FDA-approved poly(ethylene glycol)–protein conjugate drugs
Steevens N. S.
Alconcel
a,
Arnold S.
Baas
*b and
Heather D.
Maynard
*a
aDepartment of Chemistry & Biochemistry and the California NanoSystems Institute, University of California, 607 Charles E. Young Dr East, Los Angeles, CA 90095, USA. E-mail: maynard@chem.ucla.edu
bRonald Reagan UCLA Medical Center, Division of Cardiology, 757 Westwood Plaza, Los Angeles, CA 90095, USA. E-mail: abaas@mednet.ucla.edu
First published on 27th April 2011
Abstract
PEGylation or covalent attachment of poly(ethylene glycol) improves the pharmacokinetic properties of protein drugs. In vivo circulation lifetimes are increased and dosages are decreased, resulting in improved patient quality of life. PEG may be attached to proteins using a variety of different chemical reactions. This review discusses currently available FDA-approved PEGylated protein drugs, their intended use and target, and the PEG attachment chemistry utilized.
 Steevens N. S. Alconcel | Steevens N. S. Alconcel received a BS in Chemistry in 2007 from the University of Southern California, where he worked with Professor G. K. Surya Prakash on modification of Strecker and Mitsunobu reactions to synthesize fluorinated analogues. He is currently working toward his PhD in organic chemistry at the University of California, Los Angeles. Steevens is a trainee in the NIH-sponsored Chemistry–Biology Interface Training Program. His research focuses on the synthesis of protein–polymer conjugates for drug delivery. |
 Arnold S. Baas | Arnold S. Baas received his BA from Baylor University in 1985 and his MD from UT Southwestern in 1989. He was resident, followed by chief resident at the University of Michigan before training as a fellow in cardiology at the University of Washington. Baas was a clinical assistant professor there before moving to UCLA to the David Geffen School of Medicine and Division of Cardiology. Baas' interests are focused on general cardiology, cardiac transplantation, mechanical circulatory support, cardiomyopathy, atherosclerosis, cardiac rehabilitation, and cardiac imaging. |
 Heather D. Maynard | Heather D. Maynard received her BS degree from UNC-Chapel Hill in chemistry and an MS from UCSB in Materials Science. Maynard's PhD in chemistry was awarded in 2000 from Caltech, and she was an American Chemical Society postdoctoral fellow at the ETH Switzerland from 2000–2002. In 2002, Maynard moved to UCLA where she is now an Associate Professor in the Department of Chemistry and Biochemistry. Her interests include polymer synthesis, biohybrid materials, and surface modification. |
Introduction to PEGylation
PEGylation, or covalent attachment of poly(ethylene glycol) (PEG) to proteins, was introduced by Abuchowski and coworkers in 1977.1 They reported on the superior immunogenic properties of bovine serum albumin (BSA)–PEG conjugates compared to the unmodified biomolecule. In later work Abuchowski and coworkers were able to demonstrate that PEGylation resulted in enhanced circulation lifetimes, as well as reduced immunogenicities relative to native proteins.2 Since that time, many researchers have demonstrated that PEGylation improves pharmacological properties of proteins. As a result, several FDA-approved protein–polymer drugs currently exist, and many others are currently being studied in clinical trials.3 This review focuses on the synthesis and mode of action of protein–PEG conjugates that are already FDA-approved for use to treat diseases in humans.The main objective of using polymers in drug delivery is to stabilize and improve the therapeutic activity of the standalone drug.4–6 The non-ionic, hydrophilic PEG provides a steric shield for conjugates from recognition by the patient's immune system and effectively increases the size of the biomolecule,7 thereby reducing clearance from the bloodstream.8 By increasing the half-life of the protein drug, the dosage frequency is reduced.9 Since most protein drugs need to be injected, this result is significant. Increased duration of pharmacological activity, reduction of toxic side effects, and increased quality of life due to controlled, timed release are several positive results attributed to PEGylation. The detailed pharmacokinetics of PEGylated drugs have been recently reviewed.10 The dosage schedule and biological activity for each of the FDA-approved protein-PEG drugs are described in this review.
PEGylation may also decrease parent drug activity by changing conformation, sterically interfering, and altering electrostatic binding properties.3 Notably, the shape and number of PEG chains conjugated to a protein affects the stability and efficacy of the drug conjugate. Chain location is also critical to activity, as nonspecific PEGylation near an active site or to a region of a protein that causes a conformational change deleteriously affects the activity of the intended drug.9 As of today, there are well-established methods of PEGylation available,7 with a large array of different linking chemistries used.3,4,6,7,11,12 There has been a lot of recent effort placed on strategies that result in the site-specific conjugation.13 However, for many FDA-approved drugs, non-specific attachment strategies are used; these are described below and summarized in Table 1.
| Drug | PEG attachment | PEG size | Protein name | Site(s) of attachment | Native half-life | Conjugate half-life |
|---|---|---|---|---|---|---|
| Adagen |
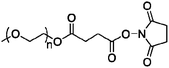
|
5 kDa | Adenosine deaminase | Nonspecific, lysine, serine, tyrosine, histidine | — | — |
| Oncaspar |
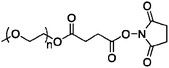
|
5 kDa | L-Asparaginase | Nonspecific, lysine, serine, tyrosine, histidine | 20 h | 357 h |
| Krystexxa |

|
10 kDa (10–11 units) | Mammalian urate oxidase (monomer) | Nonspecific, lysines | 4 h | 154–331 h |
| PEGASYS |
|
40 kDa | Interferon alfa-2a | Lysines 31, 121, 131, or 134 | 3–8 h | 65 h |
| PEG-INTRON |
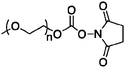
|
12 kDa | Interferon alfa-2b | Histidine 34 (major) | 7–9 h | 48–72 h |
| Cimzia |

|
40 kDa | Anti-TNFα Fab' | C-Terminal cysteine | — | — |
| Neulasta |
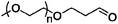
|
20 kDa | G-CSF | N-Terminal methionine | 3.5–3.8 h | 42 h |
| Mircera |
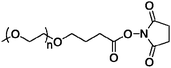
|
30 kDa | Continuous erythropoietin receptor activator | Lysine 52 or 46 | 7–20 h | 134–139 h |
| Somavert |
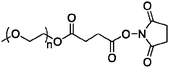
|
5 kDa (4–6 units) | hGH antagonist B2036 | Nonspecific, lysines, N-terminal phenylalanine | — | — |
FDA-approved protein–polymer drugs
I. Enzymes
Initially, PEG was synthesized by reacting mPEG (monomethoxy-PEG) of molecular weight (Mw) 5000 with cyanuric chloride, forming PEG dichlorotriazine. In this approach, one of the two remaining chlorines on PEG dichlorotriazine is displaced by nucleophilic amino acid units such as lysine, serine, tyrosine, cysteine, and histidine. The remaining chlorine is not as electrophilic, but may react to cause crosslinking of the protein. This is a nonspecific attachment process so multiple units of PEG are attached to the protein. Depending on the pH of the buffer used in the reaction, a small measure of control can be obtained to conjugate to specific nucleophilic amino acid units.1,14 This PEG was utilized at first by the company to study the drug, but is not what is used in the FDA-approved formulation.
The FDA-approved protein drug is synthesized using PEG succinimidyl succinate.7 This compound is prepared by reacting Mw 5000 mPEG with succinic anhydride. The resulting carboxylic acid is activated with N-hydroxysuccinimide (NHS) by carbodiimide coupling. The NHS group is displaced by nonspecific reaction with nucleophilic amino acid side chains.17
The protein–polymer conjugate is also synthesized by the use of PEG succinimidyl succinate. The NHS group is displaced by nucleophilic amino acid units such as lysine, serine, cysteine, tyrosine, and histidine. The PEG ester and thioesters are highly susceptible to hydrolysis and thus modification occurs primarily at the amines forming amides.7,17
The protein–polymer conjugate is synthesized by the use of a PEG p-nitrophenyl carbonate ester. The p-nitrophenol is displaced by primary amine lysine side chains, forming carbamates, which are subject to slow hydrolysis under mildly basic conditions.28 Each subunit of urate oxidase contains a total of 28–29 lysines but only approximately 12 are surface-accessible in the native tetrameric form of the complete enzyme. In fact, some of the lysine residues are sufficiently close together that PEGylation of one lysine may sterically hinder the addition of another PEG chain. In total, each subunit of urate oxidase contains an average of 10–11 PEG units after conjugation.25
II. Cytokines
A branched PEG is attached covalently to interferon alfa-2a at lysine side chains through a stable amide bond. The Mw 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 branched PEG consists of two chains, each with an average Mw of 20000. To synthesize this linker (Fig. 1), monomethoxy PEG is reacted with N,N′-disuccinimidyl carbonate, resulting in PEG N-succinimidyl carbonate. Linking the PEG is accomplished by a one pot reaction with the side chain and N-terminal amine groups of lysine, forming a bis-carbamate.32 Finally, the carboxylic acid is activated with NHS, giving the branched PEG chain linker capable of forming stable amide bonds with 11 possible lysine residues. Four main sites of PEGylation have been identified, Lys 31, 121, 131, and 134. The reaction can be manipulated to favor formation of the monosubstituted conjugate by limiting the amount of PEG chain linker used in the conjugation step. The monosubstituted conjugate is then purified using ion-exchange chromatography, with 95–99% monoPEGylated conjugates.30
000 branched PEG consists of two chains, each with an average Mw of 20000. To synthesize this linker (Fig. 1), monomethoxy PEG is reacted with N,N′-disuccinimidyl carbonate, resulting in PEG N-succinimidyl carbonate. Linking the PEG is accomplished by a one pot reaction with the side chain and N-terminal amine groups of lysine, forming a bis-carbamate.32 Finally, the carboxylic acid is activated with NHS, giving the branched PEG chain linker capable of forming stable amide bonds with 11 possible lysine residues. Four main sites of PEGylation have been identified, Lys 31, 121, 131, and 134. The reaction can be manipulated to favor formation of the monosubstituted conjugate by limiting the amount of PEG chain linker used in the conjugation step. The monosubstituted conjugate is then purified using ion-exchange chromatography, with 95–99% monoPEGylated conjugates.30
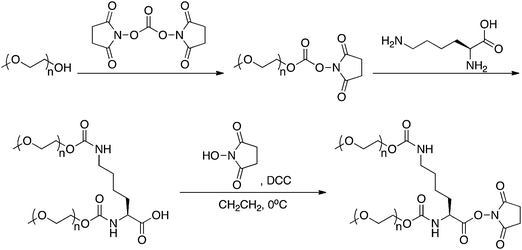 | ||
| Fig. 1 Synthesis of the branched PEG used to prepare PEGASYS. | ||
PEG-INTRON is a covalent conjugate of interferon alfa-2b linked to a single unit of Mw 12000 PEG. Monomethoxy-PEGN-succinimidyl carbonate is subject to nucleophilic attack from several possible amino acid residues.36 Histidines, serines, and lysines are possible PEG conjugation sites. In fact, there are 14 identified sites of PEGylation. The major isomer (47%) of PEG-INTRON is PEGylated at His 34.34 The His 34 conjugate is most active, perhaps because of its location in the protein sequence which is not involved in receptor recognition.37,38 In addition, the His 34 conjugate slowly hydrolyzes and releases interferon alfa-2b over time.37
III. Antibodies
The active substance of Cimzia sold by UCB is certolizumab pegol consisting of a recombinant Fab′ antibody fragment obtained and purified from E. coli expression. The fragment is covalently bound to a Mw 40000 branched PEG chain through a C-terminal cysteine reaction with maleimide.43 The synthesis of the branched PEG chain linker is as described for PEG-INTRON (vide supra). An additional step is included at the end, as an amino-maleimide linker is coupled to the lysine carboxylic acid.44
IV. Growth factors
Neulasta is a PEGylated G-CSF synthesized by covalent attachment of Mw 20000 monomethoxy-PEG aldehyde by reductive amination with the N-terminal methionine residue of the filgrastim protein. Targeting the N-terminal residue is accomplished by exploiting the lower pKa of this amine compared to the other amines such as lysines. Aldehyde-containing monomethoxy-PEG forms an imine with the N-terminal methionine residue and is subsequently reduced with sodium cyanoborohydride (Fig. 2).45,50
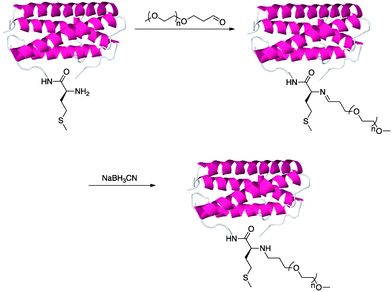 | ||
| Fig. 2 Synthesis of Neulasta by imine formation and reductive amination with sodium cyanoborohydride. | ||
The PEG–CERA conjugate is synthesized by attachment of an NHS activated monomethoxy-PEG butanoic acid to Lys 46 and 52 on EPO.52,53,55
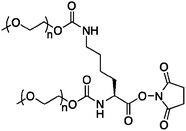
The pegvisomant conjugate sold by Pfizer is synthesized by covalent attachment of four to six Mw 5000 PEG units via NHS displacement as described for Adagen (vide supra) to several lysine residues available on hGH antagonist B2036, as well as the N-terminal phenylalanine residue. Possible sites of attachment include lysines 38, 41, 70, 115, 120, 140, 145, and 158, and phenylalanine 1.60,63–65
Conclusions and outlook
Covalent attachment of PEG to proteins produces conjugates with significantly improved pharmacokinetic properties compared to the unmodified proteins. Specifically, PEG reduces clearance, slows enzymatic degradation and provides an effective shield from the immune system. Proteins must be administered by subcutaneous or intravenous injection, and longer half-lives lead to fewer injections. As a result there are a number of FDA-approved PEG–protein conjugates that have been reviewed herein with many other PEGylated drugs currently in phase I, II, and III trials that were not described.3,6,43 There are several chemistries that are utilized in FDA-approved PEG drugs, including amidation, reductive amination, and Michael addition, although most target multiple residues.Nonspecific reaction of PEG with proteins leads to heterogeneous mixtures of multiple PEGs attached at different sites. This can lead to reductions in protein bioactivity. As a result, there has been a lot of effort placed on developing specific modification chemistries. An obvious choice is covalent attachment to free cysteines, as described above for Cimzia. The free cysteines can be naturally occurring or recombinantly placed away from the active site of a protein. PEGs with activated disulfide, maleimide, and vinyl sulfone end groups have been utilized for this purpose. In addition, proteins have either been modified chemically or produced recombinantly with artificial amino acids to present groups that react chemoselectively with PEGs.67,68 The field of incorporating a non-canonical amino acid and subsequent modification with PEG is still in the early stages of investigation. However, examples of chemistries that have been utilized are oxime bond formation and azide/alkyne click chemistries.69–71 These strategies produce homogeneous conjugates that retain bioactivity and are likely to play an increasing role in the protein–drug area.
PEG is classified to be Generally Regarded As Safe (GRAS) by the FDA and has been shown to be removed by renal clearance. Human studies show urinary excretion of PEG units up to 20 kDa, with up to 190 kDa PEG seen excreted in mouse and rat studies.3,66 However, there are potential limitations of PEG including its non-degradability in the body, and these have been recently reviewed.3 It has been discussed that PEG of molecular weights below 40–60 kDa are required to minimize accumulation in the liver72 making it advantageous to utilize smaller branched PEGs, cleavable by hydrolysis, instead of larger, nondegradable linear PEGs to form the conjugates. Branched PEG chains may provide improved shielding from the immune system, reducing immunogenicity and antigenicity further, thus minimizing the size of the PEG required. In recent years controlled radical polymerization (CRP) techniques have been increasingly utilized to synthesize these branched PEG polymers. In particular, atom transfer radical polymerization (ATRP)73–75 and reversible addition–fragmentation chain transfer (RAFT) polymerization76–78 have been employed to make end chain reactive branched PEGs12,79,80 and to form bioconjugates directly by polymerization from modified proteins.81,82 In fact, recent studies have exploited the latter approach to polymerize branched poly(ethylene glycol) methyl ether methacrylate (PEGMA) in situ from therapeutic proteins such as recombinant human growth hormone (rh-GH)83 and an intein fusion protein.84 Importantly, this grafting from approach retains protein activity in many cases and eliminates the need for postpolymerization conjugation. These strategies lead to conjugates that in some cases are superior to their PEGylated analogs.85 Recent reviews have summarized CRP approaches to bioconjugation.3,79,80,86 It is anticipated that CRPs will be increasingly utilized in the future to synthesize protein–polymer drugs.
Acknowledgements
The authors appreciate the National Institutes of Health (NCI R21 CA 137506-02) for funding. S.N.S.A. thanks the NIH sponsored Chemistry and Biology Interface (CBI) Training Program (2T32GM008496-16) for a fellowship.References
- A. Abuchowski, T. Vanes, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578–3581 CAS.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. Vanes and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS.
- J. M. Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539–551 CrossRef CAS.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS.
- G. Pasut and F. M. Veronese, Isr. J. Chem., 2010, 50, 151–159 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- C. S. Fishburn, J. Pharm. Sci., 2008, 97, 4167–4183 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- S. Davis, A. Abuchowski, Y. K. Park and F. F. Davis, Clin. Exp. Immunol., 1981, 46, 649–652 CAS.
- B. E. Bax, M. D. Bain, L. D. Fairbanks, A. D. B. Webster and R. A. Chalmers, Br. J. Haematol., 2000, 109, 549–554 CrossRef CAS.
- M. S. Hershfield, R. H. Buckley, M. L. Greenberg, A. L. Melton, R. Schiff, C. Hatem, J. Kurtzberg, M. L. Markert, R. H. Kobayashi, A. L. Kobayashi and A. Abuchowski, N. Engl. J. Med., 1987, 316, 589–596 CAS.
- F. F. Davis, T. Van Es and N. C. Palczuk, US Pat., 4179337, 1979.
- H. F. Oettgen, L. J. Old, E. A. Boyse, H. A. Campbell, F. S. Philips, B. D. Clarkson, L. Tallal, R. D. Leeper, M. K. Schwartz and J. H. Kim, Cancer Res., 1967, 27, 2619 CAS.
- L. J. Ettinger, J. Kurtzberg, P. A. Voute, H. Jurgens and S. L. Halpern, Cancer, 1995, 75, 1176–1181 CrossRef CAS.
- P. A. Dinndorf, J. Gootenberg, M. H. Cohen, P. Keegan and R. Pazdur, Oncologist, 2007, 12, 991–998 CrossRef CAS.
- J. D. Broome, Nature, 1961, 191, 1114 CrossRef CAS.
- D. H. Ho, N. S. Brown, A. Yen, R. Holmes, M. Keating, A. Abuchowski, R. A. Newman and I. H. Krakoff, Drug Metab. Dispos., 1986, 14, 349–352 CAS.
- N. Schlesinger, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2011, 10, 17–18 CrossRef CAS.
- M. S. Hershfield, J. S. Sundy, N. J. Ganson and S. J. Kelly, in PEGylated Protein Drugs: Basic Science and Clinical Applications, 2009, pp. 217–227 Search PubMed.
- M. R. Sherman, M. G. P. Saifer and F. Perez-Ruiz, Adv. Drug Delivery Rev., 2008, 60, 59–68 CrossRef CAS.
- J. S. Sundy, M. A. Becker, H. S. B. Baraf, A. Barkhuizen, L. W. Moreland, W. Huang, R. W. Waltrip, A. N. Maroli, Z. Horowitz and Pegloticase Phase 2 Study Investigators, Arthritis Rheum., 2008, 58, 2882–2891 CrossRef.
- J. S. Sundy, N. J. Ganson, S. J. Kelly, E. L. Scarlett, C. D. Rehrig, W. Huang and M. S. Hershfield, Arthritis Rheum., 2007, 56, 1021–1028 CrossRef CAS.
- L. D. Williams, M. S. Hershfield, S. J. Kelly, M. G. P. Saifer and M. R. Sherman, US Pat., 7723089, 2010.
- M. W. Fried, M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Goncales, D. Haussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman and J. Yu, N. Engl. J. Med., 2002, 347, 975–982 CrossRef CAS.
- K. R. Reddy, M. W. Modi and S. Pedder, Adv. Drug Delivery Rev., 2002, 54, 571–586 CrossRef.
- S. Zeuzem, S. V. Feinman, J. Rasenack, E. J. Heathcote, M. Y. Lai, E. Gane, J. O'Grady, J. Reichen, M. Diago, A. Lin, J. Hoffman and M. J. Brunda, N. Engl. J. Med., 2000, 343, 1666–1672 CrossRef CAS.
- J. M. Harris, F. M. Veronese, P. Caliceti and O. Schiavon, US Pat., 5932462, 1999.
- M. P. Manns, J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. H. Ling and J. K. Albrecht, Lancet, 2001, 358, 958–965 CrossRef CAS.
- Y. S. Wang, S. Youngster, M. Grace, J. Bausch, R. Bordens and D. F. Wyss, Adv. Drug Delivery Rev., 2002, 54, 547–570 CrossRef CAS.
- R. M. Bukowski, C. Tendler, D. Cutler, E. Rose, M. M. Laughlin and P. Statkevich, Cancer, 2002, 95, 389–396 CrossRef CAS.
- C. W. Gilbert and M. Park-Cho, US Pat., 5951974, 1999.
- G. Pasut, in PEGylated Protein Drugs: Basic Science and Clinical Applications, 2009, pp. 205–216 Search PubMed.
- M. Grace, S. Youngster, G. Gitlin, W. Sydor, L. Xie, L. Westreich, S. Jacobs, D. Brassard, J. Bausch and R. Bordens, J. Interferon Cytokine Res., 2001, 21, 1103–1115 CrossRef CAS.
- A. Nesbitt, G. Fossati, M. Bergin, P. Stephens, S. Stephens, R. Foulkes, D. Brown, M. Robinson and T. Bourne, Inflammatory Bowel Dis., 2007, 13, 1323–1332 Search PubMed.
- W. J. Sandborn, B. G. Feagan, S. Stoinov, P. J. Honiball, P. Rutgeerts, D. Mason, R. Bloomfield, S. Schreiber and P. S. Investigators, N. Engl. J. Med., 2007, 357, 228–238 CrossRef CAS.
- S. Schreiber, P. Rutgeerts, R. N. Fedorak, M. Khaliq-Kareemi, M. A. Kamm, M. Boivin, C. N. Bernstein, M. Staun, O. O. Thomsen, A. Innes and C. D. P. C. D. S. Grp, Gastroenterology, 2005, 129, 807–818 CrossRef CAS.
- S. Schreiber, M. Khaliq-Kareemi, I. C. Lawrance, O. O. Thomsen, S. B. Hanauer, J. McColm, R. Bloomfield, W. J. Sandborn and P. S. Investigators, N. Engl. J. Med., 2007, 357, 239–250 CrossRef CAS.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 Search PubMed.
- D. S. Athwal, D. Brown, A. N. C. Weir, A. G. Popplewell, A. P. Chapman and D. J. King, US Pat., 7012135, 2006.
- G. Molineux, Curr. Pharm. Des., 2004, 10, 1235–1244 CrossRef CAS.
- C. L. Vogel, M. Z. Wojtukiewicz, R. R. Carroll, S. A. Tjulandin, L. J. Barajas-Figueroa, B. L. Wiens, T. A. Neumann and L. S. Schwartzberg, J. Clin. Oncol., 2005, 23, 1178–1184 CrossRef CAS.
- M. D. Green, H. Koelbl, J. Baselga, A. Galid, V. Guillem, P. Gascon, S. Siena, R. I. Lalisang, H. Samonigg, M. R. Clemens, V. Zani, B. C. Liang, J. Renwick, M. J. Piccart and International Pegfilgrastim 749 Study Group, Ann. Oncol., 2003, 14, 29–35 CrossRef CAS.
- F. A. Holmes, J. A. O'Shaughnessy, S. Vukelja, S. E. Jones, J. Shogan, M. Savin, J. Glaspy, M. Moore, L. Meza, I. Wiznitzer, T. A. Neumann, L. R. Hill and B. C. Liang, J. Clin. Oncol., 2002, 20, 727–731 CrossRef CAS.
- K. Welte, J. Gabrilove, M. H. Bronchud, E. Platzer and G. Morstyn, Blood, 1996, 88, 1907–1929 CAS.
- O. B. Kinstler, N. E. Gabriel, C. E. Farrar and R. B. DePrince, US Pat., 5824784, 1998.
- F. Locatelli, G. Villa, A. L. M. de Francisco, A. Albertazzi, H. J. Adrogue, F. C. Dougherty and U. Beyer, Curr. Med. Res. Opin., 2007, 23, 969–979 CrossRef CAS.
- I. C. Macdougall and K. U. Eckardt, Lancet, 2006, 368, 947–953 CrossRef CAS.
- I. C. Macdougall, R. Robson, S. Opatrna, X. Liogier, A. Pannier, P. Jordan, F. C. Dougherty and B. Reigner, Clin. J. Am. Soc. Nephrol., 2006, 1, 1211–1215 Search PubMed.
- J. Hertel, H. Locay, D. Scarlata, L. Jackson, R. Prathikanti and P. Audhya, Am. J. Nephrol., 2006, 26, 149–156 CrossRef CAS.
- I. C. Macdougall, P. Bailon, N. Tare, W. Pahlke and J. Pill, J. Am. Soc. Nephrol., 2003, 14, 769A.
- W. Y. Chen, D. C. Wight, T. E. Wagner and J. J. Kopchick, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5061–5065 CrossRef CAS.
- J. J. Kopchick and W. Y. Chen, US Pat., 5958879, 1999.
- J. J. Kopchick and W. Y. Chen, US Pat., 6583115, 2003 Search PubMed.
- R. J. M. Ross, K. C. Leung, M. Maamra, W. Bennett, N. Doyle, M. J. Waters and K. K. Y. Ho, J. Clin. Endocrinol. Metab., 2001, 86, 1716–1723 CrossRef CAS.
- S. Pradhananga, I. Wilkinson and R. J. M. Ross, J. Mol. Endocrinol., 2002, 29, 11–14 Search PubMed.
- P. J. Trainer, W. M. Drake, L. Katznelson, P. U. Freda, V. Herman-Bonert, A. J. van der Lely, E. V. Dimaraki, P. M. Stewart, K. E. Friend, M. L. Vance, G. M. Besser, J. A. Scarlett, M. O. Thorner, C. Parkinson, A. Klibanski, J. S. Powell, A. L. Barkan, M. C. Sheppard, M. Maldonado, D. R. Rose, D. R. Clemmons, G. Johannson, B. A. Bengtsson, S. Stavrou, D. L. Kleinberg, D. M. Cook, L. S. Phillips, M. Bidlingmaier, C. J. Strasburger, S. Hackett, K. Zib, W. F. Bennett and R. J. Davis, N. Engl. J. Med., 2000, 342, 1171–1177 CrossRef CAS.
- A. J. van der Lely, R. K. Hutson, P. J. Trainer, G. M. Besser, A. L. Barkan, L. Katznelson, A. Klibanski, V. Herman-Bonert, S. Melmed, M. L. Vance, P. U. Freda, P. M. Stewart, K. E. Friend, D. R. Clemmons, G. Johannsson, S. Stavrou, D. M. Cook, L. S. Phillips, C. J. Strasburger, S. Hacker, K. A. Zib, R. J. Davis, J. A. Scarlett and M. Thorner, Lancet, 2001, 358, 1754–1759 CrossRef.
- G. Fuh, B. C. Cunningham, R. Fukunaga, S. Nagata, D. V. Goeddel and J. A. Wells, Science, 1992, 256, 1677–1680 CrossRef CAS.
- B. C. Cunningham, H. B. Lowman, J. A. Wells, R. G. Clark, K. Olson and G. G. Fuh, US Pat., 5849535, 1998.
- B. C. Cunningham, H. B. Lowman, J. A. Wells, R. G. Clark, K. Olson and G. G. Fuh, US Pat., 6057292, 2000.
- R. Webster, E. Didier, P. Harris, N. Siegel, J. Stadler, L. Tilbury and D. Smith, Drug Metab. Dispos., 2007, 35, 9–16 CAS.
- R. E. Connor and D. A. Tirrell, Polym. Rev., 2007, 47, 9–28 Search PubMed.
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039–11044 CrossRef CAS.
- G. G. Kochendoerfer, S. Y. Chen, F. Mao, S. Cressman, S. Traviglia, H. Y. Shao, C. L. Hunter, D. W. Low, E. N. Cagle, M. Carnevali, V. Gueriguian, P. J. Keogh, H. Porter, S. M. Stratton, M. C. Wiedeke, J. Wilken, J. Tang, J. J. Levy, L. P. Miranda, M. M. Crnogorac, S. Kalbag, P. Botti, J. Schindler-Horvat, L. Savatski, J. W. Adamson, A. Kung, S. B. H. Kent and J. A. Bradburne, Science, 2003, 299, 884–887 CrossRef CAS.
- R. A. Scheck, M. T. Dedeo, A. T. Lavarone and M. B. Francis, J. Am. Chem. Soc., 2008, 130, 11762–11770 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- K. Matyjaszewski, T. E. Patten and J. H. Xia, J. Am. Chem. Soc., 1997, 119, 674–680 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- G. N. Grover and H. D. Maynard, Curr. Opin. Chem. Biol., 2011, 14, 818–827.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS.
- J. Q. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- W. P. Gao, W. G. Liu, T. Christensen, M. R. Zalutsky and A. Chilkoti, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16432–16437 CrossRef CAS.
- V. Depp, A. Alikhani, V. Grammer and B. S. Lele, Acta Biomater., 2009, 5, 560–569 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
| This journal is © The Royal Society of Chemistry 2011 |