Intermolecular radical 1,2-addition of the BlocBuilder MA alkoxyamine onto activated olefins: a versatile tool for the synthesis of complex macromolecular architecture
Didier
Gigmes
*,
Pierre-Emmanuel
Dufils
,
David
Glé
,
Denis
Bertin
,
Catherine
Lefay
* and
Yohann
Guillaneuf
*
Universités d'Aix-Marseille I, II et III—UMR 6264, Laboratoire Chimie Provence Equipe CROPS—Case 542, Av. Escadrille Normandie Niemen, 13397, Marseille Cedex 20, France. E-mail: didier.gigmes@univ-provence.fr; catherine.lefay@univ-provence.fr; yohann.guillaneuf@univ-provence.fr; Fax: +33(0)4 9128 8758; Tel: +33(0)4 9128 8083
First published on 24th March 2011
Abstract
This review described the potential of the intermolecular radical 1,2-addition from the commercially available BlocBuilder MA alkoxyamine onto activated olefins to synthesize either new functionalized alkoxyamines or various macromolecular architectures. Following this approach, diblock, triblock copolymers, star polymers and hybrid materials are then easily prepared. The various applications of such architectures will be briefly reviewed. Interestingly this new synthetic tool widely expands the range of complex macromolecular architectures which could be obtained by the nitroxide-mediated polymerization (NMP) process.
 Didier Gigmes | Didier Gigmes received a PhD in 1998 in organic chemistry under the guidance of Prof. Paul Tordo (Marseille, France). Then he completed a first postdoctoral fellowship at Elf-Atochem, North America in Pennsylvania (USA) under the supervision of Dr Gary Silverman. In 2001 he obtained a position of researcher at CNRS to develop the nitroxide-mediated polymerization (NMP). In 2004 he was the co-founder with Prof. Denis Bertin of the team “Chimie Radicalaire Organique et Polymère de Spécialité (CROPS)” that belongs to the “Laboratoire Chimie Provence”. In 2008 he defended his Habilitation at the University of Provence and since June 2008 he is the group leader of the CROPS team. Since October 2010, he is appointed Research Director at the CNRS. During the past few years he was working on the development of NMP and especially in the design of highly efficient SG1-based alkoxyamines. After a significant contribution to the comprehension of the mechanisms involved in NMP, one of his main concerns is now to promote NMP in material science for various applications such as biomaterials, environment and energy. |
 Pierre-Emmanuel Dufils | Pierre-Emmanuel Dufils graduated from the National School of Chemistry of Mulhouse, France, in 2002. He obtained his PhD degree in 2005 at the University of Provence in Marseille, working in the laboratory of Professor Denis Bertin. His research, funded by ARKEMA, covered Nitroxide Mediated Polymerization and the use of the alkoxyamine BlocBuilder MA for the synthesis of functional block copolymers. In 2006, he obtained a postdoctoral position in the laboratory of Prof. B. Boutevin (IAM, Montpellier, France) in collaboration with RHODIA. In 2007, he joined SOLVAY, where he is currently working as a research manager on emulsion polymerization for food and pharmaceutical packaging. |
 David Glé | David Glé studied chemical engineering at the Ecole Normale Supérieure de Chimie de Montpellier (ENSCM, France) from which he graduated in 2008. He completed his formation with a Master of Science in polymer materials at the University of Montpellier 2. He started his PhD in January 2009 under the supervision of Dr Didier Gigmes and Dr Trang Phan at the University of Provence (Marseille, France). The focus of his research is the synthesis of block copolymers with complex architectures for the application in lithium batteries. |
 Denis Bertin | Denis Bertin obtained a PhD in Material Science under the guidance of Prof. Bernard Boutevin (Montpellier, France). He started his career in Elf-Atochem company (Serquigny, France), where he was in charge of the material process service. In 1999, he joined the University of Provence (Marseille, France) as an associate Professor. He obtained in 2003 at the University of Provence his Habilitation and became full Professor the same year in this University. His research interests are focused on controlled radical polymerization and the design of complex polymer architectures for applications in the field of health, environment and energy. He created in 2004 with Dr Didier Gigmes the team “Chimie Radicalaire Organique et Polymère de Spécialité (CROPS)” in the “Laboratoire Chimie Provence”. Since January 2008 he is the Vice-Director of the “Laboratoire Chimie Provence” and since June 2008 he is the Dean of Science of the University of Provence. |
 Catherine Lefay | Catherine Lefay obtained her PhD in 2006 in the field of nitroxide-mediated controlled radical polymerization (NMP) and polymerization in (mini)emulsion in the group of Prof. B. Charleux at the University Pierre and Marie Curie (Paris, France). In 2006, she joined the group of Prof. C. Barner-Kowollik at the University of New South Wales (Sydney, Australia) as a research associate to study the synthesis by Ring Opening Polymerization (ROP) and Reversible-Addition Fragmentation chain Transfer (RAFT) polymerization of copolymers for hernia repair. In 2007, she then moved to the Centre for Nutrition and Food Sciences (Brisbane, Australia) to work as a research associate with Prof. R. G. Gilbert and Prof. M. Gidley on the characterization of natural polymers. Since late 2007, she works as a lecturer at the Université Aix-Marseille I (Marseille, France) and her current research activities are dedicated to modification of natural polymers by (controlled) radical polymerization and/or ROP for biomedical, packaging and batteries applications. |
 Yohann Guillaneuf | Yohann Guillaneuf obtained his PhD in 2006 under the supervision of Prof. Denis Bertin at the University of Provence (Marseille, France), where he studied the kinetics of nitroxide mediated polymerization (NMP). He then joined the group of Prof. Robert G. Gilbert at the “Key Centre for Polymer Colloids (KCPC)” (University of Sydney, Australia) for a post-doctoral fellowship on the synthesis of amphiphilic copolymers by RAFT polymerization. Then, he moved with Prof. Robert G. Gilbert to the University of Queensland (Brisbane, Australia) to work on the characterization of polysaccharides. In 2008, he came back to the University of Provence to work with Dr Didier Gigmes on the nitroxide-mediated photopolymerization (NMP2). In 2010 he was appointed as a permanent CNRS researcher in the group of Dr Didier Gigmes (University of Provence, France), where his current research activities are focused on the mechanisms of NMP and the development of both NMP2 and controlled radical ring opening polymerization methods based on nitroxide (NMrROP). |
1 Introduction
During the past decades, emerging technologies such as biomaterials, optics or microelectronics need more and more sophisticated materials with enhanced mechanical, thermal or solubility properties. A breakthrough was witnessed in that topic by the emergence of controlled polymerization techniques combined now with highly efficient coupling reactions.1,2 Three main controlled radical polymerization (CRP) procedures have emerged: the nitroxide-mediated polymerization (NMP),3–6 the atom transfer radical polymerization (ATRP),7–12 and the reversible addition fragmentation/transfer (RAFT).13–17 Among these techniques, the NMP process has many advantages since it is only governed by a thermal process and does not require any transition metal catalysts or bimolecular exchange with sulfur-based compounds. The development of the acyclic β-phosphorylated nitroxide N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl) named SG1 by Tordo et al.18,19 and the derived alkoxyamine (N-(2-methylpropyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxylprop-2-yl) hydroxylamine,20–23 commercially available from ARKEMA under the trademark BlocBuilder MA24 (Scheme 1), represents a significant advance in NMP. Indeed this compound allows for example improvement of the quality of the control of the polymerization,22 offers the possibility to perform polymerization reactions in aqueous25 and aqueous dispersed media26 and the presence of carboxyl acid on the initiating radical enables the coupling reaction with amines.27–29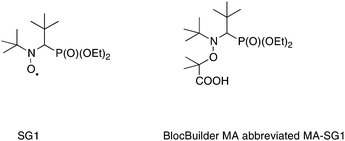 | ||
| Scheme 1 Structure of the SG1 nitroxide and its corresponding BlocBuilder MA alkoxyamine. | ||
Even if the BlocBuilder MA is a promising precursor for macromolecular engineering, the development of complex and/or functionalized materials requires generally multi-step syntheses and this could become a major constraint for the further development at an industrial scale. This assessment led our group to develop new strategies to prepare complex/functionalized initiator/macromolecules in a straightforward manner.
Intermolecular radical 1,2-addition (IRA) onto olefins is often used in organic chemistry to introduce a functional group. For example, the addition of halogenated compounds to alkenes or alkynes through a radical process was first reported by Kharasch30,31 in the 1940s and is now called atom transfer radical addition32,33 (ATRA). Zard and Quiclet-Sire34,35 developed also a very powerful methodology based on xanthate derivatives to perform both inter- and intra-molecular radical additions. Recently Barner-Kowollik et al.36 reported that non-activated olefins can also—and just once—be neatly inserted into (macro)RAFT agents through a photolytic β-cleavage of the thiocarbonyl thio moiety.
Studer (Scheme 2) was the first to perform such a reaction using TEMPO-based alkoxyamines.37 The Persistent Radical Effect (PRE) which allows to prolong the life-time of the stabilized but transient initiating alkyl radical38,39 enables to perform such reaction even for slow intermolecular radical addition steps. Indeed, stabilized radicals are in general not reactive in intermolecular addition reactions. To avoid any multiple additions, only non-activated olefins, such as octene for example, were used. This ensures the efficient trapping of the adduct radical and in the same time leads to a strong C–O bond, which cannot be cleaved under homolytic pathway anymore.37
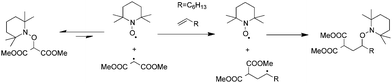 | ||
| Scheme 2 Intermolecular radical 1,2-addition onto non-activated olefins. | ||
As our goal was to develop a straightforward method to prepare “second generation” functionalized NMP initiators, we focused on the extension of this concept to activated olefins. The aim of this highlight is to demonstrate that 1,2-IRA of the BlocBuilder MA onto activated olefins is a versatile tool to produce easily new functionalized alkoxyamines and complex macromolecular architectures. After the description of the mechanism of 1,2-IRA onto olefins, the application of IRA will be illustrated with various examples.
2 Mechanism of intermolecular radical 1,2-addition (1,2-IRA)
The occurrence of the persistent radical effect38,39 during the decomposition of alkoxyamines prompted Studer37 to perform clean additions onto non-activated olefins. The success of such reaction relies on the fact that the adduct (i.e. the alkoxyamine linked to a non-stabilized alkyl moiety) is thermally stable. The intermolecular 1,2-addition onto activated olefins is more challenging since the adduct could also undergo a dissociation and lead to an unwanted multiple addition/polymerization process.To get a clean intramolecular 1,2-addition, the starting alkoxyamine has to be specially tuned to have a lower Bond Dissociation Energy BDEC–O than the adduct (Scheme 3). It has to be noticed that in that case the BDEC–O (=Ea,d − Ea,c) is roughly similar to the activation energy Ea of the dissociation rate constant Ea,d since the one of the recombination40Ea,c is close to zero.
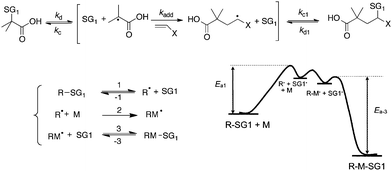 | ||
| Scheme 3 Mechanism of the intermolecular radical 1,2-addition of the BlocBuilder MA onto activated olefins. | ||
This was obtained by using the BlocBuilder MA alkoxyamine, whose homolysis produces the transient tertiary radical 1-carboxy-1-methyl ethyl (MA radical) and the persistent radical SG1. Indeed, the ground state energy of the BlocBuilder MA with the tertiary alkyl radical is higher than the one of the secondary alkyl radical adduct even if the addition of the 1-carboxy-1-methyl ethyl group increases the steric hindrance compared to the model secondary radical that is the 1-carboxy-ethyl group. At the same time, the released alkyl radical is tertiary instead of secondary that leads to a lower final state. The combination of the two effects leads to a strong decrease of the BDE (and therefore a higher dissociation rate constant kd) for the BlocBuilder MA alkoxyamine compared to the radical adduct. Due to this difference, the BlocBuilder MA is expected to lead to a clean 1,2-IRA onto activated olefins by choosing the proper reaction time and temperature.
The mechanism can then be described as follows: once the PRE is established, the tertiary alkyl radical may either react with the activated olefin CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2–X to provide the radical adduct or recombine with the SG1 to give the starting alkoxyamine. The radical adduct is then trapped by the SG1 to provide the novel alkoxyamine. At the temperature used, the obtained alkoxyamine is more stable than the BlocBuilder MA and this avoids the occurrence of multiple additions. The optimal conditions were determined with n-butyl acrylate (BA) as a model olefin and using the simple kinetic scheme already presented in Scheme 3, extended to the double and triple addition adduct and then implemented in the PREDICI software.41
CH2–X to provide the radical adduct or recombine with the SG1 to give the starting alkoxyamine. The radical adduct is then trapped by the SG1 to provide the novel alkoxyamine. At the temperature used, the obtained alkoxyamine is more stable than the BlocBuilder MA and this avoids the occurrence of multiple additions. The optimal conditions were determined with n-butyl acrylate (BA) as a model olefin and using the simple kinetic scheme already presented in Scheme 3, extended to the double and triple addition adduct and then implemented in the PREDICI software.41
The results of the radical 1,2-addition was examined in terms of the yield of the reaction but also in terms of the purity, that is the ratio between the concentration of the monoadduct alkoxyamine and the concentration of all the alkoxyamines (mono, double and triple adduct). The parameter investigated was the concentration of BlocBuilder MA, the duration of the experiment to obtain the maximum yield and the temperature used, with the concentration of the olefin being set at 1 M in tert-butanol. The results shown in Fig. 1 led to the following optimized conditions: one hour reaction at 100 °C with an initial concentration of BlocBuilder MA of 1 M. Indeed only a slight increase in yield and in purity could be obtained at lower temperature (i.e. 90 °C) but the duration of the experiment will increase by a factor above 10.
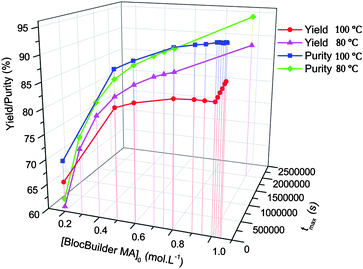 | ||
| Fig. 1 Theoretical evolution of the yield and the relative purity of the 1,2-IRA of BlocBuilder MA on n-butyl acrylate obtained using the PREDICI software. | ||
3 Applications of intermolecular radical 1,2-addition (1,2-IRA)
3.1 Synthesis of functionalized alkoxyamines
Thanks to the lower C–ON bond dissociation energy of BlocBuilder MA (MA–SG1) compared to the targeted product, 1,2-IRA was successfully applied to various activated olefins.Charleux et al. first used this method to synthesize a difunctional alkoxyamine called DIAMA by the addition of BlocBuilder MA onto tri(ethylene glycol) diacrylate (Scheme 4).42,43 The sodium salt counterpart (DIAMA–Na) furnished then a water-soluble initiator suitable for emulsion polymerization of styrene and n-butyl acrylate (see Section 3.2 for the preparation of the block copolymers).
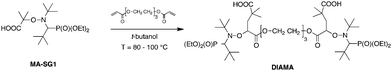 | ||
| Scheme 4 Synthesis of SG1-based difunctional alkoxyamine DIAMA. | ||
The 1,2-IRA method was then applied to functional vinylic monomers such as styrene, n-butyl acrylate, acrylic acid and 2-hydroxyethyl acrylate (HEA).23,44 In particular, the 1,2-IRA of BlocBuilder MA on HEA provided an OH-functionalized alkoxyamine44 that could be used to initiate the Ring-Opening Polymerization (ROP) of lactones. The 1,2-IRA methodology was applied to 4-vinylpyridine, sodium styrene sulfonate and N-acryloylglucosamine and furnished functionalized alkoxyamines respectively water soluble under acidic conditions, totally water soluble and glycosylated alkoxyamine (Scheme 5).23
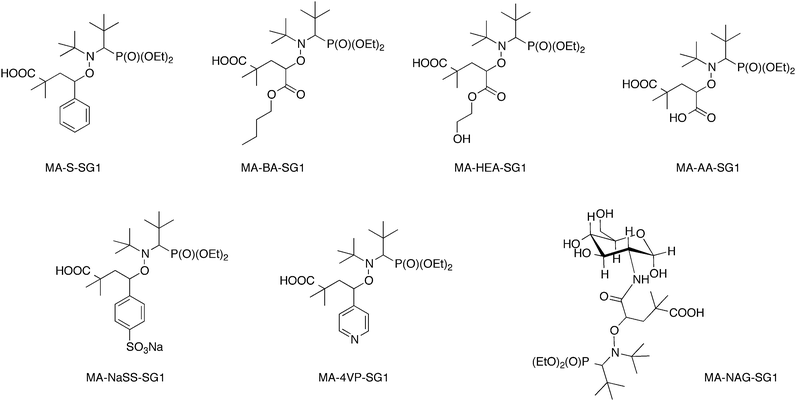 | ||
| Scheme 5 Functionalized alkoxyamines obtained by 1,2-IRA onto various olefins. | ||
The 1,2-IRA is thus a powerful and convenient modification method to synthesize new functionalized alkoxyamines from the commercially available BlocBuilder MA alkoxyamine. As the synthesis of a wide range of acrylate and acrylamide derivatives from the condensation of acryloyl chloride and amino and hydroxy compounds is well described in the literature, the combination of such reaction and 1,2-IRA is thus very attractive to prepare a library of functionalized alkoxyamines using amino or hydroxy compounds (Scheme 6). This was done for instance with 4-hydroxy and 4-amino benzophenone to prepare alkoxyamines bearing a chromophoric group.45 These alkoxyamines will thus introduce a new functionality in the final polymer or will allow to synthesize several block copolymers. This last property will be described in detail in the following part.
3.2 Towards macromolecular architectures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PDI = 1.4) was first allowed to react with acryloyl chloride before 1,2-IRA of BlocBuilder MA. The obtained PCL or PLA-b-PHE(M)A diblock copolymers and PHEMA-b-PCL-b-PHEMA triblock copolymers have then been evaluated as partially degradable scaffolds for nerve repair.
000 g mol−1, PDI = 1.4) was first allowed to react with acryloyl chloride before 1,2-IRA of BlocBuilder MA. The obtained PCL or PLA-b-PHE(M)A diblock copolymers and PHEMA-b-PCL-b-PHEMA triblock copolymers have then been evaluated as partially degradable scaffolds for nerve repair.
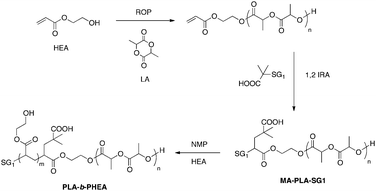 | ||
| Scheme 7 Synthesis of PLA-b-PHEA diblock copolymers. | ||
Interestingly, the 1,2-IRA allowed also to combine catalyzed chain growth (CCG) of polyethylene (PE) on magnesium and NMP of BA.47,48 Alkoxyamines can namely be synthesized by Grignard Chemistry. By instance, the reaction between C6H14MgBr and a stable nitroxyl radical furnishes a C6-alkyl based alkoxyamine.49
When transposing this methodology to the (PE)2Mg polymer chain with TEMPO or SG1 as stable nitroxides, Lopez et al.47 obtained effectively TEMPO and SG1-terminated PE chains (at a yield of 70% for TEMPO and 45% for SG1). Nevertheless, very high temperatures were necessary to cleave the C–ON bond (because of a high bond dissociation energy) that inhibits the polymerization of vinylic monomers by NMP. To succeed in using a macroalkoxyamine based on PE to perform NMP, the authors took advantage of this high bond dissociation energy and used TEMPO-based nitroxides functionalized by a cleavable alkoxyamine. The TEMPO part was used as an anchor and the SG1-based alkoxyamine as control agent for NMP of several monomers such as BA (Scheme 8).
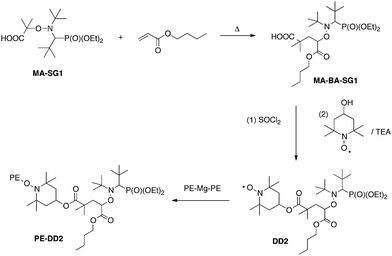 | ||
| Scheme 8 Synthesis of PE–DD2. | ||
To obtain by this method PE-b-PBA diblock copolymers, 4 steps were necessary: (1) 1,2-IRA of BlocBuilder Ma onto BA, (2) functionalization of this alkoxyamine by TEMPO providing an alkoxyamine with a free nitroxide moiety (alkoxyamine called DD2), (3) end-functionalization of a preformed PE by DD2 to afford PE-DD2, (4) polymerization by NMP of BA from the macroalkoxyamine PE-DD2. SEC and 1H NMR analyses revealed that the polymerization proceeded under control and thus that well-defined PE-b-PBA diblock copolymers were obtained. Moreover, thanks to the 1,2-IRA technique, diblock and triblock copolymers can be easily obtained by simply modifying the commercially available homopolymer. The key point is to introduce at α and/or ω chain end an acrylate moiety (for instance by reaction of an alcohol or amine function with acryloyl chloride) to next perform the 1,2-IRA of BlocBuilder MA onto the introduced acrylate group(s). This method allowed to synthesize several diblock and triblock copolymers from α,ω-diamino polyetherimide PEI (polystyrene-b-poly(etherimide)-b-polystyrene (PS-b-PEI-b-PS)),23 from ω-hydroxy poly(ethylene oxide) PEO (PEO-b-PS),50–52 and from α,ω-dihydroxy poly(ethylene oxide) (PS-b-POE-b-PS,53,54 and PSAN-b-POE-b-PSAN,55 with PSAN a random copolymer of styrene and acrylonitrile). These diblock and triblock copolymers have been analyzed by several techniques such as Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectroscopy (MALDI-TOF-MS),51 Pulsed Gradient Spin Echo NMR,52Small Angle X-ray Scattering (SAXS),50Solid-State NMR54 or Transmission Electron Microscopy (TEM).50,53 Such block copolymers like PEO-b-PS diblock copolymers revealed very interesting properties for the elaboration of nanomaterials (Fig. 2).
 | ||
| Fig. 2 TEM images of calcinated mesoporous silicas prepared using PEO114-b-PS19 (with 114 and 19 the degree of polymerization of the PEO and PS blocks respectively). | ||
In particular, the PS-b-PEO-b-PS triblock copolymers have been used as templates to prepare mesoporous silicas with large and tunable accessible pores (from 4 to 40 nm) by the sol–gel method.53 Amphiphilic triblock copolymers such as PS-b-PEO-b-PS have namely the advantage to self-organize into micelles and this ordering property can be finely tuned by adjusting the solvent and the copolymer composition. The porous structure of the silica (in terms of mesoporous size, size distribution and microporous volume) can thus be controlled by the hydrophobic and hydrophilic block length ratio. In the case of PS-b-PEO-b-PS triblock copolymers, Hornebecq et al.53 proved that the mesopore size increases with the PS hydrophobic block length (while keeping the PEO block length constant) whereas the micropore volume increases with the PEO hydrophilic block length (while keeping the PS block length constant). These results were explained by the fact that the micelles are composed of a PS core surrounded by a corona of PEO forming a loop.
The nanostructuration of the PSAN-b-PEO-b-PSAN triblock copolymers has been extensively studied too and revealed very interesting results in terms of morphology and material properties.55 In particular, the micelles of PSAN-b-PEO-b-PSAN self-assemble during the spin coating. By varying the ratio of DMF/toluene of the solvent system, different micelles can be obtained. Spherical micelles are observed with DMF/toluene 50/50 and form a nanoparticle film whereas worm-like and “Y” micelles are formed with DMF/toluene 25/75 giving rise to “spider web” films (Fig. 3).
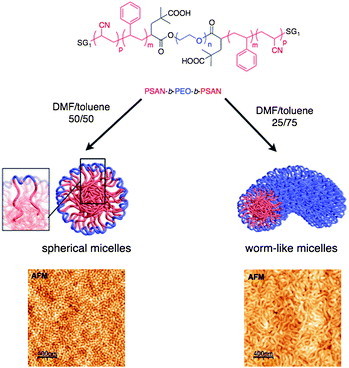 | ||
| Fig. 3 Self-assembling of PSAN-b-PEO-b-PSAN micelles. | ||
The 1,2-IRA has been used recently by Charleux and Nicolas26 to synthesize by NMP in emulsion polymerization triblock copolymers PS-b-PBA-b-PS, thanks to their previous work on the DIAMA–Na alkoxyamine (see above). This latter was used to initiate the polymerization of BA or styrene in a two-step emulsion polymerization process because as with the sodium salt of MA–SG1 (Na/MA–SG1) as an initiator, an ab initio batch emulsion polymerization initiated by DIAMA–Na failed. More precisely, various living PBA seeds were extended by a one shot monomer addition (styrene or BA) to reach higher solid contents. Fast polymerizations were observed (90% of conversion in 8H) and proceeded under control (linear evolution of Mn with conversion). It is here worth noticing that the use of DIAMA–NA instead of Na/MA–SG1 alkoxyamine permitted to significantly decrease the average particle diameter (for a PBA seed at 2.2 wt% surfactant/monomer, Dz = 660 and 170 nm with respectively Na/MA–SG1 and DIAMA–Na) and to narrow the particle size distribution by avoiding secondary nucleation. By this technique, PS-b-PBA-b-PS triblock copolymers of molar masses in good agreement with the theoretical values (51000 g mol−1) and final PDI close to 1.64 were obtained. The formation of block copolymers was in addition attested by Liquid Adsorption Chromatography (LAC).
Finally the 1,2-IRA has proven to be a powerful technique to synthesize complex macromolecular architecture such as 3 or 4-arms star polymers.441,2-IRA of BlocBuilder MA was performed onto tri- or tetra-acrylate compounds previously prepared by reaction of acryloyl chloride onto the corresponding tri- or tetrahydroxy monomers. These multialkoxyamines were then used to achieve star-like macromolecular architectures by NMP of styrene (Scheme 9). To check the star architecture of the polymers, the ester functions of the core were hydrolyzed in alkaline conditions at 70 °C. After 7 days of hydrolysis, the linear polystyrene chains were recovered. The SEC analysis of the cleaved arms revealed a good agreement between the experimental and theoretical Mn values, i.e. a Mn value of the polystyrene arms almost a quarter of the 4-star initial polystyrene one (the Mn of the star was prior corrected taking into account the g' ratio between the intrinsic viscosities of a star polymer and a linear one). Based on these results more complicated architectures could now be envisioned.
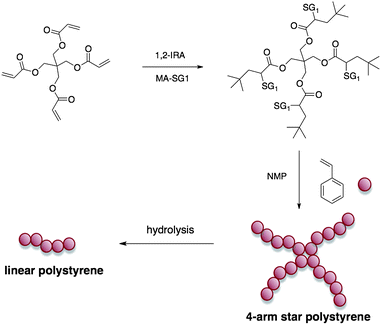 | ||
| Scheme 9 Synthesis of 4-arm star polystyrene by the 1,2-IRA technique and hydrolysis of the ester functions of the star core. | ||
Billon et al.56 developed a “grafting from” method based on NMP to modify silica particle surfaces. More precisely they used the 1,2-IRA method to prepare PBA-grafted Stöber-silica nanoparticles in 3 steps: (1) they first performed the 1,2-IRA of BlocBuilder MA (MA–SG1) onto an acrylate coupling agent (3-(trimethoxysilyl)propyl acrylate, TPMA); (2) the obtained alkoxyamine was then grafted on silica nanoparticles by condensation of its alkoxysilane function; (3) BA was polymerized from silica nanoparticles surface by NMP (Scheme 10).
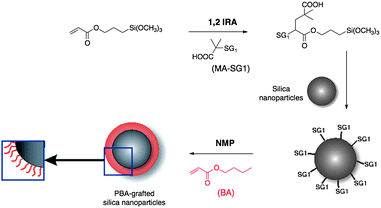 | ||
| Scheme 10 Modification of silica particle surfaces by a “grafting from” method based on NMP and 1,2-IRA. | ||
Transmission electron microscopy (TEM) and small-angle neutron scattering (SANS) confirmed the absence of particle aggregates. The analysis of the grafted PBA chains by SEC (after cleavage of the grafted polymer layer by hydrolysis of the initiator-ester function) proved that it is possible to efficiently control the molecular weight and density of grafted chains. This “grafting from” method based on 1,2-IRA and BlocBuilder MA opens thus the way to a large range of potential hybrid materials and hairy inorganic particles by simply modifying the grafted polymer layer (type of monomers, grafting density or thickness of the polymer corona). In addition, it is here worth noticing that an important advantage of the “grafting from” method based on NMP and 1,2-IRA compared to the one based on ATRP is the limited number of purification steps.
Conclusions
This paper highlighted the potential of 1,2-IRA to synthesize new functionalized alkoxyamines from the commercially available BlocBuilder MA alkoxyamine. It has to be noted that only one example is reported for the synthesis of a functionalized RAFT agent by such olefin addition/insertion mechanism57 and to our knowledge there is no example for the synthesis of the functionalized ATRP initiator. The 1,2-IRA of BlocBuilder MA onto activated olefin represents then an important advance for the development of the NMP process.By using the same versatile and efficient method, various macromolecular architectures were obtained such as diblock and triblock copolymers, star polymers and hybrid materials. In particular, 1,2-IRA is a powerful technique to easily combine different polymerization methods such as Ring-Opening Polymerization (ROP), Catalyzed Chain Growth polymerization (CCG) on Magnesium and NMP. This new synthetic tool widely extends the range of complex macromolecular architecture, which could be obtained by the NMP process. A summary of the possibility offered by the 1,2-IRA using BlocBuilder MA is depicted in Scheme 11. The polymeric materials obtained after 1,2-IRA are promising candidates for the elaboration of nanomaterials and partially (bio)degradable materials for biomedical applications.
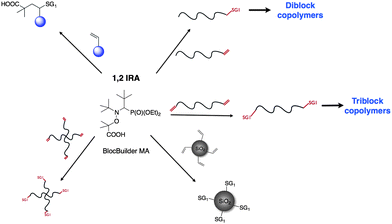 | ||
| Scheme 11 Overview of 1,2-IRA applications. | ||
Acknowledgements
The authors would like to thank the University of Provence, CNRS and Arkema for financial support.References
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, US. Pat., 4,581,429, 1985.
- D. H. Solomon, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5748–5764 Search PubMed.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, G. Moad, C. L. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653–692 CrossRef CAS.
- Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, 2008 Search PubMed.
- D. Benoit, S. Grimaldi, S. Robin, J. P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- S. Grimaldi, J.-P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33, 1141–1147 CrossRef CAS.
- D. Gigmes, S. Marque, O. Guerret, J.-L. Couturier, F. Chauvin, P.-E. Dufils, D. Bertin and P. Tordo, , 2004 Search PubMed.
- F. Chauvin, J.-L. Couturier, P.-E. Dufils, P. Gérard, D. Gigmes, O. Guerret, Y. Guillaneuf, S. R. A. Marque, D. Bertin and P. Tordo, ACS Symp. Ser., 2006, 944, 326 Search PubMed.
- F. Chauvin, P. E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- D. Gigmes, J. Vinas, N. Chagneux, C. Lefay, T. N. T. Phan, T. Trimaille, P.-E. Dufils, Y. Guillaneuf, G. Carrot, F. Boue and D. Bertin, ACS Symp. Ser., 2009, 1024, 245–262 Search PubMed.
- For more information see http://www.blocbuilder.com/ or contact mailto:laurence.couvreur@arkema.com.
- T. N. T. Phan and D. Bertin, Macromolecules, 2008, 41, 1886–1895 CrossRef CAS.
- B. Charleux and J. Nicolas, Polymer, 2007, 48, 5813–5833 CrossRef CAS.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639–3647 CrossRef CAS.
- T. Trimaille, K. Mabrouk, V. Monnier, L. Charles, D. Bertin and D. Gigmes, Macromolecules, 2010, 43, 4864–4870 CrossRef CAS.
- M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291–9303 CrossRef CAS.
- M. S. Kharasch, Science, 1945, 102, 128 CAS.
- M. S. Kharasch, J. Am. Chem. Soc., 1945, 67, 1626 CrossRef CAS.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- A. J. Clark, Chem. Soc. Rev., 2002, 31, 1–11 RSC.
- B. Quiclet-Sire and S. Z. Zard, Chem.–Eur. J., 2006, 12, 6002–6016 CrossRef CAS.
- S. Z. Zard, Angew. Chem., Int. Ed. Engl., 1997, 36, 673–685 CAS.
- T. Gruendling, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, Macromolecules, 2011, 44, 166–174 Search PubMed.
- C. Wetter, K. Jantos, K. Woithe and A. Studer, Org. Lett., 2003, 5, 2899–2902 CrossRef CAS.
- A. Studer, Chem. Soc. Rev., 2004, 33, 267–273 RSC.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS.
- J. Sobek, R. Martschke and H. Fischer, J. Am. Chem. Soc., 2001, 123, 2849–2857 CrossRef CAS.
- P.-E. Dufils, PhD thesis, Université d'Aix-Marseille I, 2005.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2005, 38, 9963–9973 CrossRef CAS.
- S. Magnet, O. Guerret and J. L. Couturier, Eur. Pat., Appl. EP 1526138, 2005.
- P.-E. Dufils, N. Chagneux, D. Gigmes, T. Trimaille, S. R. A. Marque, D. Bertin and P. Tordo, Polymer, 2007, 48, 5219–5225 CrossRef CAS.
- D. L. Versace, Y. Guillaneuf, D. Bertin, J. P. Fouassier, J. Lalevee and D. Gigmes, Org. Biomol. Chem., submitted Search PubMed.
- B. Clement, T. Trimaille, O. Alluin, D. Gigmes, K. Mabrouk, F. Feron, P. Decherchi, T. Marqueste and D. Bertin, Biomacromolecules, 2009, 10, 1436–1445 Search PubMed.
- R. G. Lopez, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson, D. Gigmes and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2705–2718 CrossRef.
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793–800 RSC.
- T. Nagashima and D. P. Curran, Synlett, 1996, 330 CrossRef CAS.
- E. Bloch, P. L. Llewellyn, T. Phan, D. Bertin and V. Hornebecq, Chem. Mater., 2009, 21, 48–55 Search PubMed.
- M. Girod, M. Mazarin, T. N. T. Phan, D. Gigmes and L. Charles, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3380–3390 CrossRef CAS.
- C. Barrere, M. Mazarin, R. Giordanengo, T. Phan, A. Thevand, S. Viel and L. Charles, Anal. Chem., 2009, 81, 8054–8060 Search PubMed.
- E. Bloch, T. Phan, D. Bertin, P. Llewellyn and V. Hornebecq, Microporous Mesoporous Mater., 2008, 112, 612–620 Search PubMed.
- F. A. Bonk, S. Caldarelli, T. Phan, D. Bertin, E. R. Deazevedo, G. L. Mantovani, T. J. Bonagamba, T. S. Plivelic and I. L. Torriani, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 55–64 Search PubMed.
- D. Quemener, G. Bonniol, T. N. T. Phan, D. Gigmes, D. Bertin and A. Deratani, Macromolecules, 2010, 43, 5060–5065 CrossRef.
- R. Inoubli, S. Dagreou, M.-H. Delville, A. Lapp, J. Peyrelasse and L. Billon, Soft Matter, 2007, 3, 1014–1024 RSC.
- M. Chen, K. P. Ghiggino, A. W. H. Mau, E. Rizzardo, W. H. F. Sasse, S. H. Thang and G. J. Wilson, Macromolecules, 2004, 37, 5479–5481 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |