2, 83 RSC.
- W. M. Wan and C. Y. Pan, Polym. Chem., 2010, 1, 1475 RSC.
- H. Willcock and R. K. O'reilly, Polym. Chem., 2010, 1, 149 RSC.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, 2008, Wiley-VCH, Weinheim Search PubMed.
- X. P. Qiu and F. M. Winnik, Macromolecules, 2007, 40, 872 CrossRef CAS.
- B. Yu, J. W. Can, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493 CrossRef.
- W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. N. Cao and Z. S. An, Macromol. Rapid Commun., 2010, 31, 1444 CrossRef CAS.
- S. Harrisson, Macromolecules, 2009, 42, 897 CrossRef CAS.
- M. I. Gibson, E. Frohlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2009, 46, 4332 CrossRef.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Muller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562 CrossRef CAS.
- A. N. Zelikin, G. K. Such, A. Postma and F. Caruso, Biomacromolecules, 2007, 8, 2950 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, P. Zhang, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Muller, Macromolecules, 2009, 42, 3707 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346 CrossRef CAS.
- B. Sasso, M. Dobinson, P. Hodge and T. Wear, Macromolecules, 2010, 43, 7453 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus and T. P. Davis, Aust. J. Chem., 2009, 62, 830 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302 CrossRef CAS.
- J. Xu, L. Tao, J. Liu, V. Bulmus and T. P. Davis, Macromolecules, 2009, 42, 6893 CrossRef CAS.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009,(24), 3580 RSC.
- F. D. Jochum, P. J. Roth, D. Kessler and P. Theato, Biomacromolecules, 2010, 11, 2432 CrossRef CAS.
- K. T. Wiss, D. Kessler, T. J. Wendorff and P. Theato, Macromol. Chem. Phys., 2009, 210, 1201 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860 CrossRef CAS.
- J. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2011, 44, 299 CrossRef CAS.
- M. Barz, F. Canal, K. Koynov, R. Zentel and M. J. Vicent, Biomacromolecules, 2010, 11, 2274 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009,(40), 6029 RSC.
- H. T. T. Duong, T. L. U. Nguyen and M. H. Stenzel, Polym. Chem., 2010, 1, 171 RSC.
- K. Godula and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 9963 CrossRef CAS.
- K. Godula, D. Rabuka, K. T. Nam and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 4973 CrossRef CAS.
- A. L. Golden, C. F. Battrell, S. Pennell, A. S. Hoffman, J. J. Lai and P. S. Stayton, Bioconjugate Chem., 2010, 21, 1820 CrossRef CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238 CrossRef CAS.
- A. W. York, F. Q. Huang and C. L. McCormick, Biomacromolecules, 2010, 11, 505 CrossRef CAS.
- A. W. York, Y. L. Zhang, A. C. Holley, Y. L. Guo, F. Q. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936 CrossRef CAS.
- L. Tao, C. S. Kaddis, R. R. O. Loo, G. N. Grover, J. A. Loo and H. D. Maynard, Macromolecules, 2009, 42, 8028 CrossRef CAS.
- L. Tao, J. Q. Liu and T. P. Davis, Biomacromolecules, 2009, 10, 2847 CrossRef CAS.
- L. Tao, J. T. Xu, D. Cell and T. P. Davis, Macromolecules, 2010, 43, 3721 CrossRef CAS.
- C. W. Chang and H. D. Maynard, Macromol. Rapid Commun., 2010, 31, 1691 CrossRef CAS.
- C. L. Duvall, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Mol. Pharmaceutics, 2010, 7, 468 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360 CrossRef CAS.
- K. L. Heredia, L. Tao, G. N. Grover and H. D. Maynard, Polym. Chem., 2010, 1, 168 RSC.
- L. J. Wong, C. Boyer, Z. Jia, M. H. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934 CrossRef CAS.
- L. J. Wong, M. Kavallaris and V. Bulmus, Polym. Chem., 2011, 2, 385 RSC.
- Z. C. Deng, M. Ahmed and R. Narain, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 614 CrossRef CAS.
- Z. C. Deng, S. Q. Li, X. Z. Jiang and R. Narain, Macromolecules, 2009, 42, 6393 CrossRef CAS.
- S. M. Henry, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 1122 CrossRef CAS.
- M. Li, P. De, H. M. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854 RSC.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650 CrossRef CAS.
- A. H. Alidedeoglu, A. W. York, D. A. Rosado, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3052 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Aust. J. Chem., 2010, 63, 1169 CrossRef CAS.
- H. Kakwere, H. C. K. Y. Chun, K. A. Jolliffe, R. J. Payne and S. Perrier, Chem. Commun., 2010, 46, 2188 RSC.
- N. Kanayama, H. Shibata, A. Kimura, D. Miyamoto, T. Takarada and M. Maeda, Biomacromolecules, 2009, 10, 805 CrossRef CAS.
- K. T. Wiss and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4758 CrossRef CAS.
- Z. F. Jia, J. Q. Liu, C. Boyer, T. P. Davis and V. Bulmus, Biomacromolecules, 2009, 10, 3253 CrossRef CAS.
- Z. Jia, L.-J. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106 CrossRef CAS.
- W. H. Liu, A. B. Greytak, J. Lee, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 472 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754 CrossRef CAS.
- C. W. Scales, Y. A. Vasilieva, A. J. Convertine, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2005, 6, 1846 CrossRef CAS.
- R. Wang, C. L. McCormick and A. B. Lowe, Macromolecules, 2005, 38, 9518 CrossRef CAS.
- Y. A. Vasilieva, D. B. Thomas, C. W. Scales and C. L. McCormick, Macromolecules, 2004, 37, 2728 CrossRef CAS.
- A. Aqil, H. J. Qiu, J. F. Greisch, R. Jerome, E. De Pauw and C. Jerome, Polymer, 2008, 49, 1145 CrossRef CAS.
- K. A. Aamer and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5618 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008,(28), 3245 RSC.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009,(10), 1198 RSC.
- L. Zhang and Y. Chen, Polymer, 2006, 47, 5259 CrossRef CAS.
- D. L. Patton and R. C. Advincula, Macromolecules, 2006, 39, 8674 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063 CrossRef CAS.
- N. C. Strandwitz, A. Khan, S. W. Boettcher, A. A. Mikhailovsky, C. J. Hawker, T.-Q. Nguyen and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 8280 CrossRef CAS.
- R. Karunakaran and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4254 CrossRef CAS.
- E. Bays, L. Tao, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 1781.
- S. N. S. Alconcel, G. N. Grover, N. M. Matsumoto and H. D. Maynard, Aust. J. Chem., 2009, 62, 1496 CrossRef CAS.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, New York, 1996 Search PubMed.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. S. Hoffman, J. Controlled Release, 2003, 93, 105 CrossRef CAS.
- J. Liu, V. Bulmus, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Macromol. Rapid Commun., 2007, 28, 305 CrossRef CAS.
- H. M. Zareie, C. Boyer, V. Bulmus, E. Nateghi and T. P. Davis, ACS Nano, 2008, 2, 757 CrossRef CAS.
- C. Boyer, J. Liu, L.-J. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207 CrossRef.
- J. Liu, H. Liu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 899 CrossRef CAS.
- J. Liu, V. Bulmus, D. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641 CrossRef CAS.
- K. Gunasekaran, T. H. Nguyen, H. Maynard, T. P. Davis and V. Bulmus, Macromol. Rapid Commun., 2011, 32, 654 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889 CrossRef CAS.
- V. Lima, X. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. Van Der Linde, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 959 CrossRef CAS.
- M. Dietrich, M. Glassner, T. Gruendling, C. Schmid, J. Falkenhagen and C. Barner-Kowollik, Polym. Chem., 2010, 1, 634 RSC.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromol. Rapid Commun., 1999, 32, 4826 CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724 CrossRef CAS.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Y. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler- Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. H. Kent and J. A. Bradburne, Science, 2003, 299, 884 CrossRef CAS.
- C. Y. Hong and C.-Y. Pan, Macromolecules, 2006, 39, 3517 CrossRef CAS.
- A. F. Tominey, J. Liese, S. Wei, K. Kowski, T. Schrader and A. Kraft, Beilstein J. Org. Chem., 2010, 6, 7 Search PubMed.
- J. Hentschel, M. G. J. ten Cate and H. G. Borner, Macromolecules, 2007, 40, 9224 CrossRef CAS.
- M. G. J. Ten Cate and H. G. Borner, Macromol. Chem. Phys., 2007, 208, 1437 CrossRef CAS.
- J. Hentschel, K. Bleek, O. Ernst, J. F. Lutz and H. G. Borner, Macromolecules, 2008, 41, 1073 CrossRef CAS.
- Y. Zhao and S. Perrier, Chem. Commun., 2007, 4294 RSC.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS.
- H. Peng and H. Lin, Biomacromolecules, 2009, 10, 1804 CrossRef CAS.
- B. Apostolovic and H. A. Klok, Biomacromolecules, 2010, 11, 1891 CrossRef CAS.
- R. N. Johnson, R. S. Burke, A. J. Convertine, A. S. Hoffman, P. S. Stayton and S. H. Pun, Biomacromolecules, 2010, 11, 3007 CrossRef CAS.
- H. Kitano, A. Hayashi, H. Takakura, H. Suzuki, N. Kanayama and Y. Saruwatari, Langmuir, 2009, 25, 9361 CrossRef CAS.
- M. Hetzer, G. J. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119 CrossRef CAS.
- X. Z. Jiang, A. Housni, G. Gody, P. Boullanger, M. T. Charreyre, T. Delair and R. Narain, Bioconjugate Chem., 2010, 21, 521 CrossRef CAS.
- F. Audouin, R. J. I. Knoop, J. Huang and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4602 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |