Living/controlled polymerization of 4-methyl-1-pentene with α-diimine nickel-diethylaluminium chloride: effect of alkylaluminium cocatalysts†
Haiyang
Gao
*a,
Xiaofang
Liu
a,
Ying
Tang
a,
Jin
Pan
a and
Qing
Wu
*ab
aDSAPM Lab, Institute of Polymer Science, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou, 510275, China. E-mail: gaohy@mail.sysu.edu.cn; ceswuq@mail.sysu.edu.cn; Fax: +86-20-84114033; Tel: +86-20-84113250
bPCFM Lab, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China
First published on 2nd April 2011
Abstract
4-Methyl-1-pentene (4MP) was polymerized with a classical α-diimine nickel complex [(2,6-(iPr)2C6H3)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(acenaphthene)CN(2,6-(iPr)2C6H3))NiBr21] in the presence of various alkylaluminium compounds. Influences of cocatalysts on 4MP polymerization behavior were evaluated in detail. The different effects of trialkylaluminium cocatalysts between ethylene polymerization and 4-methyl-1-pentene polymerization were observed. Inexpensive diethylaluminium chloride (DEAC) compound could replace methylaluminoxane (MAO) as a more active cocatalyst for 4MP polymerization, and the influences of polymerization parameters including temperature and [Al]/[Ni] mole ratio were examined. At 0 °C, living/controlled polymerization of 4-methyl-1-pentene (4MP) was also achieved using inexpensive DEAC as cocatalyst, and trialkylaluminium compounds as chain transfer agents were closely relevant to achieve living/controlled polymerization. The obtained poly(4-methyl-1-pentene)s are amorphous elastomers with low glass transition temperature (Tg). Nuclear magnetic resonance (NMR) analyses showed that various branches such as methyl, isobutyl, long 2-methylalkyl branches are present in the polymer.
C(acenaphthene)CN(2,6-(iPr)2C6H3))NiBr21] in the presence of various alkylaluminium compounds. Influences of cocatalysts on 4MP polymerization behavior were evaluated in detail. The different effects of trialkylaluminium cocatalysts between ethylene polymerization and 4-methyl-1-pentene polymerization were observed. Inexpensive diethylaluminium chloride (DEAC) compound could replace methylaluminoxane (MAO) as a more active cocatalyst for 4MP polymerization, and the influences of polymerization parameters including temperature and [Al]/[Ni] mole ratio were examined. At 0 °C, living/controlled polymerization of 4-methyl-1-pentene (4MP) was also achieved using inexpensive DEAC as cocatalyst, and trialkylaluminium compounds as chain transfer agents were closely relevant to achieve living/controlled polymerization. The obtained poly(4-methyl-1-pentene)s are amorphous elastomers with low glass transition temperature (Tg). Nuclear magnetic resonance (NMR) analyses showed that various branches such as methyl, isobutyl, long 2-methylalkyl branches are present in the polymer.
Introduction
Over the past decade, α-diimine nickel complexes have attracted considerable attentions in olefin polymerization field since the precursory work of Brookhart and co-workers.1–4 In the combination with cocatalysts, these complexes have been shown to be primarily useful for ethylene polymerizations and produce a highly branched to moderately branched polyethylene because of chain walking.1–6 In contrast to ethylene polymerization, polymerization of α-olefins using α-diimine nickel catalysts generally leads to unique chain-straightened poly(α-olefin)s because of the chain straightening processes (1,ω-enchainment or 2,ω-enchainment).7–15Previous works have strongly supported that the active species of α-diimine nickel catalyst in olefin polymerization is a cationic alkyl-nickel paired with organoaluminate as the counteranion.16,17 Typically, methylaluminoxane (MAO) was used to activate α-diimine nickel complexes to form cationic alkyl-nickel active species. Alkylaluminium compounds like diethylaluminium chloride (DEAC), 1,3-dichloro-1,3-diisobutyldialuminoxane (DCDAO), trimethylaluminium (TMA), triethylaluminium (TEA) and triisobutylaluminium (TIBA) can also replace MAO as cocatalysts and have important influences on catalytic activity for ethylene polymerization and microstructure of the obtained polyethylene because of their different Lewis acidity, alkylating power, as well as interaction with nickel center and bulkiness of organoaluminate counteranion.18–25 Though a few papers on the effect of cocatalyst for ethylene polymerization were published, the uses of alkylaluminium for α-olefins polymerizations with late transition metal complexes are rare in the current literature. The reported results in the literature have shown that the catalytic behavior of α-diimine nickel complexes and the microstructure of the obtained polyolefin are fairly correlative to the cocatalyst used.10,24,25
We have recently reported that a branched α-olefin, 4-methyl-1-pentene (4MP) can be polymerized using α-diimine nickel complexes after activation of dried MAO (dMAO),26 and living/controlled polymerization can also be achieved at 0 °C.27 In this paper, we further emphatically investigated the effect of the cocatalysts on the branched α-olefin (4-methyl-1-pentene) polymerization behavior. One observed the different effect of trialkylaluminium cocatalysts between ethylene polymerization and 4-methyl-1-pentene polymerization. DEAC could more effectively activate α-diimine nickel precursor than MAO, and living/controlled polymerization of 4-methyl-1-pentene (4MP) was also achieved using inexpensive DEAC as cocatalyst. We also disclosed trialkylaluminium compounds were closely relevant to achieve living/controlled polymerization. Additionally, the NMR analyses were performed on the poly(4-methyl-1-pentene)s to gain a better understanding the influences of cocatalysts and reaction temperature on polymer microstructure.
Experimental
All manipulations involving air- and moisture- sensitive compounds were carried out under an atmosphere of dried and purified nitrogen with standard vacuum-line, Schlenk, or glovebox techniques.Materials
Toluene was dried over sodium metal and distilled under nitrogen. 4-Methyl-1-pentene (4MP) was purchased from Acros, dried over CaH2, and distilled under nitrogen before storing over molecular sieves. MAO (methylaluminoxane solution, 10 wt. % in toluene), diethylaluminium chloride (DEAC) 1.0 M in hexane), trimethylaluminium (TMA) 98%), triethylaluminium (TEA) 93%) and triisobutylaluminium (TIBA) 95%) were purchased from Acros. Dried methylaluminoxane (dMAO) was prepared by partial hydrolysis of trimethylaluminium (TMA) in toluene at 0–60 °C with Al2(SO4)3·18H2O as water source with the initial [H2O]/[TMA] molar ratio of 1.3. Volatiles were removed in vacuum at 60 °C for 5 h, until a free flowing white powder was obtained. The α-diimine nickel complex was prepared according to the reported procedures,1,2 and characterized by elemental analysis. Other commercially available reagents were purchased and used without purification.Polymerization
In a typical procedure, the appropriate alkylaluminium cocatalyst was introduced into the round-bottom Schlenk flask, and then 4MP monomer was added via a syringe. Toluene and a nickel complex solution were syringed into the well-stirred solution in that order, and the total reaction volume was kept at 10 mL. The polymerization reaction was continuously stirred for an appropriate period at the polymerization temperature. Except for 0 °C reaction, which was maintained with an ice-water bath, the other reaction temperatures were controlled with an external oil bath in polymerization experiments. The polymerizations were terminated by the addition of 200 mL of acidic ethanol (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ethanol/HCl). The resulting precipitated polymers were collected and treated by filtration, washing with ethanol several times, and drying in a vacuum at 40 °C until a constant weight was achieved.
:5 ethanol/HCl). The resulting precipitated polymers were collected and treated by filtration, washing with ethanol several times, and drying in a vacuum at 40 °C until a constant weight was achieved.
Characterization
The molecular weight distributions (PDI = Mw/Mn) of the poly(4MP)s were determined on Waters GPC2000 at 135 °C with standard polystyrene as the reference, and 1,2,4-trichlorobenzene was employed as the eluent with a flow rate of 1.0 mL min−1. DSC analysis was conducted with a Perkin Elmer DCS-7 system. The DSC curves were recorded at second heating curves from −50 °C to 150 °C at a heating rate of 10 °C min−1 and a cooling rate of 10 °C min−1. 1H NMR and 13C NMR spectra were carried out on a Varian Mercury-plus 500 MHz spectrometer at 120 °C. Sample solutions of the polymer were prepared in o-C6D4Cl2 and o-C6H4Cl2 mixture solvent (volume ratio: 20/80) in a 10 mm tube. The spectra of the quantitative 13C NMR were taken with a 74° flip angle, an acquisition time of 1.5 s, and a delay of 4.0 s. The total branching degree (BD) was calculated by the eqn (1). | (1) |
The fraction of 1,2-insertions and 2,1-insertion can be calculated by eqn (2) and (3), respectively.27
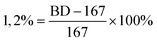 | (2) |
 | (3) |
Results and discussion
Effect of alkylaluminium compounds
Our previous results have shown that bulky α-diimine nickel complex [(2,6-(iPr)2C6H3)NC(acenaphthene)CN(2,6-(iPr)2C6H3))NiBr21] can actively polymerize 4MP to produce high molecular weight polymer,271 was therefore selected as a precursor for 4MP polymerization to investigate the effect of cocatalysts. α-Diimine nickel complex 1 was tested in 4MP polymerization using commercial MAO, dMAO, DEAC, TMA, TEA, and TIBA as cocatalysts respectively, and polymerization results with various cocatalysts are listed in Table 1.
| Entry | Cocatalyst | Yield (g) | Activity b | M n c (kg mol−1) | M w/Mnc | Efficiencyd | T g e (°C) | BDf | 2,1-ins.g |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: T = 0 °C; 10 μmol of complex; [Al]/[Ni] = 250; t = 1 h; 4MP = 2 g; solvent, toluene; total volume, 10 mL. b In units of kg (mol Ni h)−1. c Determined by GPC relative to polystyrene standards. d Activation efficiency, which is calculated by the ratio of theoretical active sites to the amount of nickel precursor used. e Determined by DSC. f BD: branching degree, the number of methyl carbon in every 1000 carbons, determined by 1H NMR. g 2,1-insertion was calculated by eqn (3). | |||||||||
| 1 | dMAO | 0.16 | 16 | 43 | 1.09 | 0.37 | −17.0 | 292 | 25 |
| 2 | MAO | 0.15 | 15 | 39 | 1.19 | 0.38 | −16.7 | 290 | 26 |
| 3 | DEAC | 0.29 | 29 | 32 | 1.07 | 0.91 | −24.2 | 253 | 48 |
| 4 | TMA | 0.09 | 10 | 8 | 4.22 | — | −25.3 | 246 | — |
| 5 | TEA | 0.07 | 7 | 4 | 3.56 | — | −24.8 | 249 | — |
| 6 | TIBA | Oligomer |
Table 1 clearly shows that the activity of the catalytic system depends on the combination of nickel precursor and cocatalysts. When MAO and dMAO were used as cocatalysts one observed the nearly same activity for 4MP polymerization. Inexpensive DEAC compound can effectively replace MAO as the cocatalyst, and DEAC in combination with 1 is more active than MAO by a factor of ∼2. When trialkylaluminium compounds including TMA, TEA, and TIBA were used instead of MAO respectively, catalytic activities for 4MP polymerization obviously decreased. The general order of catalytic activity for 4MP polymerization is DEAC > dMAO ≈ MAO > TMA > TEA > TIBA, illustrating that DEAC is the most active cocatalyst in our study. A similar general trend in catalytic activity (DEAC > MAO > trialkylaluminium) has been observed for ethylene polymerization with α-diimine complexes.21,24 It is likely that trialkylaluminium compounds as cocatalysts can provide a closer ion pair than DEAC and MAO according to olefin-separated ion-pairs model. In contrast to ethylene polymerization,21,241/TMA system exhibits a higher activity and produced poly(4MP) with a higher molecular weight than 1/TIBA. Considering the fact that TMA is a stronger chain transfer agent than TIBA, a reasonable explanation is that the bulky organoaluminate counteranion may slow down coordination and insertion of the 4MP monomer with enhanced steric bulk.
The molecular weight and molecular weight distributions (Mw/Mn) of the poly(4MP)s were shown to be strongly dependent upon the nature of the cocatalyst. Though 1/MAO or 1/dMAO showed the lower catalytic activity for 4MP polymerization than 1/DEAC, both catalytic systems produced the higher molecular-weight polymers (entries 1 and 2 vs. entry 3). It is interesting to note that narrow distributions of the polymers can be observed using dMAO (1.09), MAO (1.19), and DEAC (1.07) as cocatalysts, respectively, indicating that 4MP polymerization proceeds in a living/controlled manner under the adopted conditions. Therefore, the activation efficiency can be roughly estimated according to the ratio of theoretical active sites in the catalytic system, which is calculated gravimetrically on the basis of the weight of the produced polymer and experimental number-average molecular weights, to the amount of nickel precursor used. The calculation results show activation efficiencies of the 1/dMAO and 1/MAO catalytic systems are 0.37 and 0.38 respectively, while that of the 1/DEAC catalytic system is 0.91, demonstrating that DEAC as a cocatalyst can remarkably promote activation efficiency of α-diimine nickel precursor. This observation is similar to Peruch and Merna's investigation by UV-vis determination of nickel active species.11,13
When trialkylaluminium compounds were employed instead of MAO or DEAC, the molecular weight of the polymer significantly decreased and the molecular weight distribution (Mw/Mn) also became obviously broad. 1 in combination with TIBA gave oil product which indicates that oligomerization process was possibly occurring. This result strongly indicates that trialkylaluminium compounds are a kind of chain transfer agents for 4MP polymerization.23,24 Therefore, it is reasonable that more narrow distribution can be observed using dMAO instead of MAO as the cocatalyst because of the removal of volatiles (TMA) under vacuum. Also note that the Mw/Mn value of the polymer obtained by 1/MAO is 1.19, which is larger than that of the polymer obtained by 1/DEAC (1.07). Fig. 1 clearly shows that GPC trace of the polymer obtained by 1/MAO contains small amount of tail peak in low molecular weight fraction, but the trace of the poly(4MP) obtained by 1/DEAC is more symmetric. This result arises from the occurrence of growing chain transfer to TMA in the 1/MAO system, which is invariably contained in commercial MAO solutions.
 | ||
| Fig. 1 GPC traces of the polymers catalyzed by 1/DEAC and 1/MAO (entries 2 and 3). | ||
The glass transition temperatures (Tg) and branching degree (BD) of the polymers obtained with 1 in combination with DEAC are lower than those obtained with MAO or dMAO, indicative of higher degree of chain walking reaction in the 1/DEAC system.27
Influences of temperature and [Al]/[Ni] mole ratio
Considering the good catalytic activity and narrow molecular weight distribution of the obtained polymer, inexpensive DEAC was chosen as the cocatalyst for 1 to further investigate the influences of polymerization parameters such as temperature, and [Al]/[Ni] ratio in detail. Table 2 lists the polymerization results of 4MP with 1/DEAC catalytic system under various conditions. With an increase in the reaction temperature, the catalytic activity for 4MP polymerization increased, and then decreased. The highest activity was observed at 40 °C. Besides, raising temperature also affects molecular weight of the produced polymer and causes an increase in the molecular weight distribution (Mw/Mn) consistently. When polymerization was carried out at 0 °C, the poly(4MP) with relatively low molecular weight (Mn = 32,000) was obtained. At 20 °C, the highest molecular weight (Mn = 105,000) was produced. Higher temperatures caused a decrease in the molecular weight of the product.| Entry | T/°C | [Al]/[Ni] | Yield (g) | Activityb | M n c(kg mol−1) | M w/Mnc | T g d (°C) | BDe | 2,1-ins.f |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: 10 μmol of complex; 2 g 4MP monomer; 1 h; solvent, toluene; total volume, 10 mL. b In units of kg (mol Ni h)−1. c Determined by GPC relative to polystyrene standards. d Determined by DSC. e BD: branching degree, the number of methyl carbon in each 1000 carbons, determined by 1H NMR. f 2,1-Insertion was calculated by eqn (3). | |||||||||
| 3 | 0 | 250 | 0.29 | 29 | 32 | 1.07 | −24.2 | 253 | 48 |
| 7 | 20 | 250 | 0.82 | 82 | 105 | 1.31 | −27.6 | 248 | 51 |
| 8 | 40 | 250 | 1.05 | 105 | 97 | 1.61 | −29.2 | 242 | 54 |
| 9 | 60 | 250 | 0.34 | 34 | 24 | 1.66 | −33.8 | 240 | 56 |
| 10 | 0 | 100 | 0.27 | 27 | 32 | 1.07 | −24.0 | 255 | 47 |
| 11 | 0 | 500 | 0.33 | 33 | 34 | 1.08 | −23.8 | 257 | 46 |
| 12 | 0 | 1000 | 0.31 | 31 | 33 | 1.09 | −24.1 | 254 | 47 |
The branching degree and Tg value (see Fig. 2) were observed to decrease obviously with increasing temperature, which results from chain walking. For example, the branching degree decreases from 253 per 1000 carbons at 0 °C to 240 per 1000 carbons at 60 °C, and corresponding Tg value reduces from −24.2 to −33.8 °C. In comparison with our reported results with 1/dMAO,271/DEAC shows a same tendency in the influence of temperature on catalytic activity and microstructure of the poly(4MP), but lower branching degrees and Tg values of the poly(4MP) can be observed using DEAC as cocatalyst.
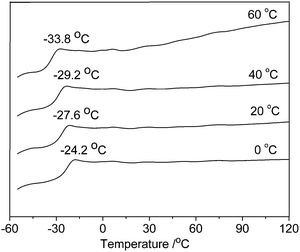 | ||
| Fig. 2 DSC thermograms of poly(4MP) prepared at different temperatures with 1/DEAC. | ||
The influence of [Al]/[Ni] mole ratio was also investigated at 0 °C. It is interesting to note that both the catalytic activity for 4MP polymerization and the molecular weight of the product are nearly invariable with respect to the [Al]/[Ni] ratio. This observation is further indicative of no occurrence of chain transfer to Al. Additionally, the influence of [Al]/[Ni] ratio on branching degree of the poly(4MP)s and insertion fashion is very slight, and all Tg of the polymers are observed around −24.0 °C, corresponding to ∼255 per 1000 carbons branching degree.
Living/controlled polymerization
Note that a narrow distribution of the polymer was observed at 0 °C, thus 4MP polymerizations with 1/DEAC were carried out at 0 °C and 250 [Al]/[Ni] mole ratio. Fig. 3 shows the GPC curves of the polymers obtained at different polymerization time, which shifts to the higher molecular weight region with the increasing reaction time. The Mn and Mw/Mn values are plotted against reaction time in Fig. 4. Number-average molecular weight of the polymers (Mn) grows linearly with reaction time and the Mw/Mn values remain very low (< 1.15), verifying that the polymerization undoubtedly proceeds in a living/controlled manner. This claim is also supported by the lack of the low molecular weight tails in the GPC traces as observed when decomposition of catalyst or chain transfer occurs during the 4MP polymerization. To the best of our knowledge, this is the first report on DEAC as a cocatalyst for living/controlled branched α-olefin polymerization with late transition metal precursors. | ||
| Fig. 3 GPC traces of poly(4MP)s prepared by 1/DEAC at different reaction time. | ||
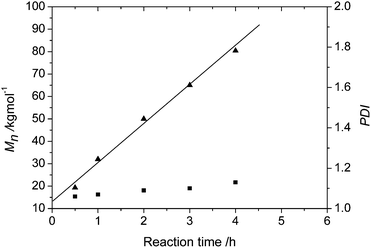 | ||
| Fig. 4 Plots of Mn (▲) and PDI (Mw/Mn) (■) as a function of reaction time for the polymerization of 4MP. | ||
Microstructure analysis of poly(4-methyl-1-pentene)
No melting points (Tm) of poly(4MP)s were detected by DSC analysis, indicating that the obtained polymers are amorphous elastomers. To have an insight into precise microstructure of the poly(4MP), investigations using 13C NMR spectroscopy were undertaken. 13C NMR of spectrum of poly(4MP) obtained by 1/DEAC is very complicated (Fig. 5) because of chain walking. Generally, this spectrum is similar to that of poly(4MP) obtained by 1/dMAO.27 On the basis of our previous resonance assignments of poly(4MP) obtained by 1/dMAO27 and with reference to model compound data found in the literature (detail resonance assignments can be seen in the ESI†), a branched structure can be safely identified including methyl branches (including isolated methyl, paired methyl, successive methyl), isobutyl, and long 2-methylalkyl branches such as 2,4-dimethylpentyl and 2-methylhexyl. There are no detectable ethyl, propyl, and butyl branches as evidenced by lack of a high field terminal methyl resonance (δ < 20 ppm). Additionally, isopropyl cannot be observed, whose methine appears at 29.1 ppm.28 The methyl on quaternary carbon at 27.3 ppm and quaternary carbon at 33.0 ppm are not present in the spectrum referring to assignment of the poly(4MP) prepared by cationic polymerization.28 These results strongly suggest that 4MP monomer cannot insert into secondary nickel-alkyl bonds. | ||
| Fig. 5 13C NMR spectra of poly(4MP) prepared by 1/DEAC and dMAO (entries 1 and 3). | ||
Like the 1/dMAO system, a set of branching types of the poly(4MP) produced by 1/DEAC system can also be explained by insertion fashions of monomer and chain walking.27 1,2-Insertion of 4MP and subsequent β-hydride elimination followed by nickel migration up to the primary carbon atom can result in 2,5-enchainment to give two short chain methyl branches in the polymer chain, while the 2,1-insertion of 4MP always results in 1,5-enchainment to give one short chain methyl branch in the polymer chain due to lack of occurrence of 4MP insertion in a secondary nickel-alkyl bond. Therefore, the fraction of 1,2-insertions can be predicted from the branching degree (methyl groups per 1000 carbons).27 Calculation results show that the actual ratio of 1,2 vs. 2,1-insertion is significantly different for both catalytic systems. One observes fewer 2,1-insertion occuring in the 1/dMAO system than in the 1/DEAC system (25% vs. 48%) under the same conditions (entry 1 vs. 3). This result may be attributed to different Lewis acidity and bulkiness of organoaluminate counteranion.23
The main difference between both spectra of the polymers obtained by 1/dMAO and DEAC is the intensity and integral of the peaks, indicating the change of the branching number. The signals in the region from 30 to 34 ppm (see Fig. 5) clearly show relatively few paired isobutyl-isobutyl units and more paired methyl-isobutyl units in the spectrum of the polymer produced by 1/DEAC, suggesting that more “straightened” polymer was produced by 1 using DEAC as the cocatalyst instead of dMAO. This result is consistent with DSC analysis and 1H NMR analysis, and can be reasonably explained by greater occurrence of 2,1-insertion in 1/DEAC.
Fig. 6 shows the dependence of branch structure of the polymer obtained by 1/DEAC on reaction temperature. An obvious trend is that isobutyl branches decrease, but long 2-methylalkyl branches increase with an increase in reaction temperature. Long branches such as 2-methylhexyl, and 2,4-dimethylpentyl are expected to arise from migration of nickel metal backward along the polymer. Besides, increasing temperature also results in an increase in 2,1-insertion of 4MP monomer from 48% to 56% (see Table 1), thus causing migration of nickel. These results support high temperature being favorable for chain walking and the 2,1-insertion of 4MP monomer.
 | ||
| Fig. 6 13C NMR spectra of poly(4MP) prepared by 1/DEAC at different temperatures (entries 6–9). | ||
Conclusions
Alkylaluminium compounds as cocatalysts have an important influence on 4MP polymerization behavior with α-diimine nickel precursor. The catalytic activity, the molecular weight, and molecular weight distributions of the obtained poly(4MP)s are shown to be strongly dependent upon the nature of the cocatalyst. Inexpensive DEAC compound can replace MAO as the more active cocatalyst for 4MP polymerization, and leads to the form of more “straightening” polymer because of more occurrence of 2,1-insertion. At low temperature (0 °C), 4MP polymerizations catalyzed by 1/DEAC can proceed in a living/controlled manner, and trialkylaluminium compounds as chain transfer agents are closely relevant to achieve living/controlled polymerization. The obtained poly(4-methyl-1-pentene)s are amorphous elastomers with various types of branches including 2,4-dimethylpentyl and 2-methylhexyl.Acknowledgements
The financial support by NSFC (Projects 20974125 and 20734004), the Science Foundation of Guangdong Province (Project 8251027501000018), the Fundamental Research Funds for the Central Universities (Projects 10lgpy10) and the Opening Fund of Laboratory Sun Yat-Sen University are gratefully acknowledged.Reference
- L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS.
- R. F. de Souza, R. S. Mauler, L. C. Simon, F. F. Nunes, D. V. S. Vescia and A. Cavagnolli, Macromol. Rapid Commun., 1997, 18, 795–800 CrossRef CAS.
- F. Liu, H. Hu, Y. Xu, L. Guo, S. Zai, K. Song, H. Gao, L. Zhang, F. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789–7796 CrossRef CAS.
- G. B. Galland, R. F. de Souza, R. S. Mauler and F. F. Nunes, Macromolecules, 1999, 32, 1620–1625 CrossRef CAS.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770–13771 CrossRef CAS.
- J. Yuan, L. C. Silva, P. T. Gomes, P. Valerga, J. M. Campos, M. R. Ribeiro, J. C. W. Chien and M. M. Marques, Polymer, 2005, 46, 2122–2132 CrossRef CAS.
- D. H. Camacho and Z. Guan, Macromolecules, 2005, 38, 2544–2546 CrossRef CAS.
- J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 4186–4187 CrossRef CAS.
- J. Merna, J. Cihlar, M. Kucera, A. Deffieux and H. Cramail, Eur. Polym. J., 2005, 41, 303–312 CrossRef CAS.
- J. Merna, Z. Hostalek, J. Peleska and J. Roda, Polymer, 2009, 50, 5016–5023 CrossRef CAS.
- F. Peruch, H. Cramail and A. Dffieux, Macromolecules, 1999, 32, 7977–7983 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410–420 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Arthur, E. B. Coughlin, S. D. Ittel, D. Tempel, L. K. Johnson, C. M. Killian and M. Brookhart, Macromolecules, 2001, 34, 362–371 CrossRef CAS.
- S. A. Svejda, L. K. Johnson and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 10634–10635 CrossRef CAS.
- D. Meinhard, P. Reuter and B. Rieger, Organometallics, 2007, 26, 751–754 CrossRef CAS.
- S. A. Svejda and M. Brookhart, Organometallics, 1999, 18, 65–74 CrossRef CAS.
- K. R. Kumar and S. Sivaram, Macromol. Chem. Phys., 2000, 201, 1513–1520 CrossRef CAS.
- L. C. Simon, R. S. Mauler and R. F. De Souza, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4656–4663 CrossRef CAS.
- G. Caroline, R. F. de Souza and K. B.-G. de Souza, Appl. Catal., A, 2007, 325, 87–90 CrossRef.
- R. S. Mauler, R. F. de Souza, D. V. V. Vesccia and L. C. Simon, Macromol. Rapid Commun., 2000, 21, 458–463 CrossRef CAS.
- R. F. de Souza, L. C. Simon, M. do Carmo and M. Alves, J. Catal., 2003, 214, 165–168 CrossRef.
- R. J. Maldanis, J. S. Wood, A. Chandrasekaran, M. D. Rausch and J. C. W. Chien, J. Organomet. Chem., 2002, 645, 158–167 CrossRef CAS.
- J. Peleska, Z. Hostalek, D. Hasalikova and J. Merna, Polymer, 2011, 52, 275–281 CrossRef CAS.
- In ref. 27, white powder MAO was used by removing volatiles in a vacuum. Herein, white powder MAO was called dried MAO (dMAO) in order to distinguish it from commercial MAO solution in toluene.
- H. Gao, J. Pan, L. Guo, D. Xiao and Q. Wu, Polymer, 2011, 52, 130–137 CrossRef CAS.
- G. Ferraris, C. Corno, A. Priola and S. Cesca, Macromolecules, 1977, 10, 188–197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00052g |
| This journal is © The Royal Society of Chemistry 2011 |