First peptide/protein PEGylation with functional polymers designed by nitroxide-mediated polymerization†
Marion
Chenal
a,
Céline
Boursier
b,
Yohann
Guillaneuf
c,
Myriam
Taverna
a,
Patrick
Couvreur
a and
Julien
Nicolas
*a
aLaboratoire de Physico-Chimie, Pharmacotechnie et Biopharmacie, Univ Paris-Sud, UMR CNRS 8612, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, F-92296, Châtenay-Malabry cedex, France. E-mail: julien.nicolas@u-psud.fr; Fax: +33 1 46 83 59 46; Tel: +33 1 46 83 58 53
bPlate-forme transcriptome-protéome, IFR 141, Univ Paris-Sud, Faculté de Pharmacie, 5, rue Jean-Baptiste Clément, F-92296, Châtenay-Malabry cedex, France
cLaboratoire Chimie Provence, Univ de Provence, UMR CNRS 6264, Avenue Escadrille Normandie-Niemen, Case 542, F-13397, Marseille cedex 20, France
First published on 21st February 2011
Abstract
Well-defined fluorescent, α-functional polymethacrylates with PEG side chains were readily designed by nitroxide-mediated polymerization (NMP) from N-hydroxysuccinimidyl (NHS) ester-containing alkoxyamines based on the nitroxide SG1. These polymers were obtained without any purification beyond a simple precipitation. They exhibited tunable reactivities towards nucleophiles depending on the nature of the alkoxyamine used. Copolymers derived from the commercially available SG1-based alkoxyamine led to quantitative coupling with small molecules and a neuroprotective peptide whereas partial conjugation was obtained with lysozyme, used here as a model protein. Complete conjugation to the latter was obtained from similar copolymers exhibiting a less sterically hindered NHS extremity, via appropriate alkoxyamine structure, which represents the very first example of peptide/protein PEGylation with functional polymers derived from NMP.
Introduction
Conjugation of synthetic polymers to proteins and peptides (termed bioconjugation) is an area of high interest. The most typical example is certainly the covalent linkage of poly(ethylene glycol) (PEG), a hydrophilic and flexible polymer, to poly(peptide)-based therapeutics, the so-called PEGylation.1–3 The resulting bioconjugates exhibit improved biodistribution and pharmacokinetics, better stability and solubility, reduced immunogenicity and longer plasma half-life due to both reduced renal filtration and proteolysis, when compared to non-PEGylated analogs.4,5The recent development of controlled/living radical polymerization (CLRP) provides the polymer chemist with efficient and easy routes for achieving various macromolecular architectures of high complexity and functionality, unavailable by other polymerization methods.6–11 Among them, nitroxide-mediated polymerization (NMP),6 atom transfer radical polymerization (ATRP)8,9,12,13 and reversible addition–fragmentation chain transfer (RAFT)7,10,11 represent the three best-known CLRP techniques. Whereas linear PEG has been historically employed for PEGylation, the fast-growth of CLRP techniques has brought about a significant renewal as new types of PEG-based and other biocompatible polymers with unique properties have been synthesized and successfully employed for bioconjugation purposes.14–17 In this field, a well-known example is the polymerization of poly(ethylene glycol) methyl ether methacrylate (MePEGMA) which leads to comb-shaped polymethacrylates with PEG side chains.18–24 In the last few years, the concept of protein PEGylation from preformed polymers (also termed the “grafting to” method) was elegantly extended to α-functional poly(MePEGMA) polymers obtained from CLRP by the Haddleton,25–29 Maynard,30 Davis31 and Lutz32 research groups. These pioneering studies used either ATRP or RAFT to design well-defined, α-functional poly(MePEGMA), followed by their conjugation to model proteins. More generally, the explosive growth in the number of publications witnessed in the past couple of years devoted to the design of bioconjugates from CLRP has almost exclusively involved ATRP and RAFT techniques.15,16 Such a supremacy can be explained by the easy access to functional ATRP initiators or RAFT agents as well as to mild reaction conditions. A great deal of effort is currently devoted to develop ATRP-based processes with lower amounts of catalyst (usually based on copper)9 or efficient RAFT end-group removal pathways33 due to potential toxicological issues. NMP could actually be considered as a relevant alternative, perhaps the technique of choice, regarding biological applications as non-cytotoxic polymers can be generated without any purification beyond a simple precipitation34 and contrary to ATRP and RAFT, it has the advantage of being only governed by a thermal process and does not require any transition metal catalysts or bimolecular exchange with sulfur-based compounds. In the bioconjugation landscape, simplicity of the synthetic and purification procedures, innocuousness of the chemical components and biocompatibility of the final structure are highly desirable.
Studies dealing with the design of bioconjugates from NMP are extremely scarce and, to the best of our knowledge, none of them reported bioconjugation to proteins or PEGylation of small-size peptides. The only publications available in the literature used a combination of solid-phase peptide synthesis (SPPS) and NMP from the solid support to construct the desired bioconjugates.35–37 Wooley and co-workers synthesized the protein transduction domain (PTD)35 of the HIV Tat protein and the antimicrobial peptide tritrpticin,36 followed by their coupling upon the resin to a TIPNO-based alkoxyaminevia the peptide terminus. Sequential polymerization of tert-butyl acrylate and methyl acrylate (or styrene) was then performed to yield the corresponding peptide–polymer bioconjugates. Similarly, Trimaille and co-workers prepared a SG1-functionalized peptide (GGGWIKVAV–SG1) by SPPS and direct coupling with a SG1-based alkoxyamine.37 Then, surface-initiated NMP of styrene yielded the bioconjugate, subsequently cleaved from the resin.
In the present study, we report a simple approach for the design by NMP of a PEGylation system based on α-functional comb-shaped polymethacrylates with PEG side chains, the efficiency of which was further demonstrated by conjugation to a neuroprotective peptide and lysozyme as a model protein (Scheme 1). The synthetic strategy relies on the polymerization of MePEGMA initiated by SG1-based alkoxyamines bearing a N-hydroxysuccinimidyl (NHS) moiety. This was achieved in the presence of a very small amount of: (i) acrylonitrile (AN) to ensure a good control38 due to the well-known inability of SG1 to control the homopolymerization of methacrylic esters39–43 and (ii) rhodamine B methacrylate (RMA)44 to confer fluorescent properties to the resulting materials. Importantly, this copolymerization system has been selected due to the lack of cytotoxicity of the corresponding copolymers at very high doses,34 which is relevant when biomedical applications are foreseen.
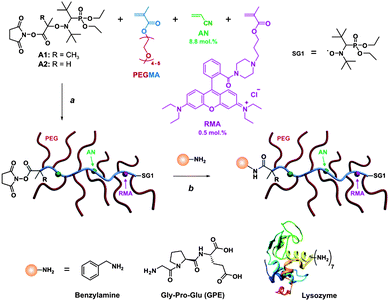 | ||
| Scheme 1 Strategy to design α-functional, fluorescent comb-shaped polymethacrylates with PEG side chains for bioconjugation. Reagent and conditions: (a) xylene, 80–95 °C; (b) DMSO/TEA (95/5; v/v) or PBS (pH 7.0), 7–24 h, 25 °C. | ||
Experimental part
Materials
Poly(ethylene glycol) methyl ether methacrylate (MePEGMA, Mn = 300 g mol−1), acrylonitrile (AN, 99+%), benzylamine (>98%), triethylamine (TEA), diethyl phosphite (98%), anhydrous dimethyl sulfoxide (DMSO, 99%), N-hydroxysuccinimide (NHS, 98%), Gly-Pro-Glu tripeptide (GPE, >98%), lysozyme from chicken egg white (molecular weight ≈ 14.3 kDa), phosphoric acid (>99%) and hexadimethrine bromide (polybrene, >95%) were purchased from Aldrich and used as received. N,N′-Dicyclohexylcarbodiimide (DCC, >99%) was obtained from Fluka. The MAMA-SG1 alkoxyamine (2-methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy] propionic acid alkoxyamine, BlocBuilder MA, 99%) and the N-tert-butyl-N-(1-diethyl phosphono-2,2-dimethylpropyl) nitroxide (SG1, 85%) were kindly supplied by Arkema. The 2-(N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy)propionic acid (AMA-SG1) was synthesized using the ATRA method as described elsewhere.452-Methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy]-N-propionyloxysuccinimide (alkoxyamine A1)
The synthesis of alkoxyamine A1 was adapted from literature.46 A 100 mL round bottom flask sealed with a rubber septum and containing a solution of NHS (2.9 g, 25 mmol) in THF (40 mL) was placed in an ice water bath at 0 °C. MAMA-SG1 alkoxyamine (8.0 g, 21 mmol) was introduced and the reaction mixture was degassed by nitrogen bubbling for 15 min. A degassed solution of DCC (4.8 g, 23 mmol) in THF (8 mL) was then added. After stirring at 0 °C for 90 min under nitrogen atmosphere, the white precipitate of N,N′-dicyclohexylurea (DCU) was filtered off and the THF was partially removed under reduced pressure. The resulting filtrate was placed at −18 °C for 3 h allowing residual DCU to precipitate and to be subsequently filtered off (this was repeated 3 times to ensure complete removal of DCU). The solution was then concentrated under reduced pressure and a precipitation was performed in pentane. The obtained solid was washed with deionized water to remove free NHS and further dried under high vacuum at ambient temperature. Alkoxyamine A1 was then recovered as a white powder. Yield: 45% (4.36 g, 9.44 mmol). 1H NMR (300 MHz, CDCl3, δ, ppm): 1.16 (s, 9H, (CH3)3CHP), 1.21 (s, 9H, (CH3)3CN), 1.24–1.34 (q, 6H, OCH2CH3), 1.82 (s, 3H, NOCHCH3), 1.89 (s, 3H, NOCHCH3), 2.83 (s, 4H, CH2CH2), 3.31 (d, J = 26 Hz, 1H, NCHP), 3.91–4.40 (m, 4H, OCH2CH3).2-[N-tert-Butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)aminoxy]-N-propionyloxysuccinimide (alkoxyamine A2)
Alkoxyamine A2 was synthesized as follows: a 100 mL round bottom flask sealed with a rubber septum and containing a solution of NHS (1.51 g, 13 mmol) in THF (19 mL) was placed in an ice water bath at 0 °C. AMA-SG1 alkoxyamine (4.17 g, 11 mmol) was introduced and the reaction mixture was degassed by nitrogen bubbling for 15 min. A degassed solution of DCC (2.52 g, 12 mmol) in THF (8 mL) was then added. After stirring at 0 °C for 90 min under nitrogen atmosphere, the white precipitate of DCU was filtered off and the THF was partially removed under reduced pressure. The resulting filtrate was placed at −18 °C for 3 h allowing residual DCU to precipitate and to be subsequently filtered off (this was repeated 3 times to ensure complete removal of DCU). The solution was then concentrated under reduced pressure and a precipitation was performed in pentane. The obtained solid was washed with deionized water to remove free NHS and further dried under high vacuum at ambient temperature. Alkoxyamine A2 was then recovered as a white powder. Yield: 62% (3.15 g, 7.02 mmol). MS (+ESI) calculated for C20H37N2O8P: 464.23; found: 487.21 ([M + Na]+). Mixture of diastereoisomers I/II; 63/37 by 31P NMR. Diastereoisomer I (R,R) or (S,S): 1H NMR (300 MHz, CDCl3, δ, ppm): 1.16 (s, 9H), 1.18 (s, 9H), 1.24–1.34 (m, 6H), 1.65 (s, 2H), 1.70 and 1.73 (d, J = 7 Hz, 3H), 2.83 (s, 4H), 3.26 and 3.35 (d, J = 25.5 Hz, 1H), 3.90–4.35 (m, 4H), 4.93 (q, J = 6.8 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 16.3 (d, J = 6.6 Hz, OCH2CH3), 16.6 (d, J = 5.0 Hz, OCH2CH3), 20.0 (s, CH3CHC(O)), 25.7 (s, (CH2)2C(O)N), 28.1 (s, (CH3)3CN), 29.8 (d, J = 4.8 Hz, (CH3)3CCHP), 35.7 (d, J = 4.8 Hz, (CH3)3CCHP), 59.1 (d, J = 7.3 Hz, OCH2CH3), 61.9 (d, J = 5.9 Hz, OCH2CH3), 62.2 (s, (CH3)3CN), 69.6 (d, J = 139.2 Hz, (CH3)3CCHN), 80.4 (s, CH3CHC(O)), 169.4 (s, NOC(O)), 168.9 (s, C(O)NC(O)). 31P NMR (121.44 MHz, CDCl3, δ, ppm): 23.88 (s, 63%). Diastereoisomer II (R,S) or (S,R): 1H NMR (300 MHz, CDCl3, δ, ppm): 1.15 (s, 9H), 1.18 (s, 9H), 1.24–1.34 (m, 6H), 1.65 (s, 2H), 1.69 and 1.72 (d, J = 7 Hz, 3H), 2.80 (s, 4H), 3.33 and 3.42 (d, J = 25.5 Hz, 1H), 3.90–4.35 (m, 4H), 5.05 (q, J = 6.8 Hz, 1H). 13C NMR (75 MHz, CDCl3, δ, ppm): 16.3 (d, J = 6.6 Hz, OCH2CH3), 16.6 (d, J = 5.0 Hz, OCH2CH3), 18.3 (s, CH3CHC(O)), 25.7 (s, (CH2)2C(O)N), 28.1 (s, (CH3)3CN), 30.4 (d, J = 5.5 Hz, (CH3)3CCHP), 35.4 (d, J = 4.6 Hz, (CH3)3CCHP), 59.5 (d, J = 7.1 Hz, OCH2CH3), 62.3 (d, J = 6.5 Hz, OCH2CH3), 61.9 (s, (CH3)3CN), 69.3 (d, J = 140.4 Hz, (CH3)3CCHN), 80.4 (s, CH3CHC(O)), 167.9 (s, NOC(O)), 168.8 (s, C(O)NC(O)). 31P NMR (121.44 MHz, CDCl3, δ, ppm): 23.14 (s, 37%).Synthesis of P(MePEGMA-co-AN)–SG1 and P(MePEGMA-co-AN-co-RMA)–SG1 copolymers
A typical synthesis (copolymer P1) was as follows: in a 250 mL three-neck round-bottom flask fitted with a reflux condenser, a magnetic stirrer, a nitrogen inlet and a thermometer, a mixture of MePEGMA (21.11 g, 0.070 mol), AN (0.360 g, 6.79 × 10−3 mol), free SG1 (38.3 mg, 1.53 × 10−4 mol) and xylene (49.5 g) was deoxygenated by nitrogen bubbling for 20 min at room temperature. The alkoxyamine initiator A1 (0.59 g, 1.23 × 10−3 mol) was added to the reaction medium. Nitrogen bubbling was carried out for 10 min and the mixture was heated at 80 °C in a preheated oil bath. The time zero of the reaction was triggered when temperature in the reactor reached 75 °C. Samples were periodically withdrawn to follow the MePEGMA conversion by 1H NMR spectroscopy. Copolymers were then precipitated twice in cold diethyl ether and dried under high vacuum overnight. The same protocol was followed for the synthesis of copolymers P2–P4 except that polymerization temperature was 90 °C, 0.5 mol% of RMA was added in the monomer feed and the solvent was xylene/DMSO (93/7; v/v) to ensure complete dissolution of RMA (Table 1). In this case, an extra dialysis step under slightly acidic conditions (pH 4, MWCO = 1000 g mol−1) was necessary to remove unreacted RMA.Conjugation to benzylamine
A solution of copolymer P1 (500 mg, 6.67 × 10−6 mol) in DMSO-d6 (0.7 mL) and TEA (35 µL) was transferred into a 5 mm NMR tube and subsequently analyzed by 1H NMR spectroscopy (time zero). Then, benzylamine (4.9 µL, 4.5 × 10−5 mol) was added in the tube which triggered the beginning of the reaction within the cavity of the spectrometer monitored by online 1H NMR spectroscopy. Acquisition was performed after different periods of time. The crude reaction medium was then dialyzed against deionized water for 5 days (MWCO = 1000 g mol−1) and dried under high vacuum overnight.Conjugation to Gly-Pro-Glu (GPE) tripeptide
In a 10 mL round bottom flask, copolymer P1 (500 mg, 6.67 × 10−6 mol) was dissolved in a solution of D2O (1.8 mL), DMSO-d6 (0.6 mL) and TEA (30 µL). GPE tripeptide (16.5 mg, 5.48 × 10−5 mol) was added and the reaction was allowed to proceed overnight under gentle stirring. The crude reaction medium was then dialyzed against deionized water for 5 days (MWCO = 1000 g mol−1) and dried under high vacuum until constant weight.Conjugation to lysozyme
Analytical techniques
The preparation of the polybrene-coated capillary was as follows: a fused-silica capillary was rinsed with water for 3 min followed by 5 min with 1 M NaOH and 3 min with water. Then coating was performed by flushing the capillary with 0.2 wt% polybrene (aqueous solution) for 5 min and then water for 2 min. The coating procedure was performed before each injection, and was followed by a 4 min rinse with the running buffer to equilibrate the capillary before injection. After each run, the capillary was rinsed by 0.1 M NaOH for 3 min and water for 2 min. Buffers and standard solutions were prepared with deionized water and were filtered through 0.2 µm pore size membrane filters. Sodium phosphate buffer (50 mM, pH 3) prepared in water was used as the separation buffer. The buffer pH was measured with a WTW series pH meter from Info Lab. Bioconjugates were dissolved in deionized water and injected at 10–30 mg mL−1.
Results and discussion
Synthesis and reactivity of α-functional polymers
N-succinimidyl alkoxyamines based on the SG1 nitroxide (A1 and A2, Scheme 1) were readily obtained in good yields from the corresponding carboxylic acid counterparts by a DCC-assisted coupling reaction. The steric hindrance around the activated ester group was modulated in order to tune the reactivity of this compound towards nucleophiles. In order to maximize the conjugation yields, a MePEGMA with relatively short PEG side chains (DPn = 4–5) has been chosen. A small library of α-NHS functional copolymers was then prepared by varying the nature of the alkoxyamine and the final molar mass (Table 1). Solution polymerizations proceeded in a controlled fashion and yielded well-defined α-NHS copolymers as attested by the linear evolution of molar masses with monomer conversion and by the low PDIs (Fig. S1†). 1H NMR spectroscopy showed an excellent correlation with the expected structure of the copolymers (Fig. 1). It gave Mn = 9600 and 4800 g mol−1 for copolymers P1 and P2, respectively, close to SEC values. The emission and excitation wavelengths were also in good agreement with the spectral properties of rhodamine B–tertiary amide derivatives.44,47,48 For instance, λex. = 568 nm and λem. = 582 nm for copolymer P2 at 1 mg mL−1 in water.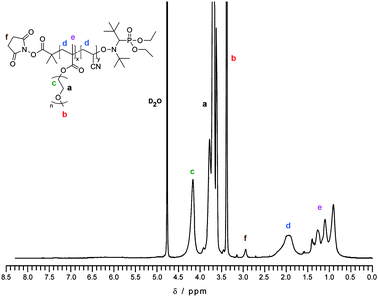 | ||
| Fig. 1 1H NMR spectrum in D2O of purified NHS–poly(MePEGMA-co-AN) copolymer (P1). | ||
The reactivity of these functional materials towards nucleophiles was estimated by monitoring the hydrolytic stability of the N-succinimidyl ester function by online 1H NMR spectroscopy in 100 mM PBS at different pHs (Fig. 2). As expected, the higher the pH, the faster the cleavage of the NHS group. For copolymer P1, hydrolysis was extremely slow at pH 6 but it gave t1/2 = 40 h and 6.5 h at pH 7 and 8, respectively. This highlighted a substantially higher reactivity of P1 compared to similar polymers synthesized by ATRP having an identical activated ester structure where t1/2 = 55 h in 100 mM PBS at pH 8.25
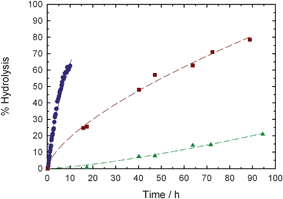 | ||
| Fig. 2 Kinetic plots for the hydrolysis of NHS–poly(MePEGMA-co-AN) copolymer (P1) in 100 mM PBS solutions at pH 6 (▲), pH 7 (■) or pH 8 (●). | ||
Copolymer P1 was then assessed for its reactivity towards benzylamine as a model amine. Conjugation yielded a quantitative coupling in ∼200 min as witnessed by the appearance of amide and methylene protons together with the conservation of the aromatic protons upon dialysis (Fig. 3 and S2†).
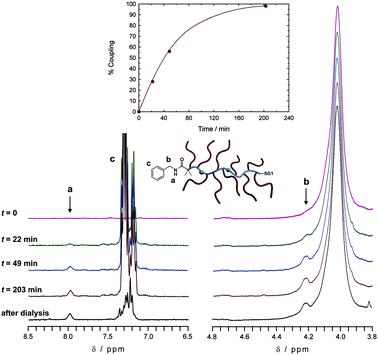 | ||
| Fig. 3 Selected 300 MHz 1H NMR spectra (left: 6.5–8.5 ppm; right: 3.8–4.8 ppm) in DMSO-d6 of the conjugation reaction between NHS–poly(MePEGMA-co-AN) copolymer (P1) and benzylamine in DMSO/TEA (95/5; v/v) mixture at various reaction times and after dialysis. Inset: kinetic plots of the conjugation following the appearance of the amide peak (a) at 7.98 ppm. | ||
PEGylation of a neuroprotective tripeptide
Insulin-like growth factor-1 (IGF-1) is a neurotrophic factor that can prevent neurons from ischemic brain injury. Protease-mediated metabolism of IGF-1 leads to the formation of the endogenous N-terminal tripeptide Gly-Pro-Glu (GPE, Scheme 1) which: (i) is able to protect different types of neurons from diverse induced injuries and (ii) exhibits neuroprotective features in different animal models of neurodegenerative diseases.49–51 Therefore, we were interested in coupling the GPE tripeptide to α-NHS functional copolymers in order to demonstrate efficient PEGylation to a biologically active peptide. Bioconjugation with copolymer P1 was performed overnight in D2O/DMSO-d6 (3/1; v/v) in the presence of triethylamine (TEA) with an eight-fold excess of GPE and followed by dialysis (Fig. 4).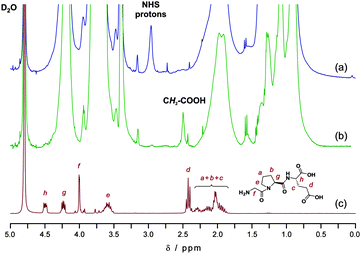 | ||
| Fig. 4 Selected 300 MHz 1H NMR spectra (0–5.0 ppm) in D2O of NHS–poly(MePEGMA-co-AN) copolymer P1 (a), GPE-poly(MePEGMA-co-AN) bioconjugate (b) and Gly-Pro-Glu (GPE) peptide (c). | ||
It yielded nearly complete coupling as attested by the quantitative appearance of the triplet assigned to the methylene in α-position of the primary carboxylic acid group (peak d), together with disappearance of NHS protons. Signals of protonsh and g in the 4.2–4.5 ppm region were also visible, even though partially overlaid with the α-ester proton signal of the MePEGMA units. Due to the ability of SG1-terminated poly(MePEGMA-co-AN) to be readily chain-extended,34 it could be therefore envisioned to synthesize GPE-decorated, PEGylated micelles or nanoparticles able to cross the blood-brain-barrier,52 together with neuroprotective features, from self-assembly of the corresponding amphiphilic block copolymer. This would also allow the very short plasma half-life of free GPE53 to be extended due to its coupling to such stealth nanocarriers.
PEGylation to lysozyme as a model protein
Lysozyme was chosen as a model substrate for polymer–protein conjugation reactions with copolymers P2–P4 (Table 1) in anhydrous DMSO/TEA mixture (95/5; v/v). SDS-PAGE was used to monitor the conjugation process. The influence of various experimental parameters over the bioconjugation was studied, such as the structure of the activated ester extremity (derived from alkoxyamine A1 or A2), the molar mass of the copolymer, the reaction duration and the copolymer-to-protein molar ratio (Fig. 5).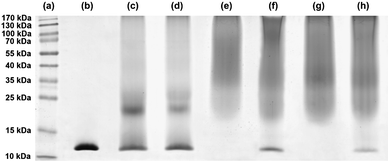 | ||
| Fig. 5 SDS-PAGE of protein standards (a), native lysozyme (b) and for the conjugation of lysozyme with NHS–poly(MePEGMA-co-AN-co-RMA) copolymer in DMSO/TEA as a function of the nature of the α-NHS copolymer, the copolymer-to-lysozyme molar ratio and the conjugation duration: P2, 30 equiv., 7 h (c); P2, 30 equiv., 24 h (d); P3, 50 equiv., 7 h (e); P3, 20 equiv, 7 h (f); P4, 50 equiv., 7 h (g) and P4, 20 equiv., 7 h (h). | ||
SDS-PAGE analysis of conjugates obtained with copolymer P2 after 7 or 24 h of reaction showed the presence of bands at higher molecular weight than with native lysozyme as well as residual lysozyme (Fig. 5, lines c and d). The molecular weight of these bands were found to be independent of the reaction duration and seemed to correspond to 2–3 polymer chains per lysozyme (lysozyme has 6 lysine residues in addition to the terminal amine). Whereas no protein–polymer bioconjugates were detected with polymers from ATRP of MePEGMA (Mn = 2.7–6.4 kg mol−1) exhibiting identical activated ester structure, a marked increase of reactivity towards lysine residues was witnessed here using a MePEGMA derivative with shorter PEG side chains, in good agreement with the higher hydrolysis rate previously observed.
By using alkoxyamine A2, complete conjugations to lysozyme were obtained from copolymers P3 and P4 after only 7 h of reaction, as assessed by the absence of the lysozyme starting material and the typical smears towards higher molecular weights (Fig. 5, lines e and g). The excess of copolymer with respect to lysozyme was then reduced (i.e., ∼3 equiv. of NHS moieties with respect to primary amine groups) which allowed the influence of the molar mass of the copolymer over the conjugation yield to be detected. As expected from a steric hindrance point of view, the lower the molar mass of the copolymer, the more efficient the bioconjugation (Fig. 5, lines f and h). Indeed, a 6.1 kg mol−1copolymer yielded an almost quantitative conjugation to lysozyme, hence confirming the high reactivity of these materials.
The bioconjugation reaction to lysozyme was also performed in water (PBS, pH 7) in order to demonstrate the flexibility of our PEGylation system. SDS-PAGE confirmed the successful formation of PEGylated lysozyme with no remaining native protein (Fig. 6, line b). According to protein standards, the bioconjugate should exhibit 1–4 polymer chains per lysozyme and is more homogeneous than the one obtained in organic medium. The efficient PEGylation was also shown by CE, which was found to be a suitable method for the analysis of PEGylated proteins by linear PEGs,54–56 although it has never been used with polymers from CLRP. The separation mechanism of CE is based on differences in the charge-to-mass ratio of the analytes. Therefore, attachment of PEG chains to a protein is expected to decrease its electrophoretic mobility and thus give a clear indication about the outcome of the PEGylation.
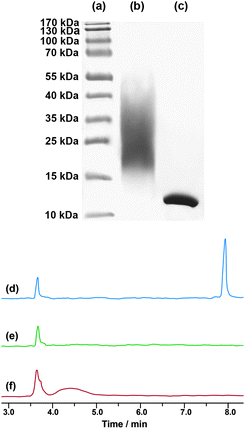 | ||
| Fig. 6 Left: SDS-PAGE of protein standards (a), for the conjugation of lysozyme with NHS–poly(MePEGMA-co-AN-co-RMA) copolymer P4 (50 equiv., 24 h) in PBS (pH 7.0, 50 mM) (b) and native lysozyme (c); right: electropherograms of native lysozyme (d), NHS–poly(MePEGMA-co-AN-co-RMA) copolymer P4 (e) and for the conjugation of lysozyme with NHS–poly(MePEGMA-co-AN-co-RMA) copolymer P4 (50 equiv., 24 h) in PBS (pH 7.0, 50 mM) (f). | ||
The charge-reversed, polybrene-coated capillary we used prevents adsorption of lysozyme which exhibits a reproducible migration time of 7.9 min, whereas the copolymer P4 is not detectable and migrates with the electroosmotic flow at 3.6 min (Fig. 6d and e). In agreement with SDS-PAGE, CE analysis of the bioconjugate revealed no remaining unmodified lysozyme. In addition, PEGylated lysozyme exhibited: (i) a reduced mobility (4.4 min) in accordance with a decrease of its charge-to-mass ratio due to PEG ligation and (ii) a relatively broad shape as a result of a multi-site attachment to lysozyme (Fig. 6f).
Bioconjugates were successfully tagged by RMA and exhibited intense fluorescence in agreement with the spectral properties of RMA. A linear evolution of the fluorescence intensity as a function of the concentration in water across a wide range of concentrations was also noticed (Fig. 7).
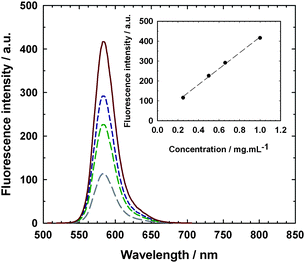 | ||
| Fig. 7 Emission spectra (λex. = 568 nm and λem. = 582 nm) of lysozyme–poly(MePEGMA-co-AN-co-RMA) fluorescent bioconjugate from P4 at various concentrations in water: 0.25 mg mL−1 (long dashed line), 0.50 mg mL−1 (medium dashed line), 0.66 mg mL−1 (short dashed line) and 1.0 mg mL−1 (solid line). Inset: evolution of the fluorescence intensity with the bioconjugate concentration. | ||
Conclusion
NMP has been successfully employed for the first time in the PEGylation landscape via the synthesis of polymer–protein/peptide bioconjugates. Well-defined and fluorescent N-succinimidyl ester-functionalized poly(MePEGMA)-based copolymers were easily synthesized under SG1-control without any purification beyond a simple precipitation. They exhibited tunable reactivities towards nucleophiles depending on the nature of the alkoxyamine used. Copolymers derived from the commercially available SG1-based alkoxyamine led to quantitative coupling with small molecules and a neuroprotective peptide whereas partial conjugation was obtained with lysozyme, used here as a model protein. Complete conjugation to the latter was obtained from similar copolymers exhibiting a less sterically hindered NHS extremity (due to the appropriate alkoxyamine structure). These α-functional copolymers are considered as an efficient platform for PEGylation purposes and represent a suitable alternative to previous systems synthesized by other CLRP techniques. More generally, this study takes advantage of the environmentally friendly feature of NMP and paves the way to innocuous, functional (co)polymers to be used in biomedical applications.Acknowledgements
The authors thank Arkema for kindly providing the SG1 nitroxide and the BlocBuilder MA alkoxyamine as well as Dr Joël Belleney (UPMC) for the phosphorous NMR analysis. CNRS and French ministry of research are warmly acknowledged for funding.References
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405 CrossRef CAS.
- F. M. Veronese and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 453 CrossRef CAS.
- A. Abuchowski, T. Van Es, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083 CrossRef CAS.
- L. Tao, J. T. Xu, D. Cell and T. P. Davis, Macromolecules, 2010, 43, 3721 CrossRef CAS.
- D. M. Haddleton, S. Perrier and S. A. F. Bon, Macromolecules, 2000, 33, 8246 CrossRef CAS.
- J. F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893 CrossRef CAS.
- J.-F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS.
- J. Nicolas, P. Couvreur and B. Charleux, Macromolecules, 2008, 41, 3758 CrossRef CAS.
- J. Nicolas, E. Khoshdel and D. M. Haddleton, Chem. Commun., 2007, 1722 RSC.
- S. Perrier, S. P. Armes, X. S. Wang, F. Malet and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1696 CrossRef CAS.
- L. Zhang, T. L. U. Nguyen, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Biomacromolecules, 2007, 8, 2890 CrossRef CAS.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004, 2026 RSC.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220 CrossRef CAS.
- S. M. Ryan, X. X. Wang, G. Mantovani, C. T. Sayers, D. M. Haddleton and D. J. Brayden, J. Controlled Release, 2009, 135, 51 CrossRef CAS.
- C. T. Sayers, G. Mantovani, S. M. Ryan, R. K. Randev, O. Keiper, O. I. Leszczyszyn, C. Blindauer, D. J. Brayden and D. M. Haddleton, Soft Matter, 2009, 5, 3038 RSC.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772 CrossRef CAS.
- L. Tao, J. Q. Liu, J. T. Xu and T. P. Davis, Org. Biomol. Chem., 2009, 7, 3481 RSC.
- Z. Zarafshani, T. Obata and J.-F. Lutz, Biomacromolecules, 2010, 11, 2130 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291 CrossRef CAS.
- M. L. Becker, J. Liu and K. L. Wooley, Chem. Commun., 2003, 180 RSC.
- M. L. Becker, J. Liu and K. L. Wooley, Biomacromolecules, 2005, 6, 220 CrossRef CAS.
- T. Trimaille, K. Mabrouk, V. Monnier, L. Charles, D. Bertin and D. Gigmes, Macromolecules, 2010, 43, 4864 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485 CrossRef CAS.
- B. Lessard, E. J. Y. Ling, M. S. T. Morin and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1033 CrossRef CAS.
- A. C. Greene and R. B. Grubbs, Macromolecules, 2010, 43, 10320 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697 RSC.
- D. Bertin, D. Gigmes, C. Le Mercier, S. R. A. Marque and P. Tordo, J. Org. Chem., 2004, 69, 4925 CrossRef CAS.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639 CrossRef CAS.
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245 CrossRef CAS.
- D. Brambilla, J. Nicolas, B. Le Droumaguet, K. Andrieux, V. Marsaud, P.-O. Couraud and P. Couvreur, Chem. Commun., 2010, 46, 2602 RSC.
- V. R. Sara, C. Carlsson-Skwirut, T. Bergman, H. Jörnvall, P. J. Roberts, M. Crawford, L. N. Håkansson, I. Civalero and A. Nordberg, Biochem. Biophys. Res. Commun., 1989, 165, 766 CrossRef CAS.
- J. Guan, R. Krishnamurthi, H. J. Waldvogel, R. L. M. Faull, R. Clark and P. Gluckman, Brain Res., 2000, 859, 286 CrossRef CAS.
- T. Alexi, P. E. Hughes, W. M. C. van Roon-Mom, R. L. M. Faull, C. E. Williams, R. G. Clark and P. D. Gluckman, Exp. Neurol., 1999, 159, 84 CrossRef CAS.
- P. Calvo, B. Gouritin, H. Chacun, D. Desmaele, J. D'Angelo, J. P. Noel, D. Georgin, E. Fattal, J. P. Andreux and P. Couvreur, Pharm. Res., 2001, 18, 1157 CrossRef CAS.
- D. C. Batchelor, H. Lin, J. Y. Wen, C. Keven, P. L. V. Zijl, B. H. Breier, P. D. Gluckman and G. B. Thomas, Anal. Biochem., 2003, 323, 156 CrossRef CAS.
- J. Bullock, S. Chowdhury and D. Johnston, Anal. Chem., 1996, 68, 3258 CrossRef CAS.
- W. Li, Y. Zhong, B. Lin and Z. Su, J. Chromatogr., A, 2001, 905, 299 CrossRef CAS.
- R. L. Cunico, V. Gruhn, L. Kresin, D. E. Nitecki and J. E. Wiktorowicz, J. Chromatogr., A, 1991, 559, 467 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical characterizations. See DOI: 10.1039/c1py00028d |
| This journal is © The Royal Society of Chemistry 2011 |