The effect of molecular weight on the supramolecular interaction between a conjugated polymer and single-walled carbon nanotubes
Patigul
Imin
a,
Fuyong
Cheng
b and
Alex
Adronov
*a
aDepartment of Chemistry and Chemical Biology, and the Brockhouse Institute for Materials Research, McMaster University, 1280 Main St. W., Hamilton, Ontario, L8S 4M1, Canada. E-mail: adronov@mcmaster.ca; Fax: +1 905 521 2773; Tel: +1 905 525 9140 ext 23514
bSteacie Institute for Molecular Sciences, National Research Council of Canada, 100 Sussex Drive, Ottawa, Ontario K1A 0R6, Canada
First published on 6th April 2011
Abstract
The synthesis and fractionation of poly[2,7-(9,9-dioctylfluorene)-alt-2,5-(3-dodecylthiophene)] resulted in the isolation of eight different molecular weight (Mw) samples ranging from Mn of 5 to 85 kg mol−1. These individual polymer samples were fully characterized by Gel Permeation Chromatography, Nuclear Magnetic Resonance, as well as absorption and fluorescence spectroscopy. Each sample was separately mixed and ultrasonicated with single-walled carbon nanotubes (SWNTs) in tetrahydrofuran, and the nanotube concentration within the resulting solutions was measured. It was found that the solubility of the polymer–SWNT complexes strongly depends on the Mw of the conjugated polymer, with a maximum concentration reached when Mw ranged between 10 and 35 kg mol−1. Higher and lower Mws resulted in substantially reduced nanotube concentrations.
Introduction
Carbon nanotubes (CNTs) have attracted significant interest in both academic and industrial research due to their exceptional mechanical, optical, and electronic properties.1,2 The potential displayed by carbon nanotubes within a variety of applications, including photovoltaic devices, field-effect transistors, and chemical sensors, has motivated the development of functionalization and processing methods that can be applied to these remarkable nanostructures.3–6 The low solubility and poor dispersability of unmodified carbon nanotubes within almost all common organic and aqueous solvents represent a significant hindrance in many of these potential applications. In an attempt to solve this problem, surface modification techniques have been developed in recent years, including covalent and non-covalent functionalizations, that enable the effective dispersion of individual carbon nanotubes within solvents and polymeric host materials.7–12 Although covalent functionalization strategies have been shown to significantly improve the solubility of CNTs in various organic and aqueous solvents, this process causes disruption of the perfectly conjugated sidewall structure of CNTs and diminishes the electronic and mechanical properties of the resulting materials.13,14 In contrast, supramolecular functionalization of carbon nanotubes with polymers or ionic surfactants provides an effective approach by which the solubility of carbon nanotubes can be enhanced while retaining all their intrinsic properties.Recently, much attention has been paid to the use of conjugated polymers as nanotube dispersing agents, owing to their ability to strongly adsorb onto the nanotube surface via π–π interactions.15–26 This approach displays a significant promise, allowing the incorporation of CNTs within a conjugated polymer matrix to produce polymer–nanotube composites displaying impressive conductivity and solution-phase processability, as well as improved mechanical and optical properties. Fluorene- and thiophene-based polymers are of particular interest with respect to the preparation of CNT composites due to their good solubility, processability, and unique opto-electronic characteristics.27–30 Several groups have reported the use of commercially available poly(fluorene) homopolymers and copolymers for the solubilization of single walled carbon nanotubes (SWNTs). In this work, it was shown that the dispersability of the carbon nanotubes is highly influenced by the length of side chains and the precise structures of the aromatic repeat units.31–39
We recently reported that both poly(fluorene) and poly(fluorene-alt-thiophene) can form strong supramolecular complexes with SWNTs, imparting an excellent solubility and solution stability in the absence of excess free polymer.40,41 In this work, it was noted that a high nanotube solubility was obtained when polymers having a molecular weight of approximately 10 kg mol−1 were used. Although it is well established that the molecular weight of a polymer directly influences its optical and physical properties, only a limited number of studies have been carried out to determine the effect of polymer molecular weight on nanotube solubility.42,43
In this study, the poly[2,7-(9,9-dioctylfluorene)-alt-2,5-(3-dodecylthiophene)] (PFT) was employed as a model compound. Using recycling preparative Gel Permeation Chromatography (GPC), a PFT sample of broad polydispersity (3.2) was fractionated to isolate eight different polymer fractions (Mn = 5–85 kg mol−1) with a relatively narrow polydispersity (∼1.7). The structural and optical properties of each fraction were characterized by 1H-NMR, absorption, and fluorescence spectroscopy. Additionally, the ability of each of these polymer fractions to effectively disperse the SWNTs in tetrahydrofuran (THF) was subsequently evaluated.
Experimental
General
Single-walled carbon nanotubes (SWNTs), prepared by the high-pressure carbon monoxide disproportionation (HiPco) process, were purchased from Carbon Nanotechnologies, Inc. (Houston, TX). All reagents and solvents were purchased from commercial suppliers and used as received. Ultrasonication was carried out in either a Branson Ultrasonics B1510 or a B2500 bath sonicator. Filtration was performed through a 450 nm pore Teflon membrane (Millipore). Polymer molecular weight and polydispersity index (PDI) were estimated from GPC analyses by using a Waters 2695 Separations Module equipped with a Waters 2996 photodiode array detector, a Waters 2414 refractive-index detector, a Waters 2475 fluorescence detector, and four Polymer Labs PL gel individual pore-size columns. Polystyrene standards were used for calibration, and THF was used as the eluent at a flow rate of 1.0 mL min−1. The concentrations of the soluble polymer-functionalized SWNTs were calculated from ultraviolet-visible (UV-Vis) absorption spectra measured using a Cary 50 UV-Vis spectrophotometer. Fluorescence spectra were measured using a Jobin-Yvon SPEX Fluorolog 3.22 equipped with a 450 W Xe lamp, double-excitation and double-emission monochromators, and a digital photon-counting photomultiplier. Slit widths were set to 2 nm band-pass on both excitation and emission. Correction for variations in lamp intensity over time and λ was achieved using a reference silicon photodiode. Nuclear Magnetic Resonance (NMR) spectra were measured on a Bruker AV 600 MHz spectrometer; the non-deuterated solvent signal was used as the internal standard for 1H-NMR spectra.Synthesis
Results and discussion
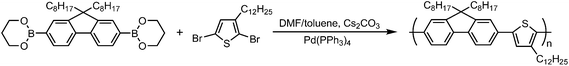
The synthesis of PFT was accomplished viapalladium-catalyzed Suzuki coupling starting from 9,9-dioctylfluorene-2,7-bis(trimethylene)borate and 2,5-dibromo-3-dodecyl-thiophene in a mixture of DMF and toluene (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), as depicted in Scheme 1.40 The resulting polymer was found to be readily soluble in common organic solvents such as THF, chloroform, dichloromethane, toluene, and dichlorobenzene. The polymerization, which proceeds via an uncontrolled step-growth process, yielded a polymer with high polydispersity. GPC analysis, using THF as the mobile phase and polystyrene standards, indicated a number average molecular weight (Mn) of 14 kg mol−1 with a polydispersity index (PDI) of 3.2. The GPC chromatogram of as-produced PFT (Fig. 1A) clearly shows the breadth of the molecular weight distribution. In order to separate this as-produced sample into a series of samples of different molecular weights, it was subjected to several rounds of recycling preparative GPC, resulting in the isolation of eight different fractions having Mn values ranging from approximately 5 to 85 kg mol−1, each having a PDI of 1.7 or lower (Table 1). The GPC chromatograms for each of these samples are shown in Fig. 1B.
:1 v/v), as depicted in Scheme 1.40 The resulting polymer was found to be readily soluble in common organic solvents such as THF, chloroform, dichloromethane, toluene, and dichlorobenzene. The polymerization, which proceeds via an uncontrolled step-growth process, yielded a polymer with high polydispersity. GPC analysis, using THF as the mobile phase and polystyrene standards, indicated a number average molecular weight (Mn) of 14 kg mol−1 with a polydispersity index (PDI) of 3.2. The GPC chromatogram of as-produced PFT (Fig. 1A) clearly shows the breadth of the molecular weight distribution. In order to separate this as-produced sample into a series of samples of different molecular weights, it was subjected to several rounds of recycling preparative GPC, resulting in the isolation of eight different fractions having Mn values ranging from approximately 5 to 85 kg mol−1, each having a PDI of 1.7 or lower (Table 1). The GPC chromatograms for each of these samples are shown in Fig. 1B.
 | ||
| Scheme 1 Synthesis of PFT. | ||
 | ||
| Fig. 1 GPC chromatogram of PFT (A) and normalized GPC chromatograms of PFT fractionations (B). | ||
| Polymer fractions (PFT) | M n/g mol−1 | PDI | UVλmax/nm |
|---|---|---|---|
| a | 83000 |
1.5 | 418 |
| b | 60000 |
1.6 | 412 |
| c | 42200 |
1.5 | 410 |
| d | 34000 |
1.7 | 408 |
| e | 19000 |
1.6 | 406 |
| f | 14000 |
1.6 | 404 |
| g | 11000 |
1.7 | 402 |
| h | 5700 | 1.6 | 398 |
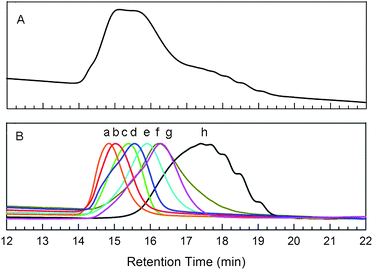
The structure of the fractionated polymer samples was further investigated by 1H-NMR spectroscopy. Fig. 2 shows the 1H-NMR spectra of the samples with highest (fraction a), intermediate (fraction e), and lowest (fraction h) molecular weights. All of the expected aliphatic and aromatic proton signals are clearly observable in these spectra. In particular, the distinct positions of the α-methylene protons of the side chains on both the thiophene monomer (2.75–2.64 ppm) and the fluorene monomer (2.05 ppm) allow the confirmation of the alternating copolymer structure. The integration of these signals provides the expected 1:2 molar ratio, consistent with the fact that the thiophene units exhibit a single aliphatic side chain, while the fluorene units are decorated with two. Additionally, small triplets (at 2.64 and 2.60 ppm) are observed adjacent to the α-methylene signal (2.75 ppm) within the thiophene units, which are attributed to the α-methylene protons in the terminal thiophene ring.44–46 The signals from the end group protons are negligible in the highest molecular weight fraction (fraction a), and the intensity of these signals increases with decreasing polymer molecular weight. The two separate triplets at different resonance frequencies in the lowest molecular weight fraction (g) are likely due to the two different regiochemical isomers possible for the terminal thiophene unit. The molecular weight of the polymer can be estimated from the integration of the α-methylene protons of the terminal thiophene to those present throughout the polymer main chain. Based on this 1H-NMR data, the molecular weight of polymersa, e and h was found to be 40.0 kg mol−1, 11.5 kg mol−1 and 5.0 kg mol−1, respectively. For the lower molecular weight fractions, these NMR values are in surprisingly good agreement with the molecular weight measurements from GPC.
 | ||
| Fig. 2 1H-NMR spectra of the PFTs, fractions a, e, and h, in THF. Insets show magnified views of the aromatic (6.8–8.0 ppm) and aliphatic (2.4–3.0 ppm) regions. | ||
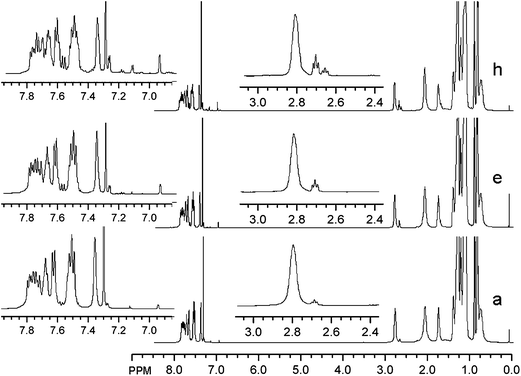
Further characterization of the separated polymer fractions by UV-Vis spectroscopy shows that, as the molecular weight increases, the λmax values shift bathochromically from 398 nm to 418 nm (Table 1 and Fig. 3A). This observation is consistent with previous reports for similar conjugated polymers.46,47 The emission spectra of the separated polymer fractions, excited at their individual absorption maxima and normalized for absorption intensity, are given in Fig. 3B. This data show no obvious changes in the emission maxima or intensities as a function of polymer molecular weight, which is again similar to what has been observed previously with comparable polymers.48
 | ||
| Fig. 3 UV-Vis absorption (A) and normalized emission spectra (B) of the individually fractionated PFT samples. | ||
The supramolecular interaction of the PFT with SWNTs was studied using our previously reported methods.40,41,49 Our prior experimental results showed that PFT can form strong supramolecular complexes with SWNTs. In order to determine the effect of polymer molecular weight on carbon nanotube solubility within the present polymer–SWNT complexes, polymer–nanotube composites containing various weight percentages of the PFT were prepared using bath sonication. In a typical procedure, 2.5 mg of carbon nanotubes were added to the polymer solution in 5 mL THF with varying polymer concentrations. This mixture was sonicated for 30 min, and the resulting solution was centrifuged at 2576g for 20 min. The vials were then kept undisturbed overnight. 1 mL of the supernatant was transferred to a 50 mL standard volumetric flask using a pipette and further diluted with THF. The solubility of the nanotubes within each sample was quantified using a spectrophotometric method in which a specific extinction coefficient for SWNTs at 700 nm, ε700 = 2.35 × 104 cm2 g−1, was used.22 The polymer–SWNT absorption at this wavelength originates solely from SWNTs, allowing estimation of the nanotube concentration without interference from polymer absorption, which was negligible at 700 nm.
In order to determine the optimal amount of polymer in the polymer–nanotube mixtures used to prepare solutions, the polymer:nanotube ratio was increased from 1:1 to 4:1 (wt/wt). As expected, the concentration of nanotubes in solution increased with increasing polymer content from the 1:1 ratio to the 3:1 ratio. However, when the polymer:nanotube ratio was increased to 4:1, quantitative flocculation and gelation of the mixture were observed. At this ratio, centrifugation resulted in complete precipitation of the nanotubes, leaving a clear, colourless polymer solution in the supernatant. The exact reason for this flocculation is unknown, and will be investigated in future studies. Since the highest nanotube solubility was obtained at a polymer:nanotube weight ratio of 3:1, this ratio was used for subsequent studies of the impact of polymer molecular weight.
Interactions of SWNTs with the individual fractionated PFT samples were investigated by mixing these components in THF, as described above. For each polymer fraction, a minimum of 3 separate solutions was prepared and measured, and the average SWNT concentration for each polymer fraction was recorded. Fig. 4 shows a plot of nanotube concentration as a function of polymer molecular weight. From this data, it is clear that the highest nanotube solubility, in the range of 500 to 800 mg L−1, was obtained with polymers having intermediate Mn values of 10 to 35 kg mol−1, while at higher or lower molecular weights the solubility was significantly lower. These results suggest that at low Mn, the π-stacking interaction between the oligomers and the nanotube surface is not strong enough to prevent re-aggregation of nanotubes into bundles. At molecular weights from 10 to 35 kg mol−1, the polymers can form strong supramolecular complexes with nanotubes and enhance nanotube solubility to a significant degree. However, at polymer molecular weights greater than 35 kg mol−1, a significant decrease of nanotube solubility was observed. This behaviour is attributed to the fact that the PFT polymer is itself not very soluble at high molecular weight. It is likely that aggregation of the higher molecular weight polymers with one another competes with the interaction of these polymers with the nanotube surface, leading to a decreased level of nanotube functionalization. However, further investigation of this phenomenon is required in order to fully understand the observed behaviour.
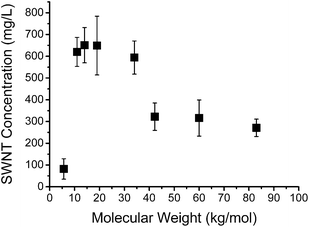 | ||
| Fig. 4 Plot of SWNT concentration as a function of PFT molecular weight in THF. Error bars represent the standard deviation from three separately prepared samples. | ||
Conclusion
PFT was synthesized, and the resulting polymer was successfully fractionated into eight different molecular weight fractions ranging from 5 to 85 kg mol−1 using recycling preparative GPC. The supramolecular interaction of these polymers with HiPco SWNTs was investigated, and it was found that the solubility of the resulting polymer–SWNT complexes strongly depends on the molecular weight of the polymer. In THF, the SWNT concentration reached a maximum when the polymer molecular weight was in the range of 10–35 kg mol−1, while higher and lower molecular weights resulted in a much lower solubility. Although this study focused upon the interactions of the PFTs with HiPco SWNTs, we believe that these results are applicable to other conjugated polymer–SWNT systems.Acknowledgements
Financial support for this work was provided by the Natural Science and Engineering Research Council of Canada (NSERC), Canada Foundation for Innovation (CFI), and Ontario Innovation Trust (OIT). PI gratefully acknowledges the Ontario Ministry of Training for an Ontario Graduate Scholarship (OGS).References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS.
- K. Bradley, J. C. P. Gabriel and G. Gruner, Nano Lett., 2003, 3, 1353–1355 CrossRef CAS.
- E. Kymakis and G. A. J. Amaratunga, Rev. Adv. Mater. Sci., 2005, 10, 300–305 CAS.
- M. M. Rahman, A. Umar and K. Sawada, Sens. Actuators, B, 2009, 137, 327–333 CrossRef.
- F. Wang, H. W. Gu and T. M. Swager, J. Am. Chem. Soc., 2008, 130, 5392–5393 CrossRef CAS.
- P. Liu, Eur. Polym. J., 2005, 41, 2693–2703 CrossRef CAS.
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637–659 RSC.
- P. M. Ajayan and J. M. Tour, Nature, 2007, 447, 1066–1068 CrossRef CAS.
- G. J. Bahun, F. Y. Cheng, C. M. Homenick, G. Lawson, J. Zhu and A. Adronov, The Polymer Chemistry of Carbon Nanotubes, in Chemistry of Carbon Nanotubes, American Scientific Publisher, Stevenson Ranch, CA, 2008 Search PubMed.
- I. Singh, P. K. Bhatnagar, P. C. Mathur, I. Kaur, L. M. Bharadwaj and R. Pandey, Carbon, 2008, 46, 1141–1144 CrossRef CAS.
- J. U. Lee, J. Huh, K. H. Kim, C. Park and W. H. Jo, Carbon, 2007, 45, 1051–1057 CrossRef CAS.
- A. Garg and S. B. Sinnott, Chem. Phys. Lett., 1998, 295, 273–278 CrossRef CAS.
- E. T. Mickelson, C. B. Huffman, A. G. Rinzler, R. E. Smalley, R. H. Hauge and J. L. Margrave, Chem. Phys. Lett., 1998, 296, 188–194 CrossRef CAS.
- B. Z. Tang and H. Y. Xu, Macromolecules, 1999, 32, 2569–2576 CrossRef CAS.
- A. Star, J. F. Stoddart, D. Steuerman, M. Diehl, A. Boukai, E. W. Wong, X. Yang, S. W. Chung, H. Choi and J. R. Heath, Angew. Chem., Int. Ed., 2001, 40, 1721–1725 CrossRef CAS.
- J. Chen, H. Y. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034–9035 CrossRef CAS.
- D. W. Steuerman, A. Star, R. Narizzano, H. Choi, R. S. Ries, C. Nicolini, J. F. Stoddart and J. R. Heath, J. Phys. Chem. B, 2002, 106, 3124–3130 CrossRef CAS.
- A. Star, Y. Liu, K. Grant, L. Ridvan, J. F. Stoddart, D. W. Steuerman, M. R. Diehl, A. Boukai and J. R. Heath, Macromolecules, 2003, 36, 553–560 CrossRef CAS.
- N. A. Rice, K. Soper, N. Z. Zhou, E. Merschrod and Y. M. Zhao, Chem. Commun., 2006, 4937–4939 RSC.
- F. Y. Cheng and A. Adronov, Chem.–Eur. J., 2006, 12, 5053–5059 CrossRef CAS.
- A. Ikeda, K. Nobusawa, T. Hamano and J. Kikuchi, Org. Lett., 2006, 8, 5489–5492 CrossRef CAS.
- J. Zou, L. Liu, H. Chen, S. Khondaker, R. McCullough, Q. Huo and L. Zhai, Adv. Mater., 2008, 20, 2055–2060 CrossRef CAS.
- X. Pang, P. Imin, I. Zhitomirsky and A. Adronov, Macromolecules, 2010, 43, 10376–10386 CrossRef CAS.
- K. M. Wu, P. Imin, A. Adronov and I. Zhitomirsky, Mater. Chem. Phys., 2010, 125, 210–218.
- A. Stuparu, C. Stroh, F. Hennrich and M. Kappes, Phys. Status Solidi B, 2010, 247, 2653–2655 CrossRef CAS.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- R. D. McCullough, J. Polym. Sci., Part A: Polym. Chem., 1998, 10, 93–116 CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS.
- F. M. Chen, B. Wang, Y. Chen and L. J. Li, Nano Lett., 2007, 7, 3013–3017 CrossRef CAS.
- A. Nish, J. Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640–646 CrossRef CAS.
- A. Nish, J.-Y. Hwang, J. Doig and R. J. Nicholas, Nanotechnology, 2008, 19, 095603 CrossRef.
- J.-Y. Hwang, A. Nish, J. Doig, S. Douven, C.-W. Chen, L.-C. Chen and R. J. Nicholas, J. Am. Chem. Soc., 2008, 130, 3543–3553 CrossRef CAS.
- N. Sturzl, F. Hennrich, S. Lebedkin and M. Kappes, J. Phys. Chem. C, 2009, 113, 14628–14632 CrossRef.
- J. Gao, M. Kwak, J. Wildeman, A. Herrmann and M. Loi, Carbon, 2010, 49, 333–338.
- T. Koyama, Y. Miyata, Y. Asada, H. Shinohara, H. Kataura and A. Nakamura, J. Phys. Chem. Lett., 2010, 1, 3243–3248 Search PubMed.
- H. Ozawa, T. Fujigaya, Y. Niidome, N. Hotta, M. Fujiki and N. Nakashima, J. Am. Chem. Soc., 2011, 133, 2651–2657 CrossRef CAS.
- F. Lemasson, T. Strunk, P. Gerstel, F. Hennrich, S. Lebedkin, C. Barner-Kowollik, W. Wenzel, M. Kappes and M. Mayor, J. Am. Chem. Soc., 2011, 133, 652–655 CrossRef CAS.
- F. Y. Cheng, P. Imin, C. Maunders, G. Botton and A. Adronov, Macromolecules, 2008, 41, 2304–2308 CrossRef CAS.
- P. Imin, F. Cheng and A. Adronov, Polym. Chem., 2011, 2, 411–416 RSC.
- G. J. Bahun, C. Wang and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1941–1951 CrossRef CAS.
- C. M. Homenick, U. Sivasubramaniam and A. Adronov, Polym. Int., 2008, 57, 1007–1011 CrossRef CAS.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- M. Jeffries-El, G. Sauve and R. D. McCullough, Macromolecules, 2005, 38, 10346–10352 CrossRef CAS.
- M. Trznadel, A. Pron and M. Zagorska, Macromolecules, 1998, 31, 5051–5058 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- D. Fujishima, T. Mori, T. Mizutani, T. Yamamoto and N. Kitamura, Jpn. J. Appl. Phys., 2005, 44, 546–550 CrossRef CAS.
- F. Y. Cheng and A. Adronov, J. Porphyrins Phthalocyanines, 2007, 11, 198–204 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |