RAFT-synthesized copolymers and conjugates designed for therapeutic delivery of siRNA†
DeeDee
Smith
,
Andrew C.
Holley
and
Charles L.
McCormick
*
Department of Polymer Science, University of Southern Mississippi, 118 College Drive, Hattiesburg, MS 39406-0001, USA. E-mail: Charles.McCormick@usm.edu
First published on 25th March 2011
Abstract
The advent of controlled radical polymerization (CRP) techniques, along with advancements in facile conjugation chemistry, now allow synthetic tailoring of precise, polymeric architectures necessary for drug/gene delivery. Reversible addition–fragmentation chain transfer (RAFT) polymerization and its aqueous counterpart (aRAFT) afford quantitative control over key synthetic parameters including block length, microstructure, and placement of structo-pendent and structo-terminal functionality for conjugation of active agents and targeting moieties. The relevance of water-soluble and amphiphilic (co)polymers synthesized by RAFT for in vivo delivery of therapeutics in biological fluids is an especially attractive feature. In many cases, polymerization, binding, conjugation, and stimulus-induced release can be accomplished directly in aqueous media. This review focuses on RAFT synthesized (co)polymers as vectors for delivery of small interfering ribonucleic acid (siRNA) and gene down-regulation via the RNA interference (RNAi) pathway. Synthetic strategies utilizing RAFT and facile side- and end-chain reaction chemistries to afford modular delivery architectures (linear, stars/hyperbranched, micelles, and hybrid (co)polymeric vehicles) are reviewed based on examples from current literature. Also, specific problems, barriers, and challenges regarding rational design of polymeric delivery systems for therapeutic siRNA are presented.
 DeeDee Smith | DeeDee Smith graduated cum laude with a BA in chemistry from Agnes Scott College in 2003. She received her PhD in organic chemistry from Northwestern University in 2009 under the direction of Prof. SonBinh T. Nguyen. Her thesis work focused on the development of well-defined amphiphilic block copolymers prepared via ring-opening metathesis polymerization (ROMP) for targeted delivery of chemotherapeutics. She is currently a post-doctoral researcher in the group of Prof. Charles McCormick at the University of Southern Mississippi. Her work involves preparation of copolymers via aqueous reversible addition–fragmentation chain transfer (RAFT) polymerization for use as siRNA delivery vehicles. |
 Andrew C. Holley | Andrew “Chris” Holley graduated magna cum laude with a BS in Biochemistry from Mississippi College in 2007. As an undergraduate, he conducted research under the direction of Prof. Reid Bishop on the statistical mechanics of DNA interchelators. Shortly thereafter, he joined the PhD program in Polymer Science at the University of Southern Mississippi under the direction of Prof. Charles McCormick. Currently in his 4th year, Chris's research is focused on the preparation of well-defined, water-soluble, amine-containing copolymers prepared via aqueous reversible addition–fragmentation chain transfer (RAFT) polymerization and subsequent targeted delivery of siRNA-polymer complexes. |
 Charles L. McCormick | Charles McCormick received his PhD in Chemistry from the University of Florida in 1973. He is a Bennett Distinguished Research Professor at the University of Southern Mississippi with appointments in the Department of Polymer Science and the Department of Chemistry and Biochemistry. His research focuses on stimuli-responsive, water-soluble and amphiphilic copolymers with precisely designed architecture as prepared by controlled “living” free radical polymerization. He has mentored 50 PhD graduates and published over 260 manuscripts in the area of Polymer Chemistry. |
Introduction
The targeted delivery of nucleic acid-based therapeutics has recently become one of the most active areas of research in the rapidly developing field of nanomedicine. Unprecedented advances in synthetic polymer chemistry over the last decade now allow for precise control of macromolecular structure and thus rational design of biologically relevant carriers with functional components engineered to aid in stabilization, trafficking and stimulus-induced release of “packaged” therapeutic agents. This review will focus exclusively on synthetic (co)polymers and their conjugates prepared via controlled radical polymerization (CRP) that have potential for therapeutic delivery of small interfering RNA (siRNA). Criteria for efficient delivery and efficient gene “knockdown” through the RNA interference (RNAi) are presented based on current understanding of cellular delivery pathways. Emphasis is placed on synthetic carrier structures and features of controllable segments which lend themselves to polyplex formation, siRNA packaging, targeting, uptake, endosomal escape and delivery to messenger RNA (mRNA) targets. Although research with siRNA/CRP polymers is in its infancy relative to more mature areas of DNA therapy, rapid growth is forecast due to fewer intra-cellular barriers to delivery and to the structural control afforded by new synthetic techniques, often accomplished directly in aqueous media. It is interesting to note from a historical perspective that discoveries of the RNAi pathway1 and reversible addition–fragmentation chain transfer (RAFT) polymerization2 were both reported in 1998. Also, the 2001 report of RNAi in mammalian cell lines3 which eventually led to the idea of RNAi-based therapies was coincidently paralleled with the first controlled homo- and copolymerizations of cationic monomers by aqueous RAFT polymerization.4,5Rational design of polymer-based delivery vehicles
The concept of tailoring synthetic polymer vehicles (vectors) for site-selected delivery and release of therapeutic agents was pioneered by Ringsdorf over 35 years ago.6 He and his colleagues suggested a simple “depot” model (Fig. 1) in which the therapeutic agent could be attached in a facile manner via a hydrolytically or enzymatically degradable spacer to a water-soluble, non-immunogenic (co)polymer backbone having requisite components for complex protection, solubility and circulation under physiological conditions. Therapeutic cargo could be trafficked either passively or actively (the latter by including targeting moieties) to specific “depot” sites for stimulus-induced release as directed by endogenous cellular components. The model was later extended for attachment of a diagnostic component to track location of the carrier, thus setting the stage for dual purpose theranostic (therapeutic/diagnostic) vehicles. The concepts of this seminal model remain at the forefront of rational design strategies for polymeric vehicles utilized for pharmaceutical delivery, including siRNA therapeutics. However, only recently have facile polymerization and orthogonal chemistries been developed that yield precise architectures, segment lengths, selected sizes, functionality, and narrow molecular weight distributions required. For example, the CRP syntheses of well-defined (co)polymers with pH-responsive, tertiary and secondary amine segments necessary for reversible polyplex formation with polynucleotides have recently been reported.7 More specifically, rationally designed siRNA polyplexes with targeting moieties have shown therapeutic potential in vitro as demonstrated by cell-specific delivery and gene silencingvia the RNAi pathway. In the sections to follow, we review the work appearing in the literature related to vector design based on CRP techniques with an emphasis on RAFT. It should be noted that despite fundamental distinctions of nucleotide structure and trafficking,8 many of the recent approaches to siRNA delivery have precedence in DNA delivery utilizing a variety of polymer types including classical, or controlled chain growth polymers, dendrimers, polymers from ring-opening polymerization, and those prepared from synthetic-bioconjugate techniques. Nano- and micro-structured particles, micelles, polymersomes, microemulsions, hydrogels, transition metal complexes, carbon nanotubes, and inorganic complexes have been reported. The reader is directed to a number of comprehensive reviews of these DNA delivery vectors.9–20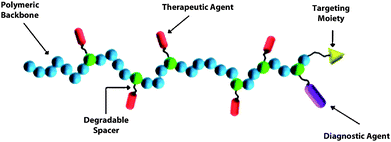 | ||
| Fig. 1 Components of the Ringsdorf “depot” model. | ||
RNA interference (RNAi)
RNA interference is a gene silencing effect induced by RNA duplexes that ultimately results in messenger RNA (mRNA) cleavage, thus inhibiting expression of target proteins. Importantly, RNAi potentially offers a pathway for therapeutic treatment of a variety of diseases including genetic, degenerative, and viral diseases as well as cancer.21 In a recent Chemical Society Review,22 Gaynor et al. present the mechanism of RNAi as currently understood. A simplified seven step pathway (Fig. 2) is detailed that starts with the formation of siRNA duplexes generally 21–23 nucleotides in length and having a two-nucleotide overhang at the 3′ end. The duplexes, which can be prepared synthetically or by endogenous processing of longer hairpin or double-stranded RNA (dsRNA) by a protein referred to as Dicer, have precise sequences such that the antisense or “guide” strand is complimentary to the target mRNA transcribed from the gene to be “silenced”. The siRNA duplex associates with cytoplasm proteins forming a “loading” complex (shown schematically in Fig. 2 as the RLC) which is trafficked to the RNA-induced silencing complex (RISC) for removal of the sense or “passenger” strand, thus activating the complex for target mRNA recognition and complementary binding. RISC contains an argonaute protein with endoribonuclease activity that cleaves the mRNA backbone specifically between nucleotides 10 and 11 relative to the 5′ end of the guide strand. Ideally, after dissociation from the cleaved mRNA, recycled RISC can then target other mRNA strands. For further discussion of kinetics, biodistribution, stabilization, cellular uptake, chemical modification, possible interferon response, and off-target effects operative in RNAi, the reader is referred to several recent reviews.21–29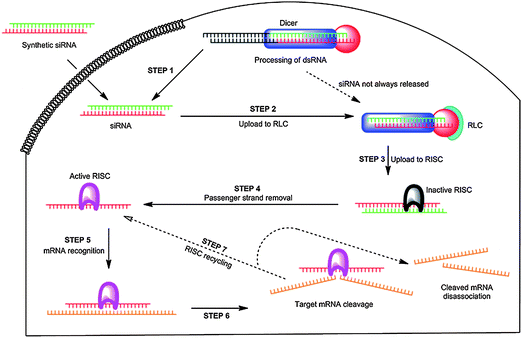 | ||
| Fig. 2 The RNA interference (RNAi) pathway. Reprinted by permission of Chemical Society Reviews.21 | ||
Barriers to siRNA and general approaches to therapeutic delivery
RNAi therapy has been highly touted due to its simplicity (relative to other forms of oligonucleotide intervention) and applicability to a large number of diseases. Advantages are that interference occurs at the translational rather than the transcriptional level, relatively small doses of siRNA are required, and lower toxicity is generally observed. However, efficient delivery necessitates overcoming significant barriers (Fig. 3) that stem from the physicochemical properties of siRNA and systemic interactions during trafficking to cytoplasm targets.23–29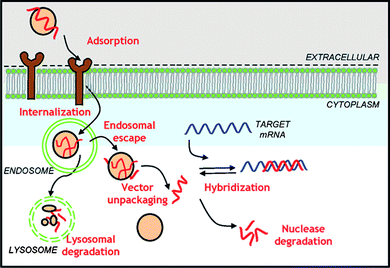 | ||
| Fig. 3 Cellular barriers to oligonucleotide delivery. Reprinted by permission of Biotechnology Progress.22 | ||
Cellular delivery of siRNA and other polynucleotide therapeutics has been accomplished utilizing a number of viral and non-viral vectors.10,12,19,20 Controlled polymer-based carriers, the focus of this review, are designed to eliminate or suppress repulsive electrostatic interactions between double stranded siRNA and the cellular surface.8,26,28 The formation of interpolyelectrolyte complexes (IPECs or polyplexes) between therapeutic RNA and cationic polymer segments, not only accomplishes this, but also may induce compaction of longer dsRNA while conferring protection from endonucleases encountered in RNAi. Cellular entry can occur by non-specific or targeted uptake of the polyplex. Wong et al. recently reviewed several pathways for endocytic uptake in polymer-mediated gene delivery: phagocytosis, macropinocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis, and clathrin- and caveolae-independent endocytosis.19 Although all of the above result in enclosure of the polyplexes within transport vesicles formed from the plasma membrane, subsequent trafficking and processing modes are different and the factors determining the specific pathway are not well elucidated at this point. Most studies so far have dealt with DNA, and there are likely to be differences between DNA and siRNA polyplex internalization based on size and interpolyelectrolyte registry. However, there are some structural similarities that may be identified. In general, non-specific endocytosis can be triggered by (a) charge-mediated interactions of cationic polyplexes with sulfated or carboxylated glucosamine residues on membrane-bound proteoglycans and/or (b) hydrophobic associations of lipophilic residues on the complex with the phospholipid layers of the cell membrane.19,28 Recently, cell-penetrating peptides, 5 to 40 amino acids in length with amphipathic and cationic segments have shown potential for binding polynucleotides and facilitating membrane translocation.30 These peptides may enter non-specifically into the phospholipid membrane by several postulated mechanisms including: formation of peptide-lined pores, direct penetration, transient uptake, and induced endocytosis.30
Targeted delivery is accomplished by incorporating ligands that bind to receptors on the cell surfaces. Receptor-mediated targeting and delivery of therapeutics to specific cell lines have been widely studied utilizing a plethora of targeting ligands including endogenous folate and transferrin and exogenous peptides, glycoproteins, and antibodies.21,28,29 To date, however, only a few of these have been reported for targeted delivery of siRNA utilizing CRP carriers.22,23,25
Once cellular uptake has occurred, the resulting polyplex-bearing vesicle may be recycled to the surface, routed to cellular organelles, or participate in the endo-lysosomal pathway.28 As illustrated in Fig. 3, the polyplex must escape the endosome vesicles before the latter mature to lysosomes in order to avoid enzymatic degradation. The maturation of the early endosomes to lysosomes is accompanied by a pH reduction from ∼6 to ∼5.22 As such, polymers with pH responsive amine segments (pKa values from 5 to 7) have been utilized in the polyplex to act as a “proton sponge”. Protonation results in osmotic swelling and disruption of the endosome and release or “escape” of the polyplex to the cytoplasm.22,28Peptides that undergo coil-to-helix transformations as pH is lowered have also been conjugated to polymeric carriers to induce disruption of the endosomes.21,23,25 After endosomal escape, polyplex dissociation must occur in order to enter the RNAi pathway.28Reduction of strength of interpolyelectrolyte complexes has been the most common method of choice to date. The polymer architecture (linear, star, dendrimer, branched, etc.), registry and spacing of charges, molecular weight, amphiphilicity, reversible cross-linking and other structural characteristics can, in principle, be altered to effect facile dissociation.21–23,28
While many laboratory and clinical studies have reported remarkable advances in nucleic acid delivery using both classical polymers and those with designed, precise architectures, there is a clear need for tailored siRNA delivery vehicles that are dynamic, responding to in situ stimuli encountered in delivery to specific cell types. Given the large number of structural variables and possible cellular interactions identified in the literature to date, Roth28 has proposed an “integrated mathematical modeling” approach to rational design of delivery vehicles. His approach involves development of kinetic models of antisense and siRNA silencing by first describing oligonucleotides and their targets in discrete locations or “compartments” within the cells. Differential equations based on conservation of mass are then derived for each of the relevant species, for example polymer-bound, free and mRNA-hybridized siRNA in either endosome or cytoplasmic environments. Roth has typically employed 10 species and 20 parameters, the latter measured or derived from literature reports, to develop his models. Some knockdown experiments in specific cell lines appear to validate these models.30 Successful implementation of such approaches can be anticipated in the future and will be aided by rapidly evolving analytical methods capable of elucidating cellular entry and trafficking mechanisms.
Controlled radical polymerization (CRP) and therapeutic vehicle design
Carrier vehicles designed with criteria consistent with the Ringsdorf “depot” model, previously introduced and shown in Fig. 1, possess essential modular components: a water-soluble or amphiphilic polymer backbone, therapeutic agent binding sites, and pendent targeting and/or diagnostic moieties. Initial progress toward optimal design based on such models was restricted by the absence of sufficiently facile polymerization techniques affording control over molecular weight, molecular weight distribution, placement of structo-pendent or structo-terminal reactive functional groups, polymer architecture (blocks, stars, etc.), and solubility. As controlled radical polymerization techniques, including stable free radical polymerization (SFRP),30 atom transfer radical polymerization (ATRP),31 and RAFT,32–34 have evolved during the last few years, additional tools for rational design of delivery vectors with the above requisite parameters have become available. RAFT and more specifically its aqueous counterpart, often referred to as aqueous reversible addition–fragmentation chain transfer (aRAFT)4,5,35 are arguably the most relevant of the known CRP techniques for preparing delivery vehicles for use in biological fluids. Not only are these particularly versatile for polymerizing a wide range of charged and reactive monomers, in many cases polymerization, conjugation, therapeutic loading, targeted delivery, and stimuli-responsive release can all be accomplished directly in aqueous media.35Key steps in the RAFT polymerization process, first proposed by the CSIRO group2 and later adapted specifically for water,4,5 are shown in Fig. 4 in order to illustrate the method's utility for synthesizing modular, water-soluble therapeutic delivery vehicles discussed in this review. RAFT polymerization is a degenerative chain transfer process that begins in step I, Fig. 4 when an initiator, for example a water-soluble diazo initiator, decomposes to yield a primary radical, R˙, which then adds to a water-soluble monomer 1. In step II, a chain transfer agent (CTA), a thiocarbonylthio compound comprised of an R group and a Z group, reacts with the primary oligomeric radical to produce an intermediate radical that subsequently eliminates R˙ which in turn adds to monomer 1. The CTA is key to control of molecular weight and “livingness” of the RAFT process and various thiocarbonylthio species including dithioesters, xanthates, dithiocarbamates, and trithiocarbonates have been utilized.32–34 The degree of stabilization of the intermediate radicals in steps II and III of Fig. 4 depends on the nature of Z as does the rate of R˙ or kinetic chain scission from the intermediate for subsequent addition to monomer 1. Active (propagating) radicals eventually reach equilibrium with dormant chains, termed macroCTAs. The reversible nature of the degenerative process, the equilibrium favoring the dormant chains, and the apparent inability of the intermediate radical to add to monomer result in the polymerization having a “living” nature with degree of polymerization increasing linearly with conversion. MacroCTAs can be extended, for example, by the sequential addition of a second water-soluble monomer as shown in step III of Fig. 4, resulting in the formation of stimuli-responsive block copolymers.7,35
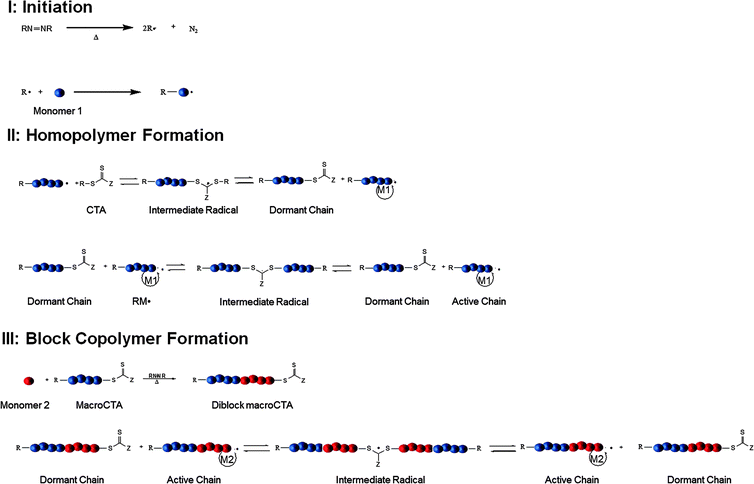 | ||
| Fig. 4 Key steps in formation of homo- and block copolymersviaRAFT polymerization. | ||
An important advantage of RAFT and other CRP polymerization techniques is the ability to prepare well-defined polymers with specified architectures (Fig. 5). Of interest to siRNA therapeutics are diblock, triblock, brush, and star architectures prepared from amine-functional monomers that can be synthesized via aRAFT directly in water without the need for protecting groups.7 R and Z functionality can be altered to allow structural variation as well as facile post-polymerization reactions.35 The nature of the segments and the molecular weight-dependent hydrodynamic volume under aqueous conditions, in principle, can be critically controlled by selecting from a wide variety of water-soluble monomers,7,35 designing CTAs and their conjugates,36 and specifying the monomer/CTA ratios.7,33–36 As detailed later, packaging and protection from enzymatic degradation have recently been accomplished by direct, molecular level complexation of siRNA with specified segments (for example those with cationic charges) or those presented on nano size, self-assembled complexes (e.g. micelles, vesicles, polymersomes and their conjugates and crosslinked versions). For example, interpolyelectrolyte complexes between siRNA and PDMAPMA (poly(3–(N,N-dimethylamino)-propyl methacrylamide)) (or other protonated tertiary amine segments of block copolymers)37–44 can be prepared with varied nitrogen-to-phosphate (N/P) ratios, affecting not only solubility but also cell entry by non-specific endocytosis and eventual release from the endosome. Neutral, water-soluble monomers such as N,N-dimethylacrylamide (DMA)45–48 or N-(2-hydroxypropyl)-methacrylamide (HPMA)14,49–62 can be copolymerized statistically or as blocks in order to maintain water solubility and prevent rapid clearance from the blood stream. Amphiphilicity can be adjusted by adding hydrophobic monomers (e.g.butyl acrylate38,39,63) or those responsive to temperature (N-isopropylacrylamide (NIPAM))64 or pH (secondary and tertiary amine-containing monomers).37–44 AB diblock or ABA triblock systems of the type, shown in Fig. 5, can be prepared utilizing dithioester or trithiocarbonate CTAs that have near-perfect end group functionality by making the R group of the initiator and that of the CTA identical. Such telechelic end groups can play important roles in attaching moieties for targeted delivery or for attaching diagnostic agents. Reactive functionalities, such as active esters, exchangeable disulfides, and primary amines, can also be readily accommodated by copolymerizing functional monomers via RAFT.7,35 A number of recent papers and reviews discuss facile chemical or bioconjugation methods utilizing reactive monomers or CTAs with appropriate functionality (Fig. 6).13,43,65–71 Notable milestones allowing the construction of delivery vehicles were the first controlled polymer syntheses by Scales, Convertine, and York et al. utilizing the hydrophilic monomers DMA, HPMA, NIPAM and the protonated (cationic) DMAEMA (2-(N,N-dimethylamino)-ethyl methacrylate), DMAPMA, and APMA (N-(3-aminopropyl) methacrylamide hydrochloride) monomers (Fig. 6) via aRAFT polymerization.45,59,72,73 Though beyond the scope of this review, the reader is referred to reports in this thematic issue as well as to recent reviews of post-polymerization modification methods including orthogonal “click” chemistries which have been or can potentially be utilized in delivery.74–90
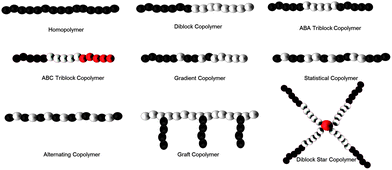 | ||
| Fig. 5 Varying architectures from CRP techniques amenable to siRNA binding. | ||
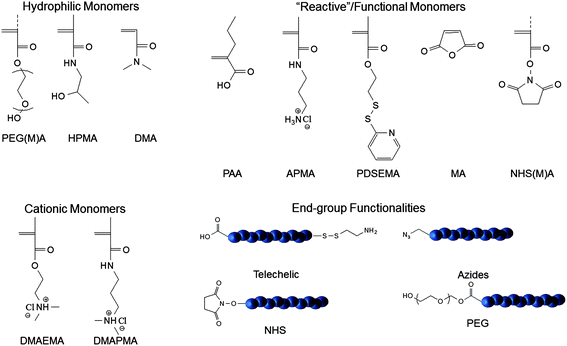 | ||
| Fig. 6 Reactive monomers and functional groups utilized for delivery vehicles prepared viaRAFT Polymerization. | ||
Linear polymer vehicles
Literature reports of linear homopolymers with controlled structures utilized for gene delivery are almost exclusively on P(DMAEMA) synthesized via RAFT or ATRP.91–98 However, as previously mentioned, complexes must be cationically charged overall to maintain particle dispersion. Although this and other favorable interactions at the cell surface promote uptake, problems of nucleotide release from the interpolyelectrolyte complex later in the process affect efficiency of delivery.99,100Perhaps the most widely studied systems for the delivery of siRNA have been block copolymers comprised of hydrophilic and cationic segments.99,101–103 These copolymers can form near-neutral interpolyelectrolyte complexes with negatively charged siRNA and have the added benefit of remaining soluble and stable in solution. However, while the hydrophilic block can increase stability/shielding and circulation time, it has also been shown to decrease uptake as compared to its positively charged counterparts.99,104 To combat this problem, York et al. utilized targeting entities for receptor-mediated endocytosis.43Copolymers containing a cationic block of DMAPMA and a hydrophilic block of HPMA-s-APMA were synthesized for siRNA delivery (Fig. 7). The APMA monomer has primary amine functionality for attaching a targeting group. These monomers were polymerized under buffered, aqueous RAFT conditions yielding copolymers of ∼50 kDa with narrow polydispersity index (PDI) values of ∼1.1. Folic acid, a well-known and extensively studied targeting group moiety,105–109 was conjugated to the polymersvia post-polymerization modification of the primary amines of APMA with efficiencies of >80%. The resulting targeted copolymer was then complexed with siRNA to yield near-neutral sterically protected complexes ∼15 nm in diameter. These complexes were incubated with cell lines that expressed both high levels (KB cells) and low levels (A549 cells) of folic acid receptors. The targeted complexes showed high selectivity for KB cells and 60% knockdown of the gene coding for human survival.
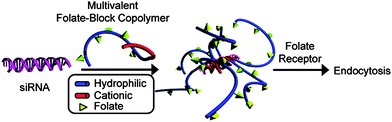 | ||
| Fig. 7 (HPMA-statistical-APMA)-block-DMAPMA copolymers complexed with siRNA for targeted gene delivery. Reprinted with permission from Biomacromolecules.43 | ||
One of the most significant bottlenecks in the delivery of siRNA to cells is release from endosomal compartments after uptake.16,110–112 One strategy has focused on the post-polymerization conjugation of endolytic agents to the hydrophilic block of these copolymers.112 However, even with the advent of facile and quantitative chemistries in recent years,75,79,85,89,113,114 such post-polymerization modifications involving macromolecular species can be inefficient and produce undesired by-products. Both quantifying yields and product characterization can prove difficult.115 Therefore, direct polymerization of CRP monomers capable of undergoing stimuli responsive transitions to “unmask” endolytic properties, such as proton sponges or hydrophobic, membrane-disrupting moieties, is an attractive approach.
Convertine et al. designed an endolytic copolymer delivery system that contained a block of DMAEMA for complexation with siRNA and an ampholyte block containing butyl methacrylate (BMA), DMAEMA, and poly(propylacrylic acid) (PAA) to act as both a shielding/stabilizing and pH-responsive block.38 The RAFT-synthesized terpolymers ranged in molecular weight from 16 kDa to 20 kDa with moderate PDIs (1.45–1.58). Despite the statistical nature of the hydrophobic block, DMAEMA and PAA were chosen to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for neutrality at pH 7.4, while BMA was varied to modify the hydrophobic content. At physiological pH, no higher ordered structures were formed prior to the addition of siRNA. At N:P ratios of 1:1, only large aggregates were identified by dynamic light scattering (DLS), with a reduction in particle size at increasing charge ratios. Particle sizes ranged from 85–236 nm with the highest BMA content (48 mol%) giving rise to the smallest particles at N:P ratios of 4:1. Despite the higher nitrogen content, surface charge of these particles remained near neutral. The endolytic activities of these copolymers and IPECs were investigated using a red blood cell hemolysis assay at varying pH values, representing physiological conditions, early endosome, and late endosome. At physiological pH, no endolytic activity was observed due to the charge neutrality of the ampholyte block. Polymers containing the most BMA content demonstrated the greatest degree of hemoglobin release at both early and late endosomal pH. Upon acidification of the endosomal compartment, it is predicted that the carboxylic acid residues are protonated, resulting in a more hydrophobic block with an overall net positive charge that is likely to interact with the membrane. Knockdown of GAPDH (glyceraldehyde 3-phosphate dehydrogenase—an enzyme that catalyzes glycolysis and is implicated in several non-metabolic processes, including transcription activation, initiation of apoptosis, and ER to Golgi vesicle shuttling) was investigated in HeLa cells and ranged between 50 and 80% depending on the polymer and formulation.
:1 for neutrality at pH 7.4, while BMA was varied to modify the hydrophobic content. At physiological pH, no higher ordered structures were formed prior to the addition of siRNA. At N:P ratios of 1:1, only large aggregates were identified by dynamic light scattering (DLS), with a reduction in particle size at increasing charge ratios. Particle sizes ranged from 85–236 nm with the highest BMA content (48 mol%) giving rise to the smallest particles at N:P ratios of 4:1. Despite the higher nitrogen content, surface charge of these particles remained near neutral. The endolytic activities of these copolymers and IPECs were investigated using a red blood cell hemolysis assay at varying pH values, representing physiological conditions, early endosome, and late endosome. At physiological pH, no endolytic activity was observed due to the charge neutrality of the ampholyte block. Polymers containing the most BMA content demonstrated the greatest degree of hemoglobin release at both early and late endosomal pH. Upon acidification of the endosomal compartment, it is predicted that the carboxylic acid residues are protonated, resulting in a more hydrophobic block with an overall net positive charge that is likely to interact with the membrane. Knockdown of GAPDH (glyceraldehyde 3-phosphate dehydrogenase—an enzyme that catalyzes glycolysis and is implicated in several non-metabolic processes, including transcription activation, initiation of apoptosis, and ER to Golgi vesicle shuttling) was investigated in HeLa cells and ranged between 50 and 80% depending on the polymer and formulation.
Polymer micelle vehicles
The complexation of siRNA with cationic polymers results in spontaneous assembly of IPECs. It is known from the literature that the resultant size and distribution of these complexes is highly dependent upon the length of the charged segments, the total length of the polymer, and the conditions under which they are prepared.7,116–123 Scales et al. have shown that in particular instances, a single siRNA molecule can be surrounded by multiple polymers, significantly limiting the therapeutic loading of these types of vectors.41 Ideally, the incorporation of multiple siRNAs per polymer complex is desirable to increase the therapeutic efficiency while maintaining control over the size, shape, and distribution of the carrier.One way to accomplish this is by the formation of the vehicle before loading the therapeutic agent. The formation of well-defined micelles and vesicles from polymers prepared via controlled polymerization methods is well established.7,67,73,101,116–121,123–139 In addition to allowing for control over the size and size distribution of the vehicle, the use of micelles/vesicles as gene delivery vehicles can also mitigate toxicity. Typically cationic (co)polymers exhibit in vitro toxicity that increases with molecular weight and cationic charge density, both of which also increase complexation and gene knockdown ability. Utilizing lower molecular weight components, higher ordered structures can degrade/dissociate into less toxic components. Though limited by the dynamic nature of such associated entities, it has been reported that the use of nucleic acids as crosslinking polyanions can result in sufficiently stable complexes.39,44,63,101,121,136
In an extension of the work by Stayton and coworkers mentioned previously, micelles capable of complexing with siRNA in the corona while containing an endosomal release component in the core were prepared viaRAFT polymerization.39 The corona consisted of DMAEMA while the core block was composed of statistically incorporated BMA, PAA, and DMAEMA (Fig. 8). The hydrophobic block was synthesized to be approximately 2.5 times larger than the hydrophilic block, and the optimum incorporation of BMA was found to be approximately 50% in the hydrophobic block. Particle sizes of the resulting micelles were 45 nm at pH 7.4, and upon the addition of siRNA (N:P = 4), particle sizes remained fairly consistent at 47 nm.
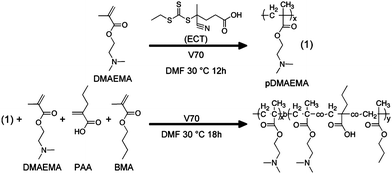 | ||
| Fig. 8 RAFT synthesis of hydrophilic, cationic DMAEMA macroCTA and subsequent chain extension for the preparation of an endolytic and polyampholyte block (which incorporates DMAEMA, PAA, and BMA). Reprinted with permission from Biomacromolecules.39 | ||
Membrane destabilizing ability was investigated using a red blood cell hemolysis assay. Three pH conditions were chosen to mimic cellular internalization: pH = 7.4 (extracellular), pH = 6.6 (early endosome), and pH 5.8 (late endosome). No significant hemolytic activity was observed at pH 7.4, while decreasing the pH below 7.4 resulted in increasing hemolytic activity. siRNA was complexed at various N:P ratios to account for any change that might be displayed in hemolytic activity due to charge–charge interactions, and none was observed even at high N:P ratios (N:P = 8). Additionally, differences in hemolytic activity between linear and assembled polymer solutions were negligible. Gene knockdown experiments were performed in HeLa cells with varying polymer (above and below the critical micelle concentration) and siRNA concentrations (12.5–100 nM). The greatest knockdown was observed with micelles at the highest siRNA concentrations, <90%. Investigation of uptake using FAM-labeled (6-carboxyfluorescein) siRNA revealed that almost 100% of cells were transfected by micelles, compared to only 25% in the previous study with the linear polymer system.38
The use of higher ordered structures such as micelles and vesicles also present the additional option of incorporating multiple therapeutic agents. Micelles with hydrophobic cores can accommodate poorly soluble drug payloads, while vesicles with hydrophilic cores can accommodate water-soluble drugs. Taking advantage of this option, Park and coworkers prepared biodegradable cationic micelles for co-delivery of siRNA and paclitaxel.44ε-Caprolactone was polymerized (7 kDa, PDI = 1.26) and subsequently N,N′-dicyclohexylcarbodiimide (DCC)-coupled to 4-cyanopentanoic acid dithionaphthalenoate (CPADN) to prepare a macroCTA for RAFT polymerization of DMAEMA (Fig. 9). Molecular weights ranged from 9.1 kDa to 21.1 kDa with PDIs ranging from 1.15–1.42. Micelles were prepared by direct dissolution into sodium acetate/acetic acid buffer (pH = 5.0) overnight and had CMC values of 15.8–24.0 mg L−1. Particle sizes ranged from 54 nm to 132 nm, and possessed moderately positive surface charges (+29.3 mV to +35.5 mV). Complexation with siRNA was investigated using a gel retardation assay, and complete retardation was observed at N:P > 4:1. siRNA transfection was monitored in MDA-MB-435-GFP cells. The experiments were carried out utilizing N:P = 36:1 and N:P = 12:1 for the micelles, with PEI (poly(ethylene imine)) (25 kDa) and DMAEMA homopolymers utilized as positive controls. The micelles had much higher transfection ability as compared to the positive controls. Cytotoxicity of the micelles (loaded with paclitaxel) was investigated in PC3 cells. The micelles were loaded with paclitaxel (6.8 wt%), and possessed a higher drug efficacy than free paclitaxel that the authors attributed to enhanced endocytosis of the micelles. Cells incubated with the free drug had viabilities of around 70%, whereas cells incubated with drug-loaded micelles had viabilities of approximately 50%, independent of the micelle concentration used. Co-delivery was investigated in PC3 cells with paclitaxel-loaded micelles complexed with siRNA at N:P = 24. Co-delivery resulted in 90% down-regulation of VEGF (vascular endothelial growth factor) expression whereas branched PEI exhibited 75% down-regulation. Interestingly, co-delivery of the therapeutic agents was slightly more efficient (90% vs. 80%) than micelle delivery of siRNA alone.
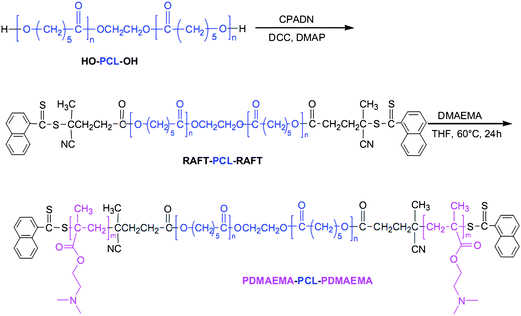 | ||
| Fig. 9 Synthetic pathway for the preparation of PDMAEMA–PCL–PDMAEMA degradable copolymers. Reprinted with permission from Biomaterials.44 | ||
Stayton and coworkers have also developed a multitherapeutic micellar system consisting of doxorubicin, an anthracycline utilized in many cancers, and siRNA against plk1, which has been shown to cause cell-cycle arrest and apoptosis in cancer cell lines as well as increased sensitivity to chemotherapeutics.63 The complexes they report consist of three basic components: (1) cationic micelles composed of P(DMAEMA-b-BMA) diblock copolymers (pDbB), (2) siRNA, and (3) anionic, pH-sensitive poly(styrene-alt-maleic anhydride) (pSMA) polymers that mediate endosomal escape (Fig. 10). pDbB was synthesized via RAFT using dodecyl cyanovaleric trithiocarbonate (DCT) CTA and 2,2′-azobis(2-methylpropionitrile) (AIBN) radical source. The overall Mw of the copolymer was 9.4 kDa with a PDI of 1.2. The pSMA was also synthesized via RAFT using 4-cyanopentanoic acid dithiobenzoate (CTP) and AIBN. A molecular weight of 48.5 kDa was targeted based on endolytic activity from previous studies.140
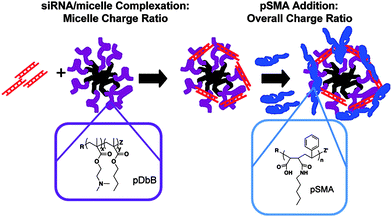 | ||
| Fig. 10 Ternary complex formation: siRNA (red), pBMA (black), pDbB (purple), and pSMA (blue). Reprinted with permission from Molecular Pharmaceutics.63 | ||
pDbB can form micelles at pH 7.4 such that the protonated DMAEMA block acts as the corona and the hydrophobic BMA block serves as the hydrophobic core. These micelles are positively charged with mean diameters of 37 nm. Both anionic siRNA and pSMA can be used to complex with the positively charged DMAEMA corona. Depending on the ratio of the three components, micelles with diameters varying from 30–100 nm can be obtained, all maintaining a solution-stable, slightly positively charged complex (ζ-potentials ranging from 0–1.2 mV). Micelles could be co-loaded with doxorubicin up to 10 wt% with a slight increase in micelle size. The gene knockdown ability and cell viability towards doxorubicin were investigated in both drug-sensitive OVCAR8 and multidrug-resistant NCI/ADR-REDS ovarian cancer cell lines. The ternary micelle complexes were able to achieve ∼50% knockdown (with or without doxorubicin) and effectively sensitized drug-resistant cells to doxorubicin resulting in a decrease in cell viability comparable to drug-sensitive cells.
Polymer-inorganic hybrid carriers—theranostics
Clearly the advent of controlled radical polymerization techniques has led to the dramatic increase in use of polymers for the construction of gene delivery vehicles. Still, many other materials have also found use in this burgeoning area. Calcium phosphate,141silica nanoparticles,142–144 and carbon nanotubes145–148 are just a small sampling of the many inorganic substrates that have been used in recent years as transfection agents.149,150 Indeed, the use of inorganic nanoparticles has proliferated due to their ease of synthesis, wide range of sizes, and biocompatibility.151–154 However, the application of simple inorganic nanoparticle constructs has been hindered due to their instability/aggregation, low loading efficiencies, and lack of functional “handles” for targeting group incorporation.155Initially, the placement of polymers onto metal nanoparticles was attempted to address issues of stability and the incorporation of reactive functionality.156,157 These early designs utilized ill-defined polymers, most of which were adsorbed onto the metal surface. However, developments in controlled syntheses of polymers as well as inorganic nanoparticles have led to the construction of well-defined hybrid materials. It has been through the combination of these materials that the development of “theranostics”, the incorporation of therapeutic and diagnostic agents on the same platform, has occurred.158–163 Thus, the combination of inorganic nanoparticles with organic polymers presents a class of novel delivery vehicles with enormous potential applications.
With recent advances in the synthesis of well-defined, narrowly dispersed iron oxide nanoparticles (IONPs), the use of this material as both diagnostic agent164 and a hyperthermia therapeutic has garnered increased attention.165 IONPs have also been used in the delivery of oligonucleotidesvia interpolyelectrolyte complexation with ill-defined cationic polymers, such as PEI, adsorbed to their surface.166 However, their usefulness in vitro/in vivo has been limited due to instability, aggregation, and fouling of the nanoparticle surface. Boyer and Davis have been able to combat this problem by developing a “grafting to” process utilizing polymers synthesized via RAFT containing a phosphonic acid end group that has a very high affinity for iron167 (Fig. 11). The phosphonic acid is coupled to a CTA utilized in the preparation of the polymers and ends up as a telechelic group on the terminus of the polymer chain.168 In more recent work, Boyer et al. have shown that by “co”-grafting two homopolymers, a cationic PDMAEA (poly(2-(N,N-dimethylamino)-ethyl acrylate)) and a hydrophilic POEGA (poly(oligo(ethylene glycol) acrylate)), onto the surface of pre-formed IONPs, the grafting density (0.12 chains nm−2) can be greatly increased over grafting homopolymer (PDMAEA 0.07 chains nm−2) or even a diblock copolymer (0.06 chains nm−2) alone. This increased grafting density has direct correlation to the overall stability of the NPs, making them storable for months instead of days, and highly resistant to protein absorption. Proton transverse relaxivity rates of bare NPs were compared to polymer grafted NPs and showed no marked effect. The gene knockdown of eGFP (enhanced green fluorescent protein) in human neuroblastoma cells of these constructs was investigated using flow cytometry. Compared to the lipofectamine control (40%), the fluorescence intensity of cells incubated with the co-graft IONPs was 78% which was attributed to decreased uptake due to pegylation of the particles. Interestingly, application of a magnetic field, which has been shown to effect the uptake of magnetic nanoparticles in cells,169 had a dramatic effect on the transfection efficiency, decreasing the fluorescence intensity to 50%, only slightly higher than the lipofectamine control.
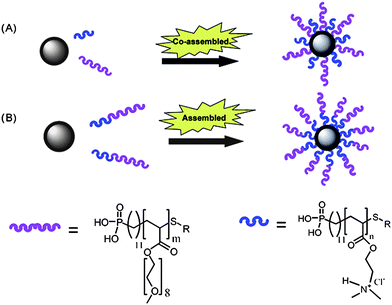 | ||
| Fig. 11 A graphical representation of the strategies used by Boyer et al. for (A) simultaneous grafting of two homopolymers, and (B) grafting of diblock copolymers. Reprinted with permission from Journal of Materials Chemistry.167 | ||
Perhaps the most successful and widely used inorganic platform is gold. Inherently non-toxic and inert, gold nanoparticles (GNPs) are easily synthesized in a range of sizes, easily functionalizable, and readily taken up by most cell lines.170–180Gold nanoparticles have also recently been approved by the FDA as formulated in the X-ray contrast agent, AuroVist™, and have the added benefit of functioning as a diagnostic agent.181,182 While the functionalization of gold nanoparticles with polymers has been widely demonstrated to increase therapeutic loading,183 incorporate targeting,184–186 and increase circulation time and stability in vivo,187 the construction of these platforms has been limited by low polymer grafting density due to inefficient ligand exchange. McCormick and coworkers have recently reported the synthesis of polymer-decorated gold nanoparticles that are formed in situ by the reduction of gold aurate by amine-containing blocks on their RAFT polymers.40,133,188,189 In particular, P(HPMA-b-DMAPMA) synthesized via aqueous RAFT was used to form and stabilize siRNA polyplexes (Fig. 12).40 The size and dispersity of these particles prior to IPEC formation with siRNA can be controlled by varying conditions including substrate concentration, amine to gold ratios, polymer block lengths, and reaction temperature. The grafting densities of the polymer-stabilized GNPs were quite high, 2.7 chains nm−2, as compared to similar systems, likely due to the extended conformation adopted by the polymer chains on the gold surface which also leads to larger diameter particles (29 nm) than predicted for random-coils (14 nm). The sizes of the GNPs before siRNA complexation were determined by TEM to be ∼6.5 nm. It should be noted that for the complexes shown in Fig. 12, the siRNA is protected against degradation by nucleases through both steric shielding conferred by the HPMA shell and by interpolyelectrolyte complexation with the protonated segments of the polymer vector. Luciferase expression was used to determine transfection efficiency of these constructs in KB cell lines. In these studies, polymer-stabilized GNPs demonstrated identical knockdown (50%) as the commercial agent Dharmafect.
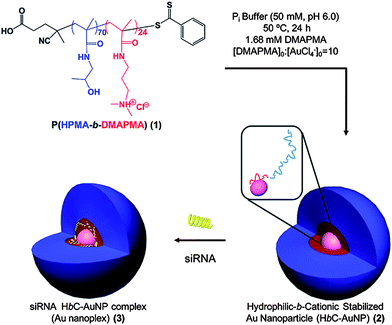 | ||
| Fig. 12 Reduction of AuCl4− in the presence of HPMA-block-DMAPMA copolymers to form hydrophilic-block-cationic polymer-stabilized AuNPs and subsequent complexation of siRNA. Reprinted with permission from Biomacromolecules.40 | ||
siRNA–polymer conjugates
Most research on siRNA delivery has focused on the formation of interpolyelectrolyte complexes with cationic polymers. However, many questions still remain concerning the binding strength of these complexes, the mechanism of siRNA release, and how these parameters affect gene knockdown efficiency.8,10,16,19,21,23,25,26,28,29,121 A number of barriers to delivery might be overcome by designing better therapeutic carriers utilizing modular concepts introduced by the original Ringsdorf model, for example, cleavable functionality to release siRNA.Much effort in the conjugation of biomolecules to RAFT polymers has focused on modification of the chain ends via functionality incorporated from the R or Z groups of the CTA agent (Fig. 6).58,66,71,190–198 However, to be applicable to the delivery of siRNA, the conjugation used must be reversible/cleavable. Bulmus and Maynard have several examples of reversible conjugation to the terminus of polymersvia a disulfide linkage.42,190,191,194,198,199 Perhaps the most promising for delivery was conjugation of siRNA to a water-soluble poly(ethylene glycol)-containing acrylate utilizing elegant RAFT chemistry.194 Simple conjugation of poly(ethylene glycol) (PEG) to siRNAvia a disulfide bond has been shown to both increase the circulation time and protect against premature degradation.200–202Pyridyl disulfide propanol was coupled to the CTA 2-(ethyl trithiocarbonate) propionic acid and used to polymerize poly(ethylene glycol) acrylate (PEGA) (Fig. 13). Linear kinetic plots were obtained in DMF at 60 °C with AIBN as an initiator after a 20 minute inhibition period. Conversions up to 80% with PDIs approaching 1.2 were obtained. Retention of the end group was confirmed by 1H NMR. Thiol exchange between the 5′-end of the siRNA and the pyridyl disulfide was accomplished with 88% yield. Complete cleavage of siRNA could be achieved under reductive conditions with dithiothreitol (DTT).
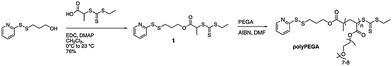 | ||
| Fig. 13 Synthesis of a pyridyl disulfide functionalized CTA and subsequent RAFT polymerization of PEGA. Reprinted with permission from Chemical Communications.179 | ||
This thiol exchange strategy, developed by Bulmus and coworkers initially for protein conjugation to RAFT polymers,191 was also utilized by York et al. for conjugation of siRNA to RAFT polymers at multiple sites along the backbone.70 Instead of conjugating one siRNA per polymer, they were able to incorporate multiple siRNAs to increase the therapeutic payload and potentially increase the efficiency (Fig. 14). HPMA was copolymerized with APMA using V-501 (4,4′-azobis(4-cyanovaleric acid)) as the initiator and CTP as the CTA. The final polymer was end-capped with AIBN due to the potential toxicity of the CTA end group.203N-Succinimidyl 3-(2-pyridyldithio) propionate (SPDP) was used to functionalize the free amines of the APMA. Thiol exchange with the sense strand of the siRNA was accomplished with 89% efficiency. Folic acid was then conjugated to the remaining free amines for targeting. The anti-sense strand was subsequently complexed to the polymer conjugate. The release of siRNA from the polymer was demonstrated under intracellular conditions with 5 mM glutathione. About 60% of the siRNA was released in a 4 h period.
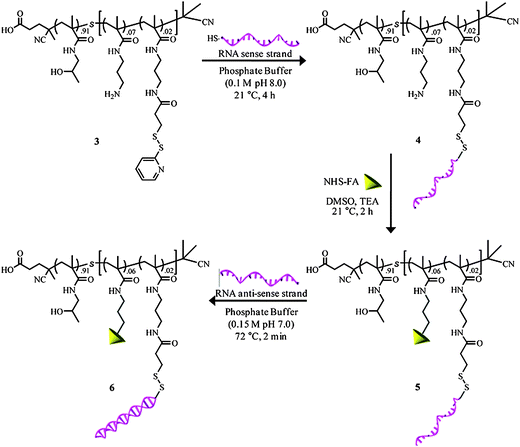 | ||
| Fig. 14 Reaction pathway for the synthesis of both RNA and folate conjugated copolymers and subsequent hybridization with RNA antisense strands. Reprinted with permission from Biomacromolecules.70 | ||
Conclusions/outlook
Development of “smart”, safe, and effective carriers for therapeutic delivery of siRNA is an area of intensive, multi-disciplinary effort, cross-cutting many fields including polymer chemistry, cell biology, pharmaceutical science, bioengineering, materials science, and nanomedicine. Controlled polymerization techniques, robust methods for placement of reactive functionality, and efficient orthogonal chemistries have been sufficiently developed during the last decade to allow for design and construction of the first phase of polymer-based, nonviral siRNA delivery modules with targeting, diagnostic, and stimuli-responsive capabilities. Such carriers specifically for siRNA, however, are still in developmental infancy and are generally based on criteria specific to DNA therapeutics, a much more mature area of experimental research. From a polymer chemistry perspective, significant differences in the physicochemical properties of siRNA and DNA alone suggest that efficient carriers (for example those to be used for interpolyelectrolyte complexation) must be designed with different criteria. From the first step of siRNA packaging and throughout the delivery, endosomal escape, unpackaging, and release to RISC for sequence specific mRNA interference, factors discussed briefly in this review including local and regional interchain charge registry, surface charge density, complex strength, hydrodynamic volume, and stimulus-responsive dissociation must be further considered in appropriate structural design. The most recent literature reports from biochemists, cell biologists, and others studying “knockdown” with current constructs convey the challenges ahead in order to design appropriate vehicles and to ultimately realize the enormous potential of polymer-based siRNA therapeutics. Clearly a more advanced level of “rational design” and synthesis must be achieved. Fortunately, experimental techniques and analytical methods for following siRNA/polymer complexes in vivo are rapidly being developed. As further understanding of mechanistic barriers to cell-specific entry and trafficking through the RNAi pathway evolve and as experimental protocols for rigorous characterization and in situ tracing of polymeric carriers advance, innovative designs based on fundamental concepts of polymer chemistry and macromolecular engineering will certainly impact the development of the next generation of siRNA therapeutics.Abbreviations
| AIBN | 2,2′-azobis(2-methylpropionitrile) |
| APMA | N-(3-aminopropyl) methacrylamide hydrochloride |
| aRAFT | aqueous reversible addition–fragmentation chain transfer |
| ATRP | atom transfer radical polymerization |
| BMA | butyl methacrylate |
| CPADN | cyanopentanoic acid dithionaphthalenoate |
| CRP | controlled radical polymerization |
| CTA | chain transfer agent |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DCT | dodecyl cyanovalerictrithiocarbonate |
| DLS | dynamic light scattering |
| DMA | N,N-dimethylacrylamide |
| DMAEA | 2-(N,N-dimethylamino)-ethyl acrylate |
| DMAEMA | 2-(N,N-dimethylamino)-ethyl methacrylate |
| DMAPMA | 3-(N,N-dimethylamino)-propyl methacrylamide |
| DNA | deoxyribonucleic acid |
| dsRNA | double stranded ribonucleic acid |
| DTT | dithiothreitol |
| FAM | 6-carboxyfluorescein |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GFP | green fluorescence protein |
| GNPs | gold nanoparticles |
| HPMA | N-(2-hydroxypropyl)-methacrylamide |
| IONPs | iron oxide nanoparticles |
| IPECs | interpolyelectrolyte complex |
| mRNA | messenger ribonucleic acid |
| N/P | nitrogen to phosphate ratio |
| NIPAM | N-isopropylacrylamide |
| OEGA | oligo(ethylene glycol) acrylate |
| PAA | poly(acrylic acid) |
| pDbB | P(DMAEMA-b-BMA) |
| PDI | polydispersity index |
| PEG | poly(ethylene glycol) |
| PEGA | poly(ethylene glycol) acrylate |
| PEI | poly(ethylene imine) |
| pSMA | poly(styrene-alt-maleic anhydride) |
| RAFT | reversible addition–fragmentation chain transfer |
| RISC | RNA-induced silencing complex |
| RNAi | ribonucleic acid interference |
| SFRP | stable free radical polymerization |
| siRNA | small interfering ribonucleic acid |
| SPDP | N-succinimidyl 3-(2-pyridyldithio) propionate |
| V-501 | 4,4′-azobis(4-cyanovaleric acid) |
| VEGF | vascular endothelial growth factor |
Acknowledgements
The authors would like to acknowledge support from the National Science Foundation through the MRSEC (DR-0213883) and NSF EPSCoR (EPS-0903787) programs. We would like to thank Eric Burden for his help preparing figures for this manuscript. Also, Brooks Abel is thanked for his suggestions and editing of the review manuscript.References
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312–325 CrossRef CAS.
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 Search PubMed.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- D. J. Gary, N. Puri and Y.-Y. Won, J. Controlled Release, 2007, 121, 64–73 CrossRef CAS.
- H. Bader, H. Ringsdorf and B. Schmidt, Angew. Makromol. Chem., 1984, 123/124, 457–485 CrossRef.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- A. D'Emanuele and D. Attwood, Adv. Drug Delivery Rev., 2005, 57, 2147–2162 CrossRef CAS.
- W. H. Heath, A. F. Senyurt, J. Layman and T. E. Long, Macromol. Chem. Phys., 2007, 208, 1243–1249 CrossRef CAS.
- M. Licciardi, Y. Tang, N. C. Billingham, S. P. Armes and A. L. Lewis, Biomacromolecules, 2005, 6, 1085–1096 CrossRef CAS.
- T. Merdan, J. Kopecek and T. Kissel, Adv. Drug Delivery Rev., 2002, 54, 715–758 CrossRef CAS.
- M. E. Napier and J. M. DeSimone, Polym. Rev., 2007, 47, 321–327 Search PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- T. G. Park, J. H. Jeong and S. W. Kim, Adv. Drug Delivery Rev., 2006, 58, 467–486 CrossRef CAS.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS.
- S. Y. Wong, J. M. Pelet and D. Putnam, Prog. Polym. Sci., 2007, 32, 799–837 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- L. Xu and T. Anchordoquy, J. Pharm. Sci., 2011, 100, 38–52 CrossRef CAS.
- J. W. Gaynor, B. J. Campbell and R. Cosstick, Chem. Soc. Rev., 2010, 39, 4169–4184 RSC.
- R. J. Christie, N. Nishiyama and K. Kataoka, Endocrinology, 2010, 151, 466–473 CrossRef CAS.
- C. L. Grigsby and K. W. Leong, J. R. Soc., Interface, 2010, 7, S67–S82 CrossRef CAS.
- Y. Higuchi, S. Kawakami and M. Hashida, BioDrugs, 2010, 24, 195–205 Search PubMed.
- Y.-K. Oh and T. G. Park, Adv. Drug Delivery Rev., 2009, 61, 850–862 CrossRef CAS.
- C. Pichon, L. Billiet and P. Midoux, Curr. Opin. Biotechnol., 2010, 21, 640–645 CrossRef CAS.
- C. M. Roth, Biotechnol. Prog., 2008, 24, 23–28 CrossRef CAS.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. S. Au, AAPS J., 2010, 12, 492–503 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Handb. RAFT Polym., 2008, 235–284 Search PubMed.
- A. H. Alidedeoglu, A. W. York, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5405–5415 CrossRef CAS.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS.
- A. J. Convertine, C. Diab, M. Prieve, A. Paschal, A. S. Hoffman, P. H. Johnson and P. S. Stayton, Biomacromolecules, 2010, 11, 2904–2911 CrossRef CAS.
- S. Kirkland-York, Y. Zhang, A. E. Smith, A. W. York, F. Huang and C. L. McCormick, Biomacromolecules, 2010, 11, 1052–1059 CrossRef CAS.
- C. W. Scales, F. Huang, N. Li, Y. A. Vasilieva, J. Ray, A. J. Convertine and C. L. McCormick, Macromolecules, 2006, 39, 6871–6881 CrossRef CAS.
- D. Valade, C. Boyer, T. P. Davis and V. Bulmus, Aust. J. Chem., 2009, 62, 1344–1350 CrossRef CAS.
- A. W. York, Y. Zhang, A. C. Holley, Y. Guo, F. Huang and C. L. McCormick, Biomacromolecules, 2009, 10, 936–943 CrossRef CAS.
- C. Zhu, S. Jung, S. Luo, F. Meng, X. Zhu, T. G. Park and Z. Zhong, Biomaterials, 2010, 31, 2408–2416 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, A. B. Lowe, C. W. Scales, L. J. Myrick and C. L. McCormick, Macromol. Rapid Commun., 2005, 26, 791–795 CrossRef CAS.
- M. S. Donovan, A. B. Lowe, B. S. Sumerlin and C. L. McCormick, Macromolecules, 2002, 35, 4123–4132 CrossRef CAS.
- M. S. Donovan, T. A. Sanford, A. B. Lowe, B. S. Sumerlin, Y. Mitsukami and C. L. McCormick, Macromolecules, 2002, 35, 4570–4572 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Aust. J. Chem., 2002, 55, 367–379 CrossRef CAS.
- A. David, P. Kopeckova, T. Minko, A. Rubinstein and J. Kopecek, Eur. J. Cancer, 2004, 40, 148–157 CrossRef CAS.
- T. Etrych, M. Jelinkova, B. Rihova and K. Ulbrich, J. Controlled Release, 2001, 73, 89–102 CrossRef CAS.
- M. Jelinkova, J. Strohalm, T. Etrych, K. Ulbrich and B. Rihova, Pharm. Res., 2003, 20, 1558–1564 CAS.
- J. Kopecek, Biomaterials, 1984, 5, 19–25 CrossRef CAS.
- M. Kovar, L. Kovar, V. Subr, T. Etrych, K. Ulbrich, T. Mrkvan, J. Loucka and B. Rihova, J. Controlled Release, 2004, 99, 301–314 CrossRef CAS.
- A. Nori, K. D. Jensen, M. Tijerina, P. Kopeckova and J. Kopecek, Bioconjugate Chem., 2003, 14, 44–50 CrossRef CAS.
- A. Nori and J. Kopecek, Adv. Drug Delivery Rev., 2005, 57, 609–636 CrossRef CAS.
- D. Putnam and J. Kopecek, Adv. Polym. Sci., 1995, 122, 55–123 CAS.
- R. Satchi-Fainaro, H. Hailu, J. W. Davies, C. Summerford and R. Duncan, Bioconjugate Chem., 2003, 14, 797–804 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- C. W. Scales, Y. A. Vasilieva, A. J. Convertine, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2005, 6, 1846–1850 CrossRef CAS.
- A. Tang, P. Kopeckova and J. Kopecek, Pharm. Res., 2003, 20, 360–367 CAS.
- K. Ulbrich, T. Etrych, P. Chytil, M. Pechar, M. Jelinkova and B. Rihova, Int. J. Pharm., 2004, 277, 63–72 CrossRef CAS.
- K. Ulbrich and V. Subr, Adv. Drug Delivery Rev., 2004, 56, 1023–1050 CrossRef CAS.
- D. S. W. Benoit, S. M. Henry, A. D. Shubin, A. S. Hoffman and P. S. Stayton, Mol. Pharmaceutics, 2010, 7, 442–455 CAS.
- Z. M. O. Rzaev, S. Dincer and E. Piskin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- C.-Y. Hong and C.-Y. Pan, Macromolecules, 2006, 39, 3517–3524 CrossRef CAS.
- M. J. Joralemon, N. L. Smith, D. Holowka, B. Baird and K. L. Wooley, Bioconjugate Chem., 2005, 16, 1246–1256 CrossRef CAS.
- D. Pan, J. L. Turner and K. L. Wooley, Macromolecules, 2004, 37, 7109–7115 CrossRef CAS.
- V. Vazquez-Dorbatt and H. D. Maynard, Biomacromolecules, 2006, 7, 2297–2302 CrossRef CAS.
- A. W. York, F. Huang and C. L. McCormick, Biomacromolecules, 2010, 11, 505–514 CrossRef CAS.
- A. W. York, C. W. Scales, F. Huang and C. L. McCormick, Biomacromolecules, 2007, 8, 2337–2341 CrossRef CAS.
- A. J. Convertine, N. Ayres, C. W. Scales, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2004, 5, 1177–1180 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- T. P. Lodge, Macromolecules, 2009, 42, 3827–3829 CrossRef CAS.
- A. B. Lowe and M. A. Harvison, Aust. J. Chem., 2010, 63, 1251–1266 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745–4750 RSC.
- J.-F. Lutz and H. G. Boerner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- J.-F. Lutz and B. S. Sumerlin, Click Chemistry for Biotechnology and Materials Science, 2009, pp. 69–88 Search PubMed.
- U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560–1598 RSC.
- H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197–2208 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Click Chemistry for Biotechnology and Materials Science, 2009, pp. 89–117 Search PubMed.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- S. Dehn, R. Chapman, K. A. Jolliffee and S. Perrier, Polym. Rev., 2011 Search PubMed , in press.
- S. C. De Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113–126 CrossRef.
- A. M. Funhoff, C. F. van Nostrum, G. A. Koning, N. M. E. Schuurmans-Nieuwenbroek, D. J. A. Crommelin and W. E. Hennink, Biomacromolecules, 2004, 5, 32–39 CrossRef CAS.
- R. A. Jones, M. H. Poniris and M. R. Wilson, J. Controlled Release, 2004, 96, 379–391 CrossRef CAS.
- T. Reschel, C. Konak, D. Oupicky, L. W. Seymour and K. Ulbrich, J. Controlled Release, 2002, 81, 201–217 CrossRef CAS.
- P. van de Wetering, J. Y. Cherng, H. Talsma, D. J. A. Crommelin and W. E. Hennink, J. Controlled Release, 1998, 53, 145–153 CrossRef.
- P. van de Wetering, J.-Y. Cherng, H. Talsma and W. E. Hennink, J. Controlled Release, 1997, 49, 59–69 CrossRef CAS.
- F. J. Verbaan, C. Oussoren, I. M. Van Dam, Y. Takakura, M. Hashida, D. J. A. Crommelin, W. E. Hennink and G. Storm, Int. J. Pharm., 2001, 214, 99–101 CrossRef CAS.
- Y.-Z. You, D. S. Manickam, Q.-H. Zhou and D. Oupicky, J. Controlled Release, 2007, 122, 217–225 CrossRef CAS.
- T. Bruckdorfer, Eur. Biopharm. Rev., 2008, 96 Search PubMed , 98, 100, 102, 104.
- H. Eliyahu, Y. Barenholz and A. J. Domb, Molecules, 2005, 10, 34–64 Search PubMed.
- K. Kataoka, K. Itaka, N. Nishiyama, Y. Yamasaki, M. Oishi and Y. Nagasaki, Nucleic Acids Symp. Ser., 2005, 17–18 Search PubMed.
- M. Oishi and Y. Nagasaki, Biotechnol.: Pharm. Aspects, 2009, 10, 35–67 Search PubMed.
- D. Oupicky, C. Konak, K. Ulbrich, M. A. Wolfert and L. W. Seymour, J. Controlled Release, 2000, 65, 149–171 CrossRef CAS.
- J. F. Tan, T. A. Hatton, K. C. Tam and H. P. Too, Biomacromolecules, 2007, 8, 448–454 CrossRef CAS.
- A. R. Hilgenbrink and P. S. Low, J. Pharm. Sci., 2005, 94, 2135–2146 CrossRef CAS.
- R. J. Lee and P. S. Low, J. Liposome Res., 1997, 7, 455–466 CrossRef CAS.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2008, 41, 120–129 CrossRef CAS.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2002, 54, 675–693 CrossRef CAS.
- Y. Lu, E. Sega, C. P. Leamon and P. S. Low, Adv. Drug Delivery Rev., 2004, 56, 1161–1176 CrossRef CAS.
- Y. W. Cho, J.-D. Kim and K. Park, J. Pharm. Pharmacol., 2003, 55, 721–734 CrossRef CAS.
- K. G. de Bruin, C. Fella, M. Ogris, E. Wagner, N. Ruthardt and C. Braeuchle, J. Controlled Release, 2008, 130, 175–182 CrossRef CAS.
- S. Oliveira, I. van Rooy, O. Kranenburg, G. Storm and R. M. Schiffelers, Int. J. Pharm., 2007, 331, 211–214 CrossRef CAS.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS.
- M. Malkoch, R. J. Thibault, E. Drockenmuller, M. Messerschmidt, B. Voit, T. P. Russell and C. J. Hawker, J. Am. Chem. Soc., 2005, 127, 14942–14949 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- J. D. Flores, X. Xu, N. J. Treat and C. L. McCormick, Macromolecules, 2009, 42, 4941–4945 CrossRef CAS.
- M. G. Kellum, A. E. Smith, S. K. York and C. L. McCormick, Macromolecules, 2010, 43, 7033–7040 CrossRef CAS.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- B. S. Lokitz, A. J. Convertine, R. G. Ezell, A. Heidenreich, Y. Li and C. L. McCormick, Macromolecules, 2006, 39, 8594–8602 CrossRef CAS.
- B. S. Lokitz, A. W. York, J. E. Stempka, N. D. Treat, Y. Li, W. L. Jarrett and C. L. McCormick, Macromolecules, 2007, 40, 6473–6480 CrossRef CAS.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- A. E. Smith, X. Xu, S. E. Kirkland-York, D. A. Savin and C. L. McCormick, Macromolecules, 2010, 43, 1210–1217 CrossRef CAS.
- K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1397–1407 CrossRef CAS.
- S. Harrisson and K. L. Wooley, Chem. Commun., 2005, 3259–3261 RSC.
- M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892–16899 CrossRef CAS.
- P. Kujawa, H. Watanabe, F. Tanaka and F. M. Winnik, Eur. Phys. J. E, 2005, 17, 129–137 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81–89 CrossRef CAS.
- B. Liu and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3643–3654 CrossRef CAS.
- A. B. Lowe, R. Wang, V. Tiriveedhi, P. Butko and C. L. McCormick, Macromol. Chem. Phys., 2007, 208, 2339–2347 CrossRef CAS.
- C. L. McCormick, S. E. Kirkland and A. W. York, Polym. Rev., 2006, 46, 421–443 Search PubMed.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- Y. Mitsukami, A. Hashidzume, S.-i. Yusa, Y. Morishima, A. B. Lowe and C. L. McCormick, Polymer, 2006, 47, 4333–4340 CrossRef CAS.
- A. E. Smith, X. Xu, T. U. Abell, S. E. Kirkland, R. M. Hensarling and C. L. McCormick, Macromolecules, 2009, 42, 2958–2964 CrossRef CAS.
- M. H. Stenzel, C. Barner-Kowollik, T. P. Davis and H. M. Dalton, Macromol. Biosci., 2004, 4, 445–453 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, D. B. Thomas, A. J. Convertine, M. S. Donovan and C. L. McCormick, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1724–1734 CrossRef CAS.
- X. Xu, A. E. Smith, S. E. Kirkland and C. L. McCormick, Macromolecules, 2008, 41, 8429–8435 CrossRef CAS.
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2003, 36, 4208–4215 CrossRef CAS.
- S. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2004, 37, 7507–7513 CrossRef CAS.
- L. Zhang, T. L. U. Nguyen, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2007, 8, 2890–2901 CrossRef CAS.
- P. S. Stayton, A. S. Hoffman, M. El-Sayed, S. Kulkarni, T. Shimoboji, N. Murthy, V. Bulmus and C. Lackey, Proc. IEEE, 2005, 93, 726–736 CrossRef CAS.
- I. Roy, S. Mitra, A. Maitra and S. Mozumdar, Int. J. Pharm., 2003, 250, 25–33 CrossRef CAS.
- G. Bardi, M. A. Malvindi, L. Gherardini, M. Costa, P. P. Pompa, R. Cingolani and T. Pizzorusso, Biomaterials, 2010, 31, 6555–6566 CrossRef CAS.
- J. E. Fuller, G. T. Zugates, L. S. Ferreira, H. S. Ow, N. N. Nguyen, U. B. Wiesner and R. S. Langer, Biomaterials, 2008, 29, 1526–1532 CrossRef CAS.
- M. Kar, P. S. Vijayakumar, B. L. V. Prasad and S. Sen Gupta, Langmuir, 2010, 26, 5772–5781 CrossRef CAS.
- H. Gao, Y. Kong, D. Cui and C. S. Ozkan, Nano Lett., 2003, 3, 471–473 CrossRef CAS.
- V. Neves, E. Heister, S. Costa, C. Tilmaciu, E. Borowiak-Palen, C. E. Giusca, E. Flahaut, B. Soula, H. M. Coley, J. McFadden and S. R. P. Silva, Adv. Funct. Mater., 2010, 20, 3272–3279 CrossRef CAS.
- A. Nunes, N. Amsharov, C. Guo, J. Van den Bossche, P. Santhosh, T. K. Karachalios, S. F. Nitodas, M. Burghard, K. Kostarelos and K. T. Al-Jamal, Small, 2010, 6, 2281–2291 CrossRef CAS.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J.-P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS.
- B. S. Sekhon and S. R. Kamboj, Nanomedicine, 2010, 6, 612–618 CAS.
- B. S. Sekhon and S. R. Kamboj, Nanomedicine, 2010, 6, 516–522 CAS.
- L. Berti and G. A. Burley, Nat. Nanotechnol., 2008, 3, 81–87 CrossRef CAS.
- I. Bilecka and M. Niederberger, Electrochim. Acta, 2010, 55, 7717–7725 CrossRef CAS.
- I. Capek, Surfactant Sci. Ser., 2010, 147, 779–861 Search PubMed.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- B. Fadeel and A. E. Garcia-Bennett, Adv. Drug Delivery Rev., 2010, 62, 362–374 CrossRef CAS.
- C. Minelli, S. B. Lowe and M. M. Stevens, Small, 2010, 6, 2336–2357 CrossRef CAS.
- F. Sonvico, C. Dubernet, P. Colombo and P. Couvreur, Curr. Pharm. Des., 2005, 11, 2095–2105.
- J. Chen, M. Yang, Q. Zhang, E. C. Cho, C. M. Cobley, C. Kim, C. Glaus, L. V. Wang, M. J. Welch and Y. Xia, Adv. Funct. Mater., 2010, 20, 3684–3694 CrossRef CAS.
- S. M. Janib, A. S. Moses and J. A. MacKay, Adv. Drug Delivery Rev., 2010, 62, 1052–1063 CrossRef CAS.
- J. A. MacKay and Z. Li, Adv. Drug Delivery Rev., 2010, 62, 1003–1004 CrossRef CAS.
- J. R. McCarthy, Adv. Drug Delivery Rev., 2010, 62, 1023–1030 CrossRef CAS.
- P. Rai, S. Mallidi, X. Zheng, R. Rahmanzadeh, Y. Mir, S. Elrington, A. Khurshid and T. Hasan, Adv. Drug Delivery Rev., 2010, 62, 1094–1124 CrossRef CAS.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS.
- J.-H. Lee, Y.-w. Jun, S.-I. Yeon, J.-S. Shin and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 8160–8162 CrossRef.
- B. Samanta, H. Yan, N. O. Fischer, J. Shi, D. J. Jerry and V. M. Rotello, J. Mater. Chem., 2008, 18, 1204–1208 RSC.
- X. Wang, L. Zhou, Y. Ma, X. Li and H. Gu, Nano Res., 2009, 2, 365–372 CrossRef CAS.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Carroll, R. Woodward and T. St Pierre, J. Mater. Chem., 2010, 20, 255–265 RSC.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111–123 RSC.
- Y.-w. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS.
- P. Alexandridis, Chem. Eng. Technol., 2011, 34, 15–28 CrossRef CAS.
- G. Kawamura and M. Nogami, J. Cryst. Growth, 2009, 311, 4462–4466 CrossRef CAS.
- S. Y. Moon, T. Sekino, T. Kusunose and S.-i. Tanaka, J. Cryst. Growth, 2009, 311, 651–656 CrossRef CAS.
- S. D. Perrault and W. C. W. Chan, J. Am. Chem. Soc., 2009, 131, 17042–17043 CrossRef CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS.
- Z. Krpetic, F. Porta, E. Caneva, V. Dal Santo and G. Scari, Langmuir, 2010, 26, 14799–14805 CrossRef CAS.
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS.
- M. D. Massich, D. A. Giljohann, A. L. Schmucker, P. C. Patel and C. A. Mirkin, ACS Nano, 2010, 4, 5641–5646 CrossRef CAS.
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Bioconjugate Chem., 2010, 21, 2250–2256 CrossRef CAS.
- N. B. Shah, J. Dong and J. C. Bischof, Mol. Pharmaceutics, 2011, 8, 176–184 CAS.
- Q.-Y. Cai, S. H. Kim, K. S. Choi, S. Y. Kim, S. J. Byun, K. W. Kim, S. H. Park, S. K. Juhng and K.-H. Yoon, Invest. Radiol., 2007, 42, 797–806 CrossRef CAS.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 Search PubMed.
- J.-H. Kim and T. R. Lee, Chem. Mater., 2004, 16, 3647–3651 CrossRef.
- D. J. Javier, N. Nitin, M. Levy, A. Ellington and R. Richards-Kortum, Bioconjugate Chem., 2008, 19, 1309–1312 CrossRef CAS.
- C. R. Patra, R. Bhattacharya, D. Mukhopadhyay and P. Mukherjee, Adv. Drug Delivery Rev., 2010, 62, 346–361 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 6065–6075 CrossRef CAS.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS.
- Y. Li, A. E. Smith, B. S. Lokitz and C. L. McCormick, Macromolecules, 2007, 40, 8524–8526 CrossRef CAS.
- A. E. Smith, X. Xu, D. A. Savin and C. L. McCormick, Polym. Chem., 2010, 1, 628–630 RSC.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- N. M. L. Hansen, D. M. Haddleton and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5770–5780 CrossRef CAS.
- S. M. Henry, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 1122–1128 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245–3247 RSC.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- J. Xu, C. Boyer, V. Bulmus and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4302–4313 CrossRef CAS.
- J. Liu, H. Liu, Z. Jia, V. Bulmus and T. P. Davis, Chem. Commun., 2008, 6582–6584 RSC.
- S. H. Kim, J. H. Jeong, S. H. Lee, S. W. Kim and T. G. Park, J. Controlled Release, 2006, 116, 123–129 CrossRef CAS.
- M. Oishi, Y. Nagasaki, K. Itaka, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2005, 127, 1624–1625 CrossRef CAS.
- D. B. Rozema, D. L. Lewis, D. H. Wakefield, S. C. Wong, J. J. Klein, P. L. Roesch, S. L. Bertin, T. W. Reppen, Q. Chu, A. V. Blokhin, J. E. Hagstrom and J. A. Wolff, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12982–12987 CrossRef CAS.
- C.-W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
Footnote |
| † Paper number 150 in a series entitled Water Soluble Polymers. |
| This journal is © The Royal Society of Chemistry 2011 |