Efficient electrochemical polymer halogenation using a thin-layered cell
Shotaro
Hayashi
,
Shinsuke
Inagi
* and
Toshio
Fuchigami
*
Department of Electronic Chemistry, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama, 226-8502, Japan. E-mail: fuchi@echem.titech.ac.jp; inagi@echem.titech.ac.jp; Fax: +81-45-924-5406; Tel: +81-45-924-5406
First published on 7th May 2011
Abstract
The electrochemical halogenation of polythiophene derivatives (PTs) such as poly(3-hexylthiophene) (PHT), poly(thiophene-alt-9,9-dioctylfluorene) (PTF) and poly(bithiophene-alt-9,9-dioctylfluorene) (PBT) was investigated using a thin-layered cell. The reactivity and the current efficiency of the electrochemical halogenation of polythiophene derivatives were markedly improved by using the thin-layered cell as compared to using a conventional beaker-type cell. Moreover, the use of the thin-layered cell reduces the amount of electrolytic solution required. In addition, the optical and electrochemical properties of the obtained polymers were significantly changed by the complete substitution of chlorine atoms onto the thiophene ring.
Introduction
The direct modification of polymer films by solid state polymer reaction is of both scientific and technological interest,1–14 so that electrochemical reaction of polymers has been developed as a highly reliable method.5–15 Moreover, recent strategies of polymer reaction have aimed to not only achieve complete modification, but also to control the properties of the resultant polymers by the degree of conversion.7,10,15–17 We have previously reported successive halogenation of polythiophene derivatives (PTs) and their optoelectronic properties.8–10 The properties of these polymers were gradually changed by the substitution ratio of chlorine atoms on the repeating thiophene unit. For instance, an increase in the degree of chlorination successively lowered the highest occupied molecular orbital (HOMO) level, extended both the optical and electrochemical band gap, and varied the photoluminescence (PL) properties; the optoelectronic properties of the polymers were controlled by precisely tuning the chlorination ratio. This chemistry is also available to create a patterned film.11 Therefore, the direct functionalization of π-conjugated polymer film on an electrode is suitable for organic semiconductor applications.However, there are several disadvantages of the electrochemical polymer reaction. (a) The polymer reaction shows limited reactivity and low current efficiency (CE); for example, the electrochemical chlorination of PTs was difficult to complete and resulted in low CE.8–10 (b) Because the volume of the polymer film expands through the doping of counter ions, film detachment from the electrode sometimes occurs and results in a film that is no longer electroactive. (c) Setups using conventional beaker-type cells (Fig. 1a) require a large amount of electrolytic solution (3–20 ml) compared to the amount of substrate (1–5 mg). Therefore, more efficient reaction systems for the electrochemical polymer reaction are still required.
 | ||
| Fig. 1 Schematic illustrations of (a) conventional beaker-type and (b) thin-layered cells. | ||
To overcome these disadvantages of the electrochemical polymer reaction, we considered a layered electrolytic cell,18 especially an electrochromic device,19,20 constructed using a sandwich structure of anode, optoelectronic materials, insoluble network polymer containing electrolytic solution, and cathode. Such a layered electrochromic device involves doping–dedoping of polymer films with a very small amount of electrolytic solution. Therefore, we fabricated a novel sandwich type layered cell for the electrochemical polymer reaction (Fig. 1b) and investigated the electrochemical halogenation of PTs and their optoelectronic properties before and after the reaction.
Experimental
Materials
Unless otherwise stated, all chemicals were used as received from commercial sources. Dry solvents were used as received. The zinc electrode was polished with Al2O3 before use.Measurements
NMR spectra were recorded on a JEOL EX-270 spectrometer. UV-vis spectra were obtained on a Shimadzu UV-1800 spectrophotometer. Photoluminescence (PL) spectra were obtained on a JASCO FP-6500 spectrophotometer. GPC analyses were performed by a Shimadzu Prominence GPC system (Shim-pack GPC 803C column), using chloroform as the eluent after calibration with polystyrene standards. Cyclic voltammetry measurements were recorded on an ALS 600A Electrochemical Analyzer. EDX analysis was performed with Genesis XM2 (Keyence).Synthesis of 2,7-bis(2-thienyl)-9,9-dioctylfluorene (1)21,22
2-Bromothiophene (815 mg, 5.0 mmol) and 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol) ester (1.12 g, 2.0 mmol), and Pd(PPh3)4 (180 mg) were dissolved in 5 ml of dry toluene under argon. To the solution, K2CO3 (aq) (2.0 M, 2.5 ml) was added. After stirring for 48 h at 100 °C, the reaction mixture was extracted with toluene. After evaporation, the resulting crude product was purified by chromatography on silica gel (eluent, CH2Cl2). After removal of solvent, 1 was obtained as a highly viscous liquid (1.06 g, 95%). 1H NMR (270 MHz, CDCl3): δ 7.66–7.70 (d, J = 7.5 Hz, 2H), 7.58–7.64 (dd, J = 3.0, 9.0 Hz, 2H), 7.54–7.58 (s, 2H); 7.37–7.42 (dd, J = 1.5, 4.5 Hz, 2H), 7.33–7.38 (dd, J = 1.5, 6.0 Hz, 2H), 7.11–7.16 (dd, J = 4.0, 6.0 Hz, 2H), 1.97–2.08 (m, 4H); 1.02–1.30 (m, 20H), 0.77–0.87 (t, J = 7.5 Hz, 6H), 0.64–0.87 (m, 4H).Synthesis of PHT8
To a stirred solution of 3-hexylthiophene (158 mg, 0.94 mmol) in CHCl3 (10 ml), FeCl3 (4.2 g, 26 mmol) was added. The reaction mixture was stirred for 24 h at room temperature. The mixture was then poured into a large amount of MeOH. The obtained powder was filtered and washed with MeOH. Dedoping of the crude polymer with hydrazine monohydrate for 6 h gave a dark-red powder (87 mg). 1H NMR (270 MHz, CDCl3, ppm): δ 7.18 (Ar–H, br), 2.8–2.4 (Ar–CH2–C5H11, br), 1.6–1.4 (Ar–-CH2–C4H8–CH3, br), 0.9 (–CH3, br). GPC (polystyrene standard): Mn = 2700, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, Mw/Mn = 3.81.
400, Mw/Mn = 3.81.
Synthesis of PTF9
2,5-Dibromothiophene (96 mg, 0.40 mmol), 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol) ester (223 mg, 0.40 mmol), and Pd(PPh3)4 (12 mg, 0.01 mmol) were dissolved in 4 ml of dry toluene under argon. To the solution, K2CO3 (aq) (2.0 M, 2.8 ml) was added. After stirring for 48 h at 100 °C, the reaction mixture was reprecipitated into a large amount of methanol. PTF was collected by filtration, dried under vacuum, and obtained as an orange powder (101 mg). 1H NMR (270 MHz, CD2Cl2): δ 7.7–7.6 (fluorene–H, br), 7.41 (thiophene–H, s), 2.03 (CH2(CH2)6CH3, br), 1.25–0.77 (CH2(CH2)6CH3, br). GPC (polystyrene standard): Mn = 4400, Mw = 6500, Mw/Mn = 1.47.Synthesis of PBT
To a stirred solution of 1 (227 mg, 0.50 mmol) in CHCl3 (5 ml), FeCl3 (2.1 g, 13 mmol) was added. The reaction mixture was stirred for 24 h at room temperature. The mixture was then poured into a large amount of MeOH. The obtained powder was filtered and washed with MeOH. Dedoping of the crude polymer with hydrazine monohydrate for 24 h gave an orange solid (265 mg, 96%). 1H NMR (270 MHz, CD2Cl2, ppm): δ 7.9–7.4 (fluorene–H, br), 7.2–7.4 (thiophene–H, br), 2.07 (Ar–CH2–C5H11, s), 1.6–1.4 (Ar–CH2–C4H8–CH3), 0.9 (–CH3). Anal. calcd. for (C37H44S2)n: C, 80.09; H, 8.36; S, 11.56. Found: C, 79.08; H, 8.20; S, 11.30%. GPC (polystyrene standard): Mn = 25100, Mw = 31300, Mw/Mn = 1.24.
General procedure for anodic halogenation using thin-layered cell
A chloroform solution containing 4 mg of PTs was cast on the ITO electrode (2 cm × 2 cm) and dried under reduced pressure. Paraffin paper was soaked in acetonitrile solution containing 100 mM Et4NX. This was sandwiched by the ITO electrode (anode) coated with polymer film and a bare zinc plate electrode (2 cm × 2 cm) as a cathode. Constant current (10 mA cm−2) was passed at room temperature with Metronix Corp. (Tokyo) constant current power supply model 5944. The amount of charge passed was monitored with Hokutodenko Coulomb/Amoperehour meter HF-201. Then the polymer film was washed with methanol, water and subsequently dried in vacuo. GPC (PHT–Cl): Mn = 3200, Mw = 13800, Mw/Mn = 4.31. GPC (PTF–Cl): Mn = 4800, Mw = 6600, Mw/Mn = 1.37. GPC (PBT–Cl): Mn = 27600, Mw = 32000, Mw/Mn = 1.16.
General procedure for anodic halogenation using beaker-type cell
A chloroform solution containing 4 mg of PTs was cast on the ITO electrode (2 cm × 2 cm) and dried under reduced pressure. A 20 ml of acetonitrile solution containing 100 mM Et4NX was prepared in an undivided cell, and de-aerated by argon bubbling. The ITO electrode coated with polymer film was introduced as an anode and a bare zinc plate electrode (2 cm × 2 cm) was used as a cathode. Constant current (10 mA cm−2) was passed at room temperature with Metronix Corp. (Tokyo) constant current power supply model 5944. The amount of charge passed was monitored with Hokutodenko Coulomb/Amoperehour meter HF-201. Then the polymer film was washed with methanol, water and subsequently dried in vacuo.Results and discussion
Synthesis of PTs
The target polymers containing thiophene unit were prepared as shown in Scheme 1. Poly(3-hexylthiophene) (PHT) was synthesized by the oxidative polymerization using FeCl3 as an oxidant. Gel permeation chromatography (GPC) molecular weights and polydispersity index for the PHT were estimated as Mn = 2700, Mw = 10400, Mw/Mn = 3.81, respectively. Poly(thiophene-alt-9,9-dioctylfluorene) (PTF) was prepared via Suzuki–Miyaura coupling polymerization of the corresponding monomers (GPC data: Mn = 4400, Mw = 6500, Mw/Mn = 1.47). Poly(bithiophene-alt-9,9-dioctylfluorene) (PBT) was obtained by the oxidative polymerization of bithiophene compound 1 (GPC data: Mn = 25100, Mw = 31300, Mw/Mn = 1.24).
 | ||
| Scheme 1 Synthesis of PTs. | ||
Cell design and electrochemical halogenation of PTs
A thin-layered cell was prepared by sandwiching paraffin paper retaining electrolyte solution (up to 0.1 ml) between an anode coated with PTs (4 mg) and a cathode. Indium tin oxide (ITO) (2 × 2 cm2) was used as the anode to monitor the doping behavior, zinc (Zn) plate (2 × 2 cm2) was used as a cathode,23 and 100 mM tetraethylammonium halide/acetonitrile was used as the electrolyte solution. The thickness between both electrodes was up to 70 μm. A conventional beaker-type cell was also set up with a fixed distance (1 cm) between an ITO anode (2 × 2 cm2) coated with polymer and a Zn cathode (2 × 2 cm2) in the same electrolyte solution (20 ml). Constant current electrolysis (10 mA cm−2) was carried out using both cells at room temperature (Scheme 2). After electrolysis, the polymer film was purified by washing with methanol and finally dried in vacuo. The PTs were then re-dissolved in an organic solvent (e.g., CDCl3) for spectroscopic analyses.24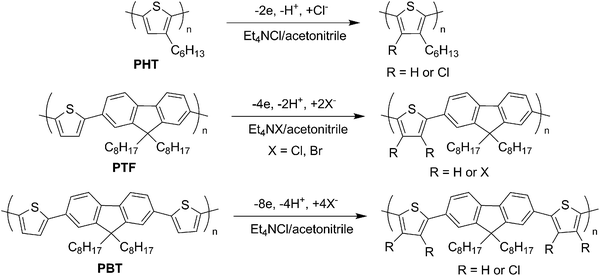 | ||
| Scheme 2 Electrochemical halogenation of PTs. | ||
To confirm the feasibility and limitations of the thin-layered cell, we investigated the chlorination of PHT (theoretical amount of electricity: 2.0 F mol−1), and the halogenation of PTF (theoretical amount of electricity: 4.0 F mol−1) and PBT (theoretical amount of electricity: 8.0 F mol−1) under various conditions. The halogenation reaction involves the doping of PTs, followed by the nucleophilic attack of the dopant chloride anion.10,25 The reaction ratio, RR (i.e., the ratio of the number of substituted chlorines per reactive points of repeating thiophene unit), was thus estimated by comparing the intensities of the protons on the thiophene ring and the methylene protons of the octyl groups in 1H NMR spectra.
Table 1 shows that the beaker-type cell and the layered cell gave almost the same reaction ratio (RR) for the anodic chlorination of PHT to produce PHT–Cl (entries 1 and 2). On the other hand, the CE of the reaction for the layered cell was significantly improved and was approximately 9 times that for the beaker-type cell. Anodic chlorination of PTF in the layered cell provided PTF–Cl in high RR and CE after 4.0–6.0 F mol−1 of charge was passed (entries 4 and 5), whereas the beaker-type cell resulted in saturation of the RR (85%) even after 30 F mol−1 of charge was passed (entry 3). Therefore, significant improvement of the CE was achieved by the layered cell. One possible reason for the limited RR (entry 3) is an increase of the oxidation potential of PTF during the course of chlorination. Under constant current conditions, the circuit including the sandwiched paper containing electrolyte between the electrodes may supply a higher potential to the anode compared to the beaker-type cell. Therefore, a high potential was applied to PTF, which caused efficient doping of nucleophiles to overcome the limitation of chlorination. In entries 6 and 7, the RRs for anodic bromination of PTF in both the beaker-type and layered cells were low. However, the CE obtained for the layered cell was much higher. Anodic iodination did not proceed, even after excess charge was passed using both cells (entries 8 and 9). Easily oxidizable iodide anion interfered the desired oxidation of PTF and resulted in recovery of the film.10
| Entry | PTs | Cell a | X− | Charge passed/F mol−1 | RR (%)b | CE (%)b |
|---|---|---|---|---|---|---|
| a A: beaker-type cell. B: thin-layered cell. b Estimated by 1H NMR. c Estimated by EDX. d Reaction was terminated by a decrease of current. | ||||||
| 1 | PHT | A | Cl − | 24 | 76 (72)c | 6 |
| 2 | PHT | B | Cl − | 2.5 | 73 (69)c | 58 |
| 3 | PTF | A | Cl − | 30 | 87 (85)c | 12 |
| 4 | PTF | B | Cl − | 6 | 100 (100)c | 67 |
| 5 | PTF | B | Cl − | 4 | 82 | 82 |
| 6 | PTF | A | Br − | 70 | 54 (56)c | 3 |
| 7 | PTF | B | Br − | 4 | 40 (34)c | 40d |
| 8 | PTF | A | I− | 70 | Trace | — |
| 9 | PTF | B | I− | 10 | Trace | — |
| 10 | PBT | A | Cl − | 80 | 71 (74)c | 7 |
| 11 | PBT | B | Cl − | 12 | 98 (99)c | 66 |
In entries 10 and 11, we examined the anodic chlorination of PBT for the first time. The results of 1H NMR and EDX analysis for the polymers are shown in Fig. 2. The chlorination ratio of PBT using the beaker-type cell was 71% (entry 10), whereas the anodic chlorination of PBT using the layered cell (12 F mol−1) reached a high RR and a high CE (entry 11). Throughout this study, the RRs, estimated by 1H NMR were in good agreement with the results from energy dispersive X-ray analysis (EDX). In particular, the completion of the chlorination reaction in the thin-layered cell (entries 4 and 11) is significant because the optical and electrochemical properties of the resultant polymers would be drastically changed compared to those of the precursor polymers. Furthermore, GPC analysis for the polymers indicated that neither propagation nor dissociation of the polymers occurred after the electrochemical reaction.
 | ||
| Fig. 2 (a) 1H NMR spectra of PBT before and after electrochemical chlorination in CD2Cl2.(b) EDX analysis of PBT and PBT–Cl on Si-wafer. | ||
The limitation of chlorine substitution for the beaker-type cell is due to the increase of the oxidation potential of the polymer with increasing chlorination ratio.9,10 To elucidate the reason why the thin-layered cell yielded high RRs and CEs for the chlorination of PTs, linear sweep voltammetry measurements were carried out for the PTF film using both beaker-type and thin-layered cells (Fig. 3). The current density of PTF using the thin-layered cell was lower than that using beaker-type cell. At the same current density, the potential applied to the working electrode of the thin-layered cell was higher than that of the beaker-type cell. The paraffin paper acts as a resistance layer as well as a medium for containment of the electrolytic solution, and as a separator of the anode and cathode. Therefore, constant current electrolysis in the thin-layered cell results in application of high potential to the polymer film on the anode, which improves the current efficiency and enables complete substitution with chlorine atoms.
 | ||
| Fig. 3 Linear sweep voltammograms of PTF measured in the solid state in 100 mM Et4NClO4/acetonitrile at a sweep rate of 50 mV s−1. | ||
The advantage of the layered cell is not only the improved reactivity, but also avoidance of technical problems. The electrochemical reaction of polymers can sometimes be terminated by detachment of the film from the electrode due to the doping by bulky counter ions. However, with the sandwiched structure, the polymer film is pressed to the electrode, which prevents film detachment; therefore, carrier paths between the polymer and the electrode can be sufficiently maintained. Moreover, the cell requires only 0.1 ml of electrolytic solution (for 4 mg of PTs), which is regarded as an atom-economical method.
Optical properties of chlorinated PTs
Fig. 4a shows that the UV-vis absorption maximum of PTF was blue-shifted after chlorination (PTF–Cl). The introduction of chlorine atoms at the 3- and 4-positions of the thiophene ring in the polymer causes steric repulsion between the adjacent thiophene and fluorene units, and as a result, the effective conjugation length is shortened.9 In our previous work, we observed a blue-shift of the absorption maximum (ca. 30 nm) when PTF was chlorinated (RR = 87%).9,10 In contrast, PTF–Cl (RR = 100%) prepared with the layered cell exhibited a larger blue shift (46 nm) due to complete substitution. | ||
| Fig. 4 (a) UV-vis absorption spectra of PTF and PTF–Cl in chloroform and in the solid state. (b) PL spectra of PTF and PTF–Cl in chloroform solution and in the solid state. (c) Photographs of PTF (RR = 100%) under UV irradiation before (left) and after (right) chlorination. | ||
The PL properties of PTF could be tuned from green to blue luminescence by electrochemical chlorination. PTF–Cl (RR = 100%) exhibited deep blue emission in dilute CHCl3 solution and in the solid state with emission maxima at 453 and 468 nm, respectively, whereas PTF exhibited green emission with emission maxima at 470, 507 nm in CHCl3 and 493, 522 nm in the solid state, respectively (Fig. 4b and c). The emission maxima of chlorinated PTF (RR = 87%) in chloroform solution and in the solid state were 467 and 482 nm, respectively.
Conclusion
We have successfully demonstrated that the electrochemical halogenation of PTs using a thin-layered cell afforded high current efficiencies. The reactivity of PTF and PBT was improved by the efficient doping of nucleophiles to the polymer film. In addition, the optical and electrochemical properties of the obtained polymers were significantly changed by the complete substitution of chlorine atoms onto the thiophene ring.Acknowledgements
This study was supported by a Grant-in-Aid for Young Scientists (no. 20850015). We thank Prof. Mahito Atobe at Yokohama National University for assistance with the EDX measurements.Notes and references
- F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Hawker, Nat. Chem., 2010, 2, 207 CrossRef CAS.
- T. S. Hansen, A. E. Daugaard, S. Hvilsted and N. B. Larsen, Adv. Mater., 2009, 21, 4483 CrossRef CAS.
- S. V. Orski, A. A. Poloukhtine, S. Arumugam, L. Mao, V. V. Popik and H. Locklin, J. Am. Chem. Soc., 2010, 132, 11024 CrossRef CAS.
- J. Bouffard, M. Watanabe, H. Takaba and K. Itami, Macromolecules, 2010, 43, 1425 CrossRef CAS.
- S. Inagi, S. Hayashi and T. Fuchigami, Chem. Commun., 2009, 1718 RSC.
- S. Hayashi, S. Inagi and T. Fuchigami, Macromolecules, 2009, 42, 3755 CrossRef CAS.
- S. Inagi, K. Koseki, S. Hayashi and T. Fuchigami, Langmuir, 2010, 26, 18631 Search PubMed.
- S. Hayashi, S. Inagi, K. Hosaka and T. Fuchigami, Synth. Met., 2009, 159, 1792 Search PubMed.
- S. Inagi, S. Hayashi, K. Hosaka and T. Fuchigami, Macromolecules, 2009, 42, 3881 Search PubMed.
- S. Inagi, K. Hosaka, S. Hayashi and T. Fuchigami, J. Electrochem. Soc., 2010, 157, E88 Search PubMed.
- S. Inagi, Y. Ishiguro, M. Atobe and T. Fuchigami, Angew. Chem., Int. Ed., 2010, 49, 10136 Search PubMed.
- Y. Li, K. Kamata, S. Asaoka and T. Iyoda, Org. Biomol. Chem., 2003, 1, 1779 RSC.
- Z. Qi, N. G. Rees and P. G. Pickup, Chem. Mater., 1996, 8, 701 CrossRef CAS.
- P. Soudau, L. Breau and D. Bélanger, Langmuir, 2000, 16, 4362 CrossRef CAS.
- E. Kim, Y. Liu, X.-W. Shi, X. Yang, W. E. Bentley and G. F. Payne, Adv. Funct. Mater., 2010, 20, 2683 Search PubMed.
- T. Michinobu, J. Am. Chem. Soc., 2008, 130, 14074 CrossRef CAS.
- Y. Li and T. Michinobu, Polym. Chem., 2010, 1, 72 RSC.
- O. Yurchenko, J. Heinze and S. Ludwigs, ChemPhysChem, 2010, 11, 1637 Search PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS.
- M. Higuchi, Polym. J., 2009, 41, 511 Search PubMed.
- L. Liu, C.-L. Ho, W.-Y. Wong, K.-Y. Cheung, M.-K. Fung, W.-T. Lan, B. Djurisic and W.-K. Chan, Adv. Funct. Mater., 2008, 18, 2824 CrossRef CAS.
- L. A. Estrada, V. A. Montes, G. Zyryanov and P. Anzenbacher, Jr, J. Phys. Chem. B, 2007, 111, 6983 CrossRef CAS.
- Zinc was the best cathode to achieve electrochemical halogenation using the thin-layered cell because of its higher overpotential for evolution of hydrogen than platinum. The generated hydrogen gas disrupts the further electrolysis.
- The product polymers were dissolved in chloroform completely. This suggests that no closs-linked material was produced by the electrode reaction.
- NMR analysis for the polymers revealed the uniformity of the reaction within the mass of the film.
| This journal is © The Royal Society of Chemistry 2011 |