Biopolymer stabilized nanoparticles as co-catalysts for photocatalytic water oxidations†
Yi-Yeoun
Kim
b,
Fiona C.
Meldrum
b and
Dominic
Walsh
*a
aSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, BS8 1TS, UK. E-mail: d.walsh@bristol.ac.uk
bSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
First published on 30th March 2011
Abstract
Dextran and carboxylated dextran biopolymer coating of a range of cobalt, iron and manganese oxide nanoparticles and also reusable biopolymer beads coated with metal oxide nanoparticles are prepared, analysed and evaluated as a means of preserving high surface area co-catalysts for aqueous photocatalytic water oxidations. Stabilization of the nanoparticles is found to inhibit aggregation and allow efficient water oxidations to take place relative to commercial uncoated cobalt oxide nanoparticles. Possible reasons for the relative rates of oxygen production found within the range of functional biopolymer–nanomaterials tested are discussed.
Introduction
Photocatalytic splitting of water into its constituent H2 and O2 provides an elegant and green means of supplying future renewable fuels. Current concerns over renewable energy sources and environmentally benign energy generation mean that there is a renewed impetus for researching this ancient means of energy production.1Oxygen-evolving natural photosynthesis became prevalent around 2400 million years ago, and uses light-absorbing porphyrin chlorophyll groups containing a magnesium ion. Associated with the photosynthetic pathway is also a manganese-based Mn4Ca cluster complex which is vital for the photooxidation of water during the light phase of the photosynthetic process.2 Artificial means of photocatalytic cleavage of water have been studied extensively for several decades, and considerable effort has been expended in identifying catalysts and conditions which use both UV and visible light as the energy source. The major hurdle in achieving this goal remains improving levels of efficiency.3–5 While production of H2 has been quite effectively accomplished,6–8 O2 production remains a continuing challenge, being based on a four-electron redox process. In terms of applications such as energy derived from fuel cells, selective H2 and O2 productions are clearly desirable.9Two main approaches have been employed for O2 production, based around the use of metal oxides as catalysts for the oxidation step. The first approach employs a photoelectrochemical method, where an applied mild overpotential (ξo(O2/H2O) = 0.89 V at pH 5.8)10 helps drive the catalytic reaction and allows direct O2 generation.11–13 Ideally, the necessary bias voltage is itself generated by an attached photovoltaic device. The second approach which is more akin to natural photosynthesis, and which is also relevant to this paper, uses a light-absorbing dye molecule to generate electrons which are then scavenged by a reducible reagent combination. The dye molecule is then regenerated by electrons collected from the metal oxide particle surface. The metal oxide then becomes a powerful oxidizing agent which is able to split water into O2 and protons, the protons generated are collected by a buffer.10,14,15 A range of metal oxides have been examined as potential candidates for large-scale water splitting applications, of which oxides of Ru and Ir are among the most efficient co-catalysts for O2 generation, however these are relatively scarce and expensive materials. Oxides of Mn and Fe are also known to be effective although less efficient, while cobalt oxide is recognised as being an excellent candidate for this process.10,16 Among other conditions, the efficiency of the photocatalysis is highly dependent on the surface area of the metal oxide particle, with the efficiency increasing markedly with increasing surface area.1,16 However, as the particle size decreases, there is also a reduction of the stability of the particles in solution. Unstabilized nanoparticles at the dimensions used here are not readily available commercially as in aqueous solutions the nanoparticles are subject to rapid phase separation into aggregates which grow into larger assemblies. This is particularly the case with magnetite nanoparticles which permanently aggregate into macroscopic agglomerations within minutes or hours of synthesis, this is in part why various polymers have previously been investigated for stabilisation of magnetite.17 Aggregation of catalyst in solution has been previously addressed by using inorganic supports such as silica or titania, or by coating of the metal oxide cores with a further inorganic layer such as chromium oxide.10,18 Whilst effective, these supports require several preparation steps and calcinations. More recently the use of porphyrin coating of nanoparticles as a means of overcoming co-catalyst aggregation has been investigated.19
In this paper we examine the relative efficiencies of Co and Fe oxide biopolymer-stabilised nanoparticles of differing core sizes and using dextrans at various molecular weights as the shell for application as functional co-catalysts. Dextran is a natural polysaccharide composed of a majority of α1–6 and a minority of α1–3 or α1–4 glycosidic linked glucose units, and readily forms stable nanosuspensions of coated nanoparticles of controllable sizes under 10 nm. Dextran-stabilised nanoparticles of Co3O4 and Fe3O4 (magnetite) were synthesised and analysed using an established protocol20 and Co3O4 coated with a carboxylated dextran (CM–dex), with carboxyl present at ∼20 mol % (or one carboxyl per 3–4 glucose units), was used as a comparison to the purely hydroxylated dextran. We also report the rapid and straightforward preparation and photocatalytic application of supported metal oxides of Co, Fe and Mn composed of commercial dextran ‘Sephadex’ beads decorated with metal oxide nanoparticles bound to the network of crosslinked dextran polymer at the bead surface. These organic-supported samples have the significant advantage that the coated beads can readily recovered by filtration after use and then re-employed. Photocatalyzed gas evolution using these materials was also compared under identical conditions to the smallest commercially available, uncoated and nanosized Co3O4 (of an average size of 26 nm and a BET of 38.7 m2 g−1) that we were able to source.
Experimental
The preparation and analysis of dextran stabilised Co3O4, Fe3O4 and Mn3O4 used our previously reported methodology.20 For the sample preparation methodology of nanoparticle coated Sephadex bead supports, photocatalytic reaction mixture methodologies and evolved gas21 and DLS analysis details please refer to the ESI†.Results and discussion
Synthesis of the nanoparticle suspensions was conducted using dextran of Mr of 6k, 70k and 500k, enabling the preparation of smaller nanoparticle cores with decreasing dextranMr,20 and allowing the effect of an increasing hydrodynamic diameter on the water oxidation reaction to be evaluated. XRD and Transmission Electron Microscopy (TEM) analysis of the dextran-stabilised metal oxide particles were conducted20,22 and Table 1 shows the measured nanoparticle core sizes and the corresponding DLS measured hydrodynamic diameter of the samples employed for the photocatalysis.| Sample (Mr) | Core particle diameter/nm | Core surface area/m2 g−1 | Hydrodynamic diameter/nm | [O2] released after 60 min/μmol |
|---|---|---|---|---|
| Dex(6k)–Co3O4 | <2 | >500 | 3.5 | 56 |
| Dex(70k)–Co3O4 | <2 | >500 | 10 | 59 |
| Dex(500k)–Co3O4 | 2.0 | 490 | 17 | 32 |
| Dex(6k)–Fe3O4 | 2.3 | 506 | 11 | 56 |
| Dex(70k)–Fe3O4 | 3.2 | 364 | 32 | 41 |
| CMdex(∼15k)–Co3O4 | 2.0 | 490 | 28 | 39 |
| Sephadex–Co3O4 | 7.3 | 135 | — | 44.4 |
| Sephadex–Fe3O4 | 11.2 | 104 | — | 36.4 |
| Sephadex–Mn3O4 | 23.4 × 5.9 (rods) | 106 | — | 31 |
| Commercial Co3O4 | 26 | 38.7 | — | 28.4 |
The prepared and dried nanoparticle-coated Sephadex beads are free-flowing, coloured powders (ESI, Fig. S1†, images of dried Sephadex powders) which rapidly hydrate into 100–200 μm beads in aqueous solution. The metal oxide nanoparticles coating the surfaces of beads were subsequently released for TEM and XRD analysis by the dextranase enzyme treatment of the beads. This results in hydrolysis of the crosslinked dextran into soluble maltoses which can readily be removed by washing.
XRD analysis of the liberated cobalt oxide gave broad reflections due to the small particle size that corresponded to the cubic Co3O4 (JCPDS 00-042-1467), however following heating the sample briefly to 550 °C sharper reflections were obtained. Iron oxide preparations were shown to be cubic Fe3O4 (JCPDS 00-019-062 (magnetite)) and manganese oxides gave reflections matching the tetragonal Mn3O4 (JCPDS 00-024-0734 (hausmannite)) (Fig. 1).
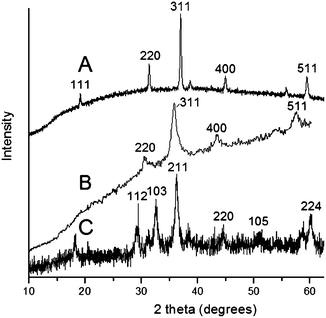 | ||
| Fig. 1 XRD diffractograms of Sephadex coated with metal oxides following removal of the organic support by the dextranase enzyme treatment (a) Co3O4 (heated to 550 °C); (b) Fe3O4 and (c) Mn3O4. | ||
TEM measurement showed that the (unheated) Co3O4 and Fe3O4 particles were roughly spherical with average sizes of 7.3 nm (σ = 1.67 nm) and 11.2 nm (σ = 3 nm) respectively. Mn3O4 was found to exist as rods of 23.4 nm (σ = 5.37 nm) in length and 5.9 nm (σ = 0.79 nm) in width (Table 1 and Fig. 2) (n = 50 in all cases). Thermogravimetric analysis (TGA) was used to determine the wt% of metal oxide present in all of the samples (ESI, Fig. S2†, table of TGA data of prepared samples). This showed that dextran and Sephadex samples were particularly rich in Fe3O4 with up to 27 wt% magnetite obtained, Co3O4 and Mn3O4 were present at lower levels ranging from 1 wt% up to a maximum of 7 wt% Co3O4 obtained with CM–dex.
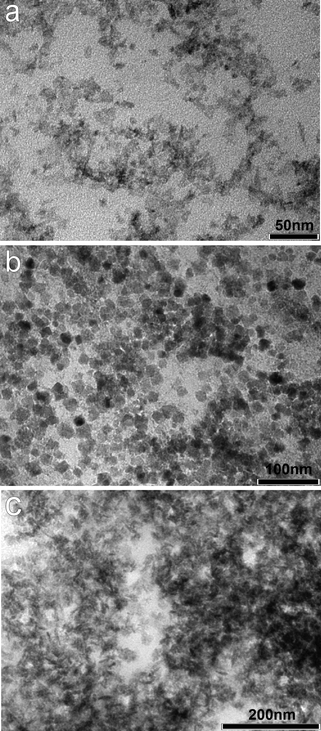 | ||
| Fig. 2 TEM images of washed material obtained from the dextranase enzyme treatment of metal oxide particle coated Sephadex bead organic supports showing nanoparticles of (a) Co3O4; (b) Fe3O4 and (c) Mn3O4. | ||
The relative efficiencies of the biopolymer-coated metal oxide nanoparticles in splitting water under visible light irradiation were evaluated, and compared with uncoated commercial Co3O4 nanoparticles using the established heterogeneous photocatalytic cycle of a [Ru(bpy)3]2+ photosensitizer, a sacrificial persulfate electron acceptor and bicarbonate buffer. The reaction mixture was buffered to pH 5.8 as more acidic conditions favour undesirable decomposition of the [Ru(bpy)3]3+.4,5,10,15,16 The metal oxide then regenerates the reduced [Ru(bpy)3]2+ and oxidation of water into O2 and H+ takes place on the metal oxide nanoparticle surface and thus a photocycle is maintained. Varying amounts of dried biopolymer or Sephadex metal oxide samples were measured such that they all contained 9 mg of the pure metal oxide for comparison with 9 mg of the uncoated commercial Co3O4 sample. The dried dextran–metal oxide materials were then re-suspended in degassed DI water for use in the reaction mixture.
All water oxidations were conducted under the same experimental conditions and the evolved gas was collected by water displacement from an attached container. A UV-vis absorption spectrum of the reaction mixture at pH 5.8 showed the [Ru(bpy)3]2+* MLCT absorption λmax to be at 452 nm (ESI, Fig. S3†, UV-vis absorption spectra). Irradiation was carried out using a blue 1W LED lamp (λmax 470 nm) and gas evolution was typically initially rapid, before it decreased and virtually ceased after approximately 30 min, due to exhaustion of the buffer and electron acceptor. Further addition of these reagents resulted in resumption of gas evolution, which was initially at levels of first experimental runs. However gas evolution decreased more rapidly indicating that some level of Ru(bpy)3 decomposition had occurred.15 Control experiments in which no metal oxide or dextran alone was used gave no observable gas evolution, indicating that decomposition of the sensitizer complex alone was not responsible for gas evolution. Use of reduced persulfate levels was found to result in the decreased gas output. Similarly, shielding of an ongoing metal oxide reaction mixture from light resulted in rapid cessation of evolved gas.
A graph of the results for the catalytic oxidations of the various dextran coated metal oxides and for Sephadex beads coated in metal oxides with a reaction course of 60 min is shown in Fig. 3a and b and the total gas volumes evolved are shown in Table 1.
![Time resolved graph of [O2] evolution from photocatalytic [Ru(bpy)3]2+ metal oxidewater oxidation reactions using (a) dextran stabilised metal oxides and (b) Sephadex beads coated with metal oxide nanoparticles. A weight of biopolymer/metal oxide was used such that 9 mg of the metal oxide component was present in all sample runs.](/image/article/2011/PY/c1py00037c/c1py00037c-f3.gif) | ||
| Fig. 3 Time resolved graph of [O2] evolution from photocatalytic [Ru(bpy)3]2+ metal oxidewater oxidation reactions using (a) dextran stabilised metal oxides and (b) Sephadex beads coated with metal oxide nanoparticles. A weight of biopolymer/metal oxide was used such that 9 mg of the metal oxide component was present in all sample runs. | ||
The composition of the evolved gases was tested using both lime water for determination of CO2 content and methylene blue dye for detection of O2. Quantitative lime water analysis showed that under the chosen reaction conditions, for both the synthesised particles and commercial Co3O4 particles employed, CO2 was present between 5 and 8 vol% of the evolved gases. The methylene blue dye analysis indicated that O2 made up the entirety of the remaining evolved gas, although it cannot be ruled out that trace quantities of CO were not also present. The presence of carbon oxide gases may result either from the use of the bicarbonate buffering system, where absorption and decomposition of carbonate or bicarbonate species occur on the metal oxide during photocatalysis, and/or from some decomposition of the bipyridyl complex.10,15 A low CO2 content does not present an obstacle to the use of the system for bulk water oxidation as CO2 can be very readily removed with soda lime or similar absorbent agents.
The results demonstrate that Co3O4-containing samples were superior in terms of the rate and the total volume of evolved gas, although the 6k dextran Fe3O4 samples were only moderately less efficient and were significantly more active than the 70k Fe3O4 dextran samples (Fig. 3a and Table 1). This is probably in part due to the lower surface area of the 70k coated magnetite. Notably, all of the prepared samples gave more efficient water oxidation than the commercial Co3O4 nanoparticles. The lower Mrdextran-stabilised metal oxides also performed at a clear step above their Sephadex-supported counterparts in terms of total gas evolved with the exception of the Co3O4 samples stabilised with the carboxylated dextran which gave a surprisingly low final evolved gas volume, considering their small core size. It was found that the biopolymerMr had a noticeable effect on the rate and final volume of gas evolved, with dextran 70k Co3O4 giving the best result. There was also a tendency for gas production to be more prolonged with higher Mr coated metal oxides. Although 6k dextran Co3O4 and Fe3O4 samples gave near identical final evolved volumes, a significantly faster initial rate of gas production was obtained with the cobalt based catalyst. Co3O4 particles stabilised using 500k dextran were one of the poorest performing catalyst combinations, and also showed a distinct lag time before the onset of the reaction such that gas evolution did not begin until 3–4 minutes after mixing of the reagents, adjustment to pH 5.8 and exposure to the light source. For all the other samples tested no such lag time was observed.
Fe3O4 is not reported as having significant activity for water oxidations, however Fe2O3 and especially Fe ions are known to be effective.14 Under the mildly acidic reaction conditions Fe2+ ions can be expected to be released from the surface of the magnetite nanoparticles and the nanoparticle surface convert to γ-Fe2O3 (maghemite).17 It is possible that this process is moderated by the biopolymer coat with more nanoparticle surface dissolution taking place with shorter biopolymer coatings. This may also contribute to the high activity of the 6k dextran–Fe3O4 sample and lower activity of the 70k dextran–Fe3O4.
DLS measurements gave the hydrodynamic diameters of the prepared samples, which showed an increase in diameter of the biopolymer-coated metal oxide particles in solution with increasing Mr. The smallest core–shell particle of 3.5 nm diameter was Co3O4 coated with 6k dextran, however the small cluster sized Co3O4 core may less efficient that the slightly larger core present with the best performing 70k Co3O4. Another possible cause for the observed differences in evolved gas volumes is that the biopolymer coat serves to bring together the various components of the photocatalytic reaction in a loosely coordinated arrangement. During a photocatalysis reaction the photosensitizer dye exists largely as the more highly charged [Ru(bpy)3]3+ and the rate-determining step in the process is believed to be the dark reactions, such as electron transfer between the metal oxide and [Ru(bpy)3]3+, charge transport within the nanoparticles, or oxygen evolution.15 Therefore, there may be an optimal thickness of the coiled dextran coat at which interactions between its pendant hydroxyl groups and the Ru3+ complexes occur. The effectiveness of electron transfer reactions decreases exponentially with distance between the reaction centres,23 and therefore a weakly bound nanoscale complex where the reactive components are held in close proximity may be more efficient.
In the case of the 500k dextran Co3O4 sample, the large dynamic diameter relative to the core size may mean that the diffusion rate to the metal oxide surface is too great, leading to a reduction in efficiency and the time lag which was experimentally observed. The carboxylated CM–dextran Co3O4 sample had one of the highest hydrodynamic diameters of the samples tested, and as with the 500k dextran sample gave relatively poor gas production. Notably, no lag phase was observed with this sample which may be attributable to the shell containing additional bound water rather than the coiled dextran chains of the 500k dextran samples. In contrast however, the 70k dextran Fe3O4 particles had slightly larger measured hydrodynamic diameters than CM–dex Co3O4 but gave a superior gas evolution. While there is certainly a relationship between the core and hydrodynamic diameters and efficiency, it is clear that additional factors are also involved, and it is possible that charged carboxyl groups interfere with the electron transfer processes. For Sephadex ‘organic’ supported samples gas volumes generated were less than for dextran coated metal oxides (Fig. 3b). This can be attributed to the lower surface area of nanoparticles and also the beads are discrete macroscopic entities distributed in the solution. Therefore some decomposition of the sensitizer directly without O2 generation may occur. Mn3O4 coated Sephadex gave a high initial rate of O2 production which may be a consequence of the rod-like morphology of the nanocrystals. However rates were not as sustained as with the biopolymer coated nanoparticles (Fig. 3b).
The theoretical maximum [O2] that could be released based on ideal photon capture and persulfate conversion is 273 μmol, efficiency therefore varies from 10.4% for the uncoated commercial Co3O4 up to 21.6% for the Dex(70k)–Co3O4 sample. Thus relative efficiency is more than doubled by use of the Co3O4 with its biopolymer preserved and discrete cores. It can be envisaged that optimization studies on the reaction conditions including reagent concentration could improve the efficiencies of these systems up to and beyond the 30–40% efficiencies for existing optimized inorganic supports. Co3O4 particles gave superior gas production as compared to the Fe3O4, with the Mn3O4 particles performing least well. Cobalt is known to be a superior co-catalyst and the surface areas of these nanoparticles were ∼35 m2 g−1 greater, resulting in greater efficiency. The surface areas of the supported Fe3O4 and Mn3O4 were comparable such that the greater effectiveness of the Fe3O4 is likely to be due to the inherent higher activity of Fe ions as compared to Mn3O4 for water oxidations under the conditions used. For Seph–Mn3O4 a possible additional factor may be the low concentration of the nanoparticle coating, and hence the relatively high concentration of beads required, which may scatter the incoming light. A positive feature of the beads however is that they could be recovered by filtration and reused for at least three repeat reactions with almost no drop in rate/volume of gas produced. The Fe3O4 coated beads were also observed to retain a strong response to an applied strong magnet, showing that there was little if any oxidation of the surface bound magnetite nanoparticles into non-superparamagnetic α-Fe2O3.
Conclusions
In conclusion biopolymer stabilisation of photocatalytic metal oxides was found to be a very effective means of preserving stable, high surface area particles which notably showed good efficiencies for water oxidation relative to one of the best commercially available non-stabilised nanoparticle Co3O4. Use of nanoparticles in solution is generally restricted due to their strong tendency to aggregate, here biopolymer templating either by coating with biopolymer or upon biopolymer beads overcomes this serious limitation. The length of the biopolymer chain was found to affect the reaction profiles and a combination of Co3O4 stabilised with dextran of Mr 70k was found to give the highest volume of evolved gas. Fe3O4 showed good activity, which may be due to release of Fe ions from the nanoparticle surfaces under the reaction conditions. Interplay between the core size, diffusion rates and possible formation of weakly bound complexes also appeared to determine overall efficiency. Cross-linked dextran Sephadex beads coated with metal oxide nanoparticles were effective supported photocatalysts that could be readily recovered for re-use without losing catalytic activity. Further research on optimization and effects on the functional biopolymer–metal oxide catalysts in water oxidations is currently underway.Acknowledgements
We thank the EPSRC (Grant No. EP/C544803/1) (DW) and Grant No. EP/E037364/1 (YYK, FCM) for financial support of this study. We also thank David Williams, COMC, University of Bristol for assistance with DLS measurements.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- N. P. Luneva, E. I. Knerelman, V. Y. Shafirovich and A. E. Shilov, New J. Chem., 1989, 13, 107–110 Search PubMed.
- A. Mills, A. Harriman and G. Porter, Anal. Chem., 1981, 53, 1254–1257 CAS.
- A. Harriman, M. C. Richoux, P. A. Christensen, S. Mosseri and P. Neta, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3001–3014 RSC.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- V. M. Aroutiounian, V. M. Arakelyan and G. E. Shahnazaryan, Sol. Energy, 2005, 78, 581–592 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- Y. Li and J. Z. Zhang, Laser Photonics Rev., 2010, 4, 517–528 Search PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 Search PubMed.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS.
- R. van de Krol, Y. Q. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311–2320 RSC.
- K. Rajeshwar, J. Appl. Electrochem., 2007, 37, 765–787 CrossRef CAS.
- N. P. Luneva, V. Y. Shafirovich and A. E. Shilov, J. Mol. Catal., 1989, 52, 49–62 CrossRef CAS.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275–5280 CrossRef CAS.
- F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920–2922 RSC.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750–7759 CrossRef CAS.
- Y. S. Nam, A. P. Magyar, D. Lee, J. W. Kim, D. S. Yun, H. Park, T. S. Pollom, D. A. Weitz and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 340–344 CrossRef CAS.
- Y. Y. Kim, K. Hore, S. R. Hall and D. Walsh, Small, 2009, 5, 913–918 CrossRef CAS.
- M. Sumitani, S. Takagi, Y. Tanamura and H. Inoue, Anal. Sci., 2004, 20, 1153–1157 CAS.
- Y. Y. Kim and D. Walsh, Nanoscale, 2010, 2, 240–247 RSC.
- W. N. Lanzilotta, V. D. Parker and L. C. Seefeldt, Biochemistry, 1998, 37, 399–407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optical image, TGA analysis, UV-vis absorption spectra. See DOI: 10.1039/c1py00037c |
| This journal is © The Royal Society of Chemistry 2011 |