Architecture effects on L-selectin shedding induced by polypeptide-based multivalent ligands†
Shuang
Liu
a and
Kristi
Kiick
*ab
aDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, Delaware 19716, USA. E-mail: lius@udel.edu
bDelaware Biotechnology Institute, 15 Innovation Way, Newark, DE 19711, USA. E-mail: kiick@udel.edu; Fax: +1-302-831-4545; Tel: +1-302-831-0201
First published on 18th April 2011
Abstract
Multivalent interactions between selectins and their ligands play key roles in mediating the rolling and tethering of leukocytes in the early steps of the inflammatory response, as well as in lymphocyte circulation. L-selectin shedding, which is the proteolytic cleavage of L-selectin, can be induced by L-selectin clustering through the binding of multivalent ligands to multiple L-selectin molecules, and it has been shown to regulate leukocyte rolling and subsequent integrin activation for firm adhesion. In this paper, we report the production of homogenous glycopolypeptides modified with a 3,6-disulfo-galactopyranoside equipped with a caproyl linker. The saccharide residue was chemically attached to various polypeptide backbones of differing architectures; the composition and purity of the sulfated glycopolypeptides was confirmed via1H-NMR spectroscopy, amino acid analysis (AAA), and electrophoretic analysis. The retention of the conformation of the polypeptide backbone was confirmed via circular dichroic spectroscopy. The shedding of L-selectin from the surface of Jurkat cells induced by these sulfated glycopolypeptides, determined via ELISA-based methods, varied based on differences in the architectures of the polypeptide scaffolds, suggesting opportunities for these strategies in probing cell-surface receptor arrays and directing cell signaling events.
Introduction
Multivalent interactions abound in the immune system, directing complex biological processes that include the detection, identification and clearance of pathogens and malfunctioning cells. Leukocyte recruitment is the first step of the inflammatory response of the immune system, in which leukocytes are first tethered to the surface of the endothelium, followed by rolling, and the firm adhesion required for transmigration to the affected tissue.1 This process is mediated by various interactions between adhesion molecules and their ligands on the surface of both endothelial cells and leukocytes, such as selectins and integrins.2 Of the selectins, L-selectin is a glycoprotein belonging to the family that includes the E- (endothelium), P- (platelet) and L- (leukocyte) selectins named according to the cells in which the gene for the molecule was identified.3L-selectin is expressed on the surface of leukocytes, and its ligands are expressed on the surface of endothelial cells, while E- and P-selectins are expressed on the surface of endothelial cells and platelets, and their receptors are expressed on the surface of monocytes, neutrophils and T lymphocytes.3L-selectin binds to its ligands with C-type lectin domain on the N-terminal through protein–carbohydrate interactions. Most of the L-selectin ligands are carbohydrate-based glycoproteins and glycolipids, expressed on vascular endothelium and post-modified through sialyation, fucosylation, sulfation and/or phosphorylation;4,5 indeed, the oligosaccharides sialyl-Lewisx (sLex), sialyl-Lewisa (sLea) and their sulfated equivalents are widely found in the O-glycans attached to multiple serine and threonine residues in these L-selectin ligands.4,6 The binding between L-selectin and its ligands plays important roles in a variety of biological events and diseases, such as leukocyte rolling and adhesion, lymphocyte circulation, signaling activation and tumor metastasis.2,3 Uniquely, L-selectin undergoes a shedding event, in which the glycoprotein is rapidly cleaved proteolytically (by a class of enzymes called sheddases) and soluble L-selectin (sL-selectin) is released into plasma.7L-selectin shedding has been shown to regulate neutrophil rolling by preventing the “jerkiness” (variable velocity) of the rolling.8,9L-selectin clustering by crosslinking antibodies has also been shown to enhance β2 integrin activation for the firm adhesion of leukocytes, and L-selectin shedding can limit this activation and thus also limit inflammation.7 The shed sL-selectin also regulates the attachment of lymphocytes to the endothelium.3,7,10 Therefore, manipulation of L-selectin shedding has significant potential in the development of anti-inflammatory therapeutics and in the attenuation of inflammatory responses to biomaterials.
L-selectin shedding can be induced by various cytokines and chemokines, such as phorbol esters (e.g. PMA), interleukin 8 (IL-8) and some nonsteroidal anti-inflammatory drugs (NSAIDs), as well as cross-linked L-selectin antibodies and L-selectin multivalent ligands.7,11,12 Studies have shown that more than one mechanism exists for L-selectin shedding, although exact mechanisms remain somewhat obscure.7 Certain proteases have shown to be able to cleave L-selectin, among them A Disintegrin and Metalloprotease 17 (ADAM 17) is the best known L-selectin sheddase, which also cleaves the precursors of other proteins, such as tumor necrosis factor-α (TNF-α). It has also been shown that L-selectin sheddase operates to cleave L-selectin in the cis but not the trans configuration.7,13L-selectin shedding induced by the crosslinking of L-selectin antibodies and multivalent ligands is suggested to occur through L-selectin clustering.7,14 Synthetic multivalent ligands have been designed to study L-selectin binding and inhibition, L-selectin clustering formation and subsequently induced signal transduction and shedding.11,15 Polymeric scaffolds have been useful in such investigations due to their sizes and lengths; the rapid development of living polymerization techniques has enabled the production of polymers with narrow molecular weight distribution and controlled architecture16 and has expanded the use of synthetic polymeric scaffolds in investigations of multivalent binding phenomena.17–19 Various saccharides and oligosaccharides have been attached to polymeric backbones for the study of L-selectin binding, and the architecture of these multivalent ligands, including backbone chain length and average ligand densities, has been shown to elicit differences in L-selectin binding and inhibition.15,20,21
However, the architecture of chemically synthesized polymeric multivalent ligands can only be controlled by varying the degree of polymerization or average degree of modification; the lack of control over placement of individual residues and chain conformation has limited the use of polymeric scaffolds in precise manipulation of cell-surface receptor landscapes. Recombinant polypeptide backbones offer multiple advantages for these kinds of studies. The placement of chemically reactive amino acids can be specified with great precision along the chain, permitting attachment of multiple ligands and exploration of the role of ligand placement. A variety of ligand/polypeptide combinations are possible, and versatility is further expanded by the suite of non-natural amino acids that can be incorporated into recombinant proteins and polypeptides.22–25 Also, the chain conformation can be controlled by sequence design, based on the conformation propensities of each amino acid residue. They thus serve as intriguing emerging candidates for engaging receptors on the cell surface.26
In this paper, the design and synthesis of polypeptide-based, sulfated glycopolymeric multivalent ligands are described. These multivalent scaffolds were prepared via a combination of protein engineering and chemical modification methods. Specifically, an N-linked 3,6-disulfo-galactopyranoside was modified via the addition of a caproyl linker and was chemically conjugated to alanine-rich polypeptide-based scaffolds of different conformations and functionality. These polypeptide-based scaffolds were suggested in previous investigations to offer advantages in the accessible presentation of ligands, the reduction of unfavorable chain entropic penalties upon binding, the control of backbone conformation, and the spacing of ligands.27–30 The composition and purity of the sulfated glycopolypeptide scaffolds were determined viaSDS-PAGE, NMR and amino acid analysis. The effects of glycopolymer architectural variables on induced L-selectin shedding were studied with a select set of glycopolypeptides and evaluated via ELISA-based assays, and indicated the opportunities to manipulate L-selectin-based signaling with these polypeptide-based scaffolds.
Experimental
Materials
N-ε-Fmoc-aminohexanoic acid (Fmoc-Ahx-OH) and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from EMD Biosciences Inc. (San Diego, CA). Dibutyltin oxide (Bu2SnO), β-D-galactosylamine, phorbol 12-myristate 13-acetate (PMA), piperidine, triethylamine, dimethyl sulfoxide (DMSO), diisopropylethylamine (DIPEA), anhydrous dimethylformamide (DMF) and trifluoroacetic acid (TFA) were obtained from Sigma-Aldrich (St Louis, MO). A poly(acrylic acid)–sialyl Lewisabiotin conjugate (sLea–PAA–biotin) was purchased from GlycoTech (Gaithersburg, MD). Deuterated solvents for NMR are from Cambridge Isotope Lab. (Andover, MA). L-selectin ELISA kits were purchased from R&D Systems, Inc. (Minneapolis, MN). 3,3′,5,5′-Tetramethylbenzidine (TMB) was purchased from Pierce (Rockford, IL). Jurkat T-lymphoma cells and RPMI-1640 media were purchased from ATCC (Manassas, VA). Fetal bovine serum (FBS), penicillin and streptomycin were purchased from Invitrogen (Rockville, MD). Ni-NTA resin was obtained from Qiagen (Valencia, CA). Isopropyl β-D-thiogalactopyranoside (IPTG), tryptone, yeast extract, ampicillin, chloramphenicol, kanamycin, acrylamide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bisacrylamide (29:1), pyridine sulfur trioxide complex (Py·SO3), sodium thiosulfate (Na2S2O3), silver nitrate (AgNO3), sodium carbonate (NaCO3) and C96 Maxisorp microtiter plates and all other chemicals were purchased from Fisher Scientific (Pittsburgh, PA).
:bisacrylamide (29:1), pyridine sulfur trioxide complex (Py·SO3), sodium thiosulfate (Na2S2O3), silver nitrate (AgNO3), sodium carbonate (NaCO3) and C96 Maxisorp microtiter plates and all other chemicals were purchased from Fisher Scientific (Pittsburgh, PA).
Synthesis of N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine
Synthesis of N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine is based on previously reported methods.11,29,31 A solution of β-D-galactosylamine (1) (1.4 mmol, 250 mg in 5 mL DMF) was added to 2 mL of Fmoc-Ahx-OH (2) solution (1.4 mmol, 500 mg), and pre-activated with HBTU (1.4 mmol, 550 mg) and DIPEA (7 mmol, 1.3 mL) for 5 min. The reaction was stirred at room temperature overnight and then was precipitated into cold diethyl ether. The precipitate was then dissolved in 35% MeOH in H2O and purified viaHPLC employing a C18 column with a H2O/MeOH mobile phase, with approximately 60% yield. The obtained product N-(ε-Fmoc-aminocaproyl)-β-D-galactosylamine (3) (160 mg, 0.312 mmol) was dissolved in 2 mL anhydrous MeOH and then Bu2SnO (80 mg, 0.312 mmol) was added and the mixture was heated to reflux. The solution became clear when the stannylation was complete (approximately 30 min). MeOH was removed under vacuum and the product was dried under vacuum overnight. Anhydrous DMF and pyridine sulfur trioxide (100 mg, 0.624 mmol) were added to the reaction flask and stirred at room temperature. The synthesis of monosulfated and disulfated products was monitored, and the products separated, via anionic exchange chromatography (on a DEAE column with a mobile phase of 50 mM Tris buffer with 1 M NaCl pH 8.0 for monitoring and 1 M triethylammonium bicarbonate (TEAB) buffer at pH 8.0 for separation, respectively). The fractions containing N-(ε-Fmoc-aminocaproyl)-3,6-disulfo-β-D-galactosylamine (4) were collected and lyophilized and characterized viaelectrospray ionization mass spectrometry (ESI-MS) and 1H NMR. The Fmoc protecting group was deprotected in 20% piperidine in DMF and the disulfated ligand N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine (5) was obtained by precipitation in cold diethyl ether (final yield of approximately 20%) and also characterized viaESI-MS and 1H NMR.Synthesis of polypeptide-based glycoconjugates
Polypeptides 17-H-3, 17-H-6, 35-H-6 and 35-RC-6 (with sequences listed in Table 1) were expressed from E. coli expression hosts, purified, and characterized as reported previously and described in the ESI†.30,32,33 The expressed and purified polypeptides were dissolved in DMSO at a concentration of 1 mg mL−1. A ten-fold excess (based on moles of carboxylic acid groups in the polypeptides) of sulfated ligand, a ten-fold excess of HBTU, and a twenty-fold excess of DIPEA were added to the reaction solution every three hours to ensure completion of the reaction. The reaction solutions were stirred at room temperature for 9 hours. The reaction mixtures were then dialyzed against PBS buffer (pH 7.4) with 150 mM NaCl (4 changes) for one day, then against H2O (4 changes) for another day, and then lyophilized. The resulting solid product was characterized viaSDS-PAGE, amino acid analysis (AAA), and 1H NMR.| Sulfated ligandsa | Polypeptide sequencesb | # of ligandsc | Approx. spacingd (Å) |
|---|---|---|---|
| a SulfoCap x-H-y: SulfoCap represents the ligand N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine, x represents the approximate distance between the adjacent glutamic acids, H represents helical conformation, RC represents random coil conformation, and y represents the number of repeats of the monomeric peptide sequence. b Only the repeating units are listed in the table. All the polypeptides have a histidine tag (MGH10S2GHIHM) at the N-terminus; 17-H-3, 17-H-6 and 35-RC-6 have GGYGGMG at the C-terminus while 35-H-6 carries only a glycine residue. c The numbers of ligands shown in the table are the numbers of glutamic acid residues in the sequence, the actual functionality of the final conjugates, which ranged from 70 to 85%, was determined via 1H NMR and is presented in the text. d The spacing between adjacent ligands of the conjugates are calculated based on energy minimization modeling for α-helical polypeptides and molecular dynamics simulations for the random-coil polypeptides. | |||
| SulfoCap17-H-3 | [AAAQEAAAAQAAAQAEAAQAAQ]3 | 6 | 17 |
| SulfoCap17-H-6 | [AAAQEAAAAQAAAQAEAAQAAQ]6 | 12 | 17 |
| SulfoCap35-H-6 | [AAAQAAQAQAAAEAAAQAAQAQ]6 | 6 | 35 |
| SulfoCap35-RC-6 | [{(AG)2PSG}2(AG)2PEG{(AG)2PSG}2]6 | 6 | 35 |
General characterization methods
ESI-MS and 1H NMR were used to characterize the N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine. A Q-Tof API-US (Micromass UK limited, Manchester, UK) instrument was used for ESI-MS; a small amount of the sample in MeOH solution was injected and the negative mode was used for detection. The 1H NMR spectra of the galactopyranosides in D2O were recorded on a Bruker NMR DRX 400 instrument with 16 scans for small molecules. The conjugation reactions were monitored viaSDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) with silver staining, and the composition was confirmed via1H NMR (in D2O with 128 scans) and AAA (University of California Davis, Davis, CA). The functionality of glycopolypeptides was determined by 1H NMR spectroscopy. The secondary structure of the sulfated glycopolypeptides was characterized viaCD spectroscopy in PBS buffer at pH 7.4, with concentrations confirmed viaAAA.Circular dichroic spectroscopy (CD)
Circular dichroic spectra were recorded on a Jasco J-810 spectropolarimeter (Jasco Inc., Easton, MD) equipped with a Jasco PTC-424S temperature controller. Background scans of buffers (PBS pH = 7.4) were recorded and subtracted automatically from the sample scans. Samples were made at a concentration of approximately 5 μM via dilution. The stock concentrations were confirmed viaAAA (University of California Davis, Davis, CA). The samples (400 μL) were loaded into a 1 mm path length quartz cuvette. Data points for the wavelength-dependent CD spectra were recorded at every nanometer with a 1 nm bandwidth and a 10 second averaging time for each data point from 190 to 250 nm. Data points for the temperature scans were recorded at 222 nm, at 1 °C intervals with an equilibration time of 1 min with a heating rate of 1 °C min−1. The mean residue ellipticity [θ]MRE (deg cm2 dmol−1) was calculated via use of the concentration, molecular weight of the samples, number of residues and cell path length, as described in the ESI†.34L-selectin shedding assay
Jurkat T-cells were grown in RPMI-1640, 10% FBS, and 100 U of the antibiotics penicillin and streptomycin. Jurkat cell cultures were collected viacentrifugation (800 × g 5 min), and the cell pellets were washed with PBS buffer pH 7.4 containing Ca2+ and Mg2+ and resuspended to 4 × 106cells per mL. The suspension was then incubated with the multivalent samples (or with controls) for 30 min at 37 °C. Sample concentrations were determined viaAAA. PMA solution (100 ng mL−1) and PBS buffer were used as positive and negative controls, respectively. Supernatants were collected for L-selectin ELISA after centrifuging the incubated mixtures for 5 min (800 × g) to remove suspended cells.L-selectin enzyme-linked immunosorbent assay (ELISA)
A standard sandwich ELISA assay was conducted in 96-well microplates according to previously reported protocols.35 Microplates were incubated at room temperature overnight with 100 μL of 4 μg mL−1L-selectin capture antibody in PBS solution (pH 7.4) in each well and then blocked with 1% BSA–PBS solution for 1 h at room temperature. The plate was washed with washing buffer (0.05% Tween 20 in PBS buffer) and then 100 μL of samples (or L-selectin standards) were loaded in separate wells. The plate was incubated at room temperature for 2 h with gentle shaking then washed with washing buffer three times. A biotin-conjugated L-selectin detection antibody (100 μL of a 1% BSA–PBS solution with a concentration of antibody of 100 ng mL−1) was added and incubated at room temperature for 2 h. After another wash step, 100 μL streptavidin–horseradish peroxidase (HRP) conjugate in 1% BSA–PBS solution (1:200) was then added and the plate incubated for 20 min at room temperature. After washing with wash buffer, the plate was developed with TMB solution for 15 min. The reaction was quenched with 2 M H2SO4 and the absorbance was monitored at 450 nm. The data were plotted and fit by a nonlinear regression equation described in the ESI†.36
L-selectin competitive binding assay
Microplates were incubated at room temperature overnight with 100 μL of 4 μg mL−1L-selectin capture antibody in PBS solution (pH 7.4) in each well and then blocked with 1% BSA–PBS solution for 1 h at room temperature. The plate was washed with washing buffer (0.05% Tween 20 in PBS buffer) and then the plate surface was equipped with L-selectinvia incubation with L-selectin solutions (2330 pg mL−1) in individual wells. The plate was incubated at room temperature for 2 h with gentle shaking then washed with washing buffer three times. A solution of sLea–PAA–biotin (at a series of concentrations for generating the standard curve), or a mixture of sLea–PAA–biotin with different concentrations of glycoconjugates (or unmodified polypeptide controls) were added and incubated at room temperature for 2 h. After another wash step, 100 μL streptavidin–HRP conjugate in 1% BSA–PBS solution (1:200) was then added and the plate incubated for 20 min at room temperature. After washing with wash buffer, the plate was developed with TMB solution for 15 min. The reaction was quenched with 2 M H2SO4 and the absorbance measured at 450 nm to determine the amount of sLea–PAA–biotin bound to the plate surface. The sLea–PAA–biotin binding standard curve was plotted and fit by a nonlinear equation. The percentage of inhibition for each glycopolypeptide concentration was then calculated based on the standard curve. The data were plotted as the percentage of inhibition versus the concentration of ligand, on a saccharide basis. Error is reported as the standard error between duplicate measurements.
Results
Synthesis of monovalent disulfated ligand
The synthetic scheme, based on previously reported methods,11,37 for synthesis of the monovalent ligand N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine is shown in Fig. 1. Galactose was chosen as the saccharide template owing to its existence in the terminal saccharides found in naturally occurring L-selectin ligands. Sulfation at the 3 and 6 positions of the galactopyranoside was chosen based on the necessary sulfation of L-selectin ligands38–40 and owing to the previous demonstration of the binding of 3,6-disulfo-galactopyranoside-modified glycopolymers to L-selectin.11 The 3,6-disulfo-galactopyranoside was modified with a 6-carbon linker arm in the present study in order to improve the accessibility of the ligand, as has been previously reported in studies of the binding of related multivalent ligands to the pentameric B5 subunit of the cholera toxin.29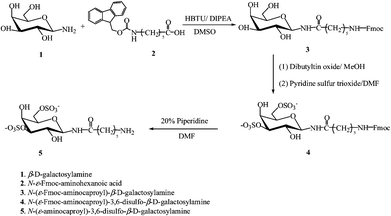 | ||
| Fig. 1 Synthesis of N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine. | ||
During sulfation, the monosulfated byproduct is also obtained, but could be separated from the reaction mixture via treatment by anionic exchange chromatography. The mono- and disulfated Fmoc-protected N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine elute from a DEAE anionic exchange column at 30% and 60% of buffer B (50 mM Tris buffer (pH 8.0) with 1 M NaCl), respectively, and the ratio of the two fractions varies from reaction to reaction. A representative chromatogram is shown in Fig. S1†. The fractions were collected separately and characterized viaelectrospray ionization mass spectrometry (ESI-MS) and 1H NMR spectroscopy. The fraction that eluted at ∼60% buffer B represented the disulfated product with a molecular mass of 452 g mol−1, as determined viaESI-MS under negative detection mode (Fig. S2†). The chemical shift of the anomeric proton of the sulfated galactopyranosides, in 1H NMR, varies for products with different sulfation patterns, which was also used to confirm the functionality of the product (Fig. S3†).
Polypeptide scaffolds
The three α-helical polypeptides 17-H-3, 17-H-6 and 35-H-6, and a random coil polypeptide 35-RC-6, synthesized viaprotein engineering methods, were investigated as scaffolds for the presentation of the N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine. In contrast to the use of small-molecule ligands, the polymeric ligands may allow the co-localization of multiple receptors, to aid in investigations of the role of cluster number in resulting cell signaling events. The sequences, number of ligands, and their approximate inter-ligand distances are listed in Table 1. For the helical 17-H-3, 17-H-6, and 35-H-6 scaffolds, the inter-ligand distances are estimated based on the known inter-residue distances for a fully helical α-helix.41–43 The polypeptide 35-RC-6, rich in glycine and proline residues, adopts a random coil conformation in solution.30 The distance between two adjacent glutamic acid residues was calculated based on a random-flight chain model, considering the number of residues between Glu residues and a characteristic ratio (Cn) of 2.0. In all of the polypeptide scaffolds, the glutamic acid residues were employed as the sites of ligand attachment.Polypeptide-based sulfated multivalent ligands
The coupling reaction of N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine to polypeptides through amide bond formation is shown in Fig. 2. To approach full functionalization of the sulfated glycopolypeptides, a large stoichiometric excess of the saccharide ligands, HBTU and DIPEA, were added during the reaction. The excess small molecules were later removed viadialysis of the product against PBS buffer with 150 mM NaCl and then H2O. The completion of the reaction was monitored viaSDS-PAGE, as shown in Fig. 3. The difference in molecular mass of the unmodified and modified polypeptides is apparent in the electrophoretic analysis, and indicates the modification of the scaffolds. | ||
| Fig. 2 Conjugation of polypeptides with N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine. | ||
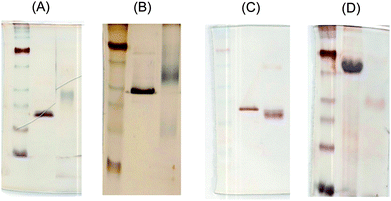 | ||
| Fig. 3 SDS-PAGE of polypeptides and disulfated glycopolypeptides with silver staining: (A)17-H-3 and SulfoCap 17-H-3, (B) 17-H-6 and SulfoCap 17-H-6, (C) 35-H-6 and SulfoCap 35-H-6 and (D) 35-RC-6 and SulfoCap 35-RC-6. | ||
As reported previously, these anionic polypeptides migrate at an anomalously high molecular mass due to differences in their binding of SDS versus that of the marker proteins.28,30,33 The exact migration of the sulfated polypeptides is complicated by multiple factors. Although the molecular mass is slightly increased after modification, which would reduce electrophoretic mobility, the charge on the scaffolds is effectively doubled, which would increase electrophoretic mobility. Furthermore, non-ideal binding of SDS by these conjugates is almost certain, and is likely variable for the different scaffolds. Thus, for the 17-H-3 and 17-H-6 scaffolds, the conjugates migrate at a greater apparent molecular mass than the unmodified scaffolds, likely as a result of both their higher molecular mass and the reduction of SDS binding from the high density of negatively charged ligands. The conjugates of 35-H-6 and 35-RC-6, in contrast, migrate at an apparently lower molecular mass than the unmodified scaffolds. This is perhaps due to the lower density of ligands, which would have less of an impact on SDS binding, thus resulting in an overall greater electrophoretic mobility despite the increase in molecular mass of the conjugates.
The characterization of the extent of functionalization of the scaffolds was determined via1H NMR (Fig. S4†). The degree of functionality can be obtained from the 1H NMR characterization based on the integration of the anomeric proton on the saccharide ligands compared to that of the β- and γ-protons of the glutamic acid and glutamine residues of the polypeptides, which appear at 2.2 and 2.4 ppm. These data indicate high levels of functionalization, with 5 out of 6 sites modified, indicating the utility of these scaffolds for evaluation of the impact of glycopolymer architecture on the L-selectin shedding event.
The scaffolds were also characterized via circular dichroic spectroscopy to confirm the expected conformations of the helical and random coil sulfated glycopolypeptides. The studies were conducted in the PBS buffer (pH 7.4) used in the L-selectin shedding assay. The concentration of samples was determined viaAAA. The mean residue ellipticity ([θ]MRE) values of samples were calculated based on the cell path length, molecular weight, number of residues and concentrations. The spectra of the glycopolypeptides are shown in Fig. 4. (The spectra of the unmodified polypeptides has been previously reported.33) For the α-helical conjugates, the spectra show two minima at 208 nm and 222 nm, and a maximum at approximately 195 nm, which is consistent with the spectrum of an α-helical polypeptide, although the sulfated glycopolypeptides have lower helicities (see below). In contrast, the 35-RC-6 and SulfoCap 35-RC-6 exhibited a minimum at 198 nm that is typical for the random coil conformation.
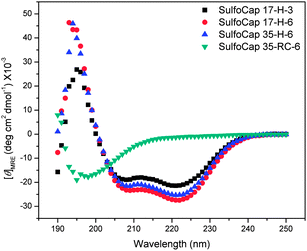 | ||
| Fig. 4 Circular dichroic spectra of the sulfated glycopolypeptides in PBS buffer (pH 7.4) at 5 °C. | ||
The thermal unfolding behavior of these samples was also evaluated via circular dichroic spectroscopy; the full wavelength spectra of the samples were monitored as a function of temperature to evaluate the conformational changes and reversibility of any unfolding. The thermal transition and reversibility of the conformational transitions of SulfoCap 17-H-6 are shown in Fig. 5 as an example. With increasing temperature from 5 to 80 °C, the minima at 208 and 222 nm decrease in absolute intensity, with the appearance of a single minimum near 198 nm at elevated temperature, which is consistent with a non-helical conformation.33,44 The isodichroic point (∼203 nm) suggests a local two-state transition at the residue level, co-operative within short range. The whole-chain thermal transition from helical to random coil is not a two-state transition, and intermediate steps may be involved.45–48 The glycopolypeptides regain their original conformations after heating and cooling, illustrated by the identical spectra at 5 °C before and after heating (Fig. 5(B)), suggesting that thermally induced unfolding does not permanently change the conformation of the polypeptides under these experimental conditions.
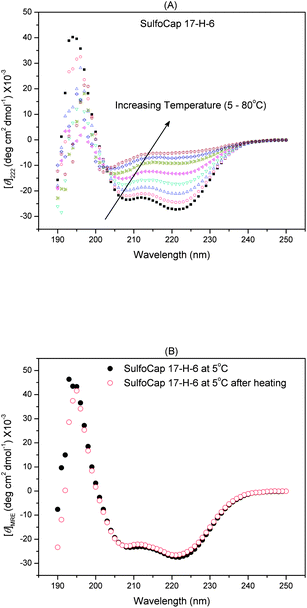 | ||
| Fig. 5 Full wavelength CD spectra of SulfoCap 17-H-6 in PBS buffer (pH = 7.4) with increasing temperature from 5 to 80 °C (A) and the spectra of SulfoCap 17-H-6 in PBS buffer (pH = 7.4) at 5 °C before and after heating (B). | ||
L-selectin shedding induced by polypeptide-based disulfated glycoconjugates
The ability to induce L-selectin shedding of multivalent sulfated glycoconjugates was tested by an L-selectin shedding assay followed by an ELISA assay. In the L-selectin shedding assay, PBS buffer and 100 ng mL−1 of PMA solution were used as negative and positive controls, respectively. The soluble L-selectin was isolated from the cell mass by centrifugation and was quantified in the supernatant solution by a standard L-selectin ELISA. The degree of shedding induced by each sample was normalized to the degree of shedding induced by PMA in the same set of experiments. The data were plotted as the normalized percentage of shedding as a function of ligand concentration (on a saccharide basis (for unmodified polypeptide backbones, the numbers of glutamic acids were used)) and are shown in Fig. 6. The consistent non-zero background of L-selectin shedding, observed when Jurkat cells were treated with only PBS buffer, results from the fact that L-selectin can be shed constantly in natural biological environments without further stimulation, thus yielding soluble L-selectin in plasma that regulates L-selectin expression and lymphocyte circulation.3,7,10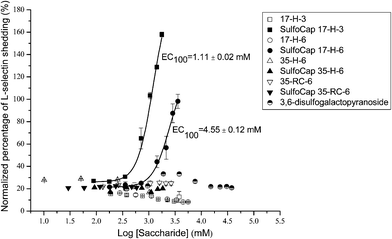 | ||
| Fig. 6 Normalized concentration of soluble L-selectin shed by monovalent ligand, polypeptides, and multivalent sulfated glycopolypeptides, normalized to that induced by PMA, as determined via ELISA. Curves shown are fits to the data by a nonlinear regression equation described in the ESI†. The EC100 values are estimated as the concentration of samples on saccharide basis at 100% of PMA-induced L-selectin shedding, based on the fitted curves by a nonlinear regression equation. The reported errors are obtained from the EC100 values determined for two different batches of samples. | ||
The L-selectin shedding induced by the monovalent sulfated ligand is near the background shedding level, and no significant L-selectin shedding was determined within the concentration range of up to 38 mM. In addition, none of the polypeptide backbones induced more than background shedding within the concentration range used for the sulfated glycoconjugates (up to approximately 300 μM polypeptide concentration (∼3.6 mM on a saccharide basis)). Within the experimental concentration range, the SulfoCap 17-H-3 and SulfoCap 17-H-6 induced L-selectin shedding similar to or greater than PMA-induced shedding, while SulfoCap 35-H-6 and SulfoCap 35-RC-6 did not induce any shedding above background shedding within a similar concentration range. Dose–response curves were fit with a nonlinear regression equation described above. Because the glycopolypeptide scaffolds yield different ultimate levels of L-selectin shedding that are greater than that of the PMA positive control, standard EC50 values (effective concentration that induces 50% of maximal effective shedding with background shedding subtracted) that were obtained for SulfoCap 17-H-3 and SulfoCap 17-H-6 do not capture the full extent of their differences in the L-selectin shedding. Thus EC100 values (effective concentration that induces 100% of the shedding observed for the PMA controls) were chosen for comparisons of the shedding induced by the SulfoCap 17-H-6 relative to the SulfoCap 17-H-3. The EC100 values obtained for SulfoCap 17-H-3 and SulfoCap 17-H-6 are 1.11 and 4.55 mM, respectively.
The binding of select glycopolypeptides to L-selectins on immobilized surfaces was studied with a competitive binding assay to indicate whether the observed differences in L-selectin shedding may be a result of differences in binding to L-selectin. L-selectin was immobilized on a plate coated with L-selectin antibody. The glycopolymer sLea–PAA–biotin, a non-biological polymer backbone (polyacrylate) bearing a known L-selectin ligand sialyl Lewis a (sLea), binds to L-selectin on the plate and the binding can be quantified via immunosorbent methods. The binding between sLea–PAA–biotin and L-selectin can be inhibited by the preferential binding of a ligand to the L-selectin on the plate. Results of experiments to compare two glycopolypeptides of similar length but with different shedding profiles (SulfoCap 17-H-6 and SulfoCap 35-H-6) are shown in Fig. 7. The data were collected over ligand concentration ranges similar to those probed in the shedding experiments, and indicate that both glycopolypeptides inhibit sLea–PAA–biotin binding to L-selectin without significant difference.
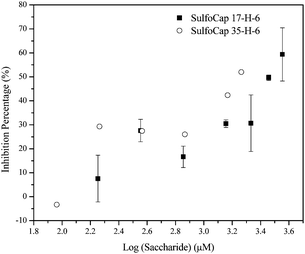 | ||
| Fig. 7 The inhibition of sLea-PAA-biotin binding, by glycopolypeptides (SulfoCap 17-H-6 and SulfoCap 35-H-6) versus the ligand concentration on a saccharide basis. | ||
Discussion
Design of polypeptide-based multivalent ligands
Sulfated glycopolypeptides have been designed as multivalent scaffolds for evaluation of the impact of glycopolymer architecture on the shedding of L-selectin, and with an eye toward the development of additional classes of architecturally distinct and modifiable scaffolds as cell biology tools. In this work, N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine has been employed as a ligand on polypeptide-based scaffolds with different backbone conformations and ligand densities. These synthetic multivalent ligands of L-selectin are designed with features similar to those of L-selectin natural ligands, which are mucin-like glycoproteins or glycolipids with a rigid mucin-like polypeptide backbone and multiple copies of O-glycans that are post-translationally modified,4 such as GlyCAM-1, PNAd, CD34, PSGL-1, and podocalyxin.4,5Sialic acids, fucoses and sulfated groups are required for the activity of many L-selectin ligands,6,49 and a simple, weak binding ligand-based on 3,6-disulfo-galactose has also been identified.50,51 In our designs here, the polypeptide chains have various stiffnesses which may be tuned to better mimic that of the natural ligand. The 3,6-disulfo-galactopyranoside was chosen owing to the importance of sulfated groups in natural L-selectin ligands in strengthening L-selectin binding,38–40,52 and given that an O-linked 3,6-disulfo galactose and sulfated tyrosine (sTyr) have both been used to modify polymer scaffolds for L-selectin binding.11,15 The use of these backbones was further motivated by our related work indicating that specific ligand densities and backbone architectures of multivalent glycopolypeptides can be used to manipulate the binding of multivalent ligands with protein receptors.27,29,30,33,53That similar recombinantly derived, polypeptide scaffolds would be useful for studies of cell-surface receptor clustering was suggested by previous work indicating that multivalent ligands with minimal chain length and valencies can be used to address cell surface receptors and control signaling. For example, a 15-mer polynorbornene-based glycopolymer containing 3,6-disulfo-galactose inhibited leukocyte rolling on the surface coated with the natural ligand GlyCAM-1, although with higher IC50 values than a glycopolymer containing a strong-binding oligosaccharide ligand, 3′,6-disulfo-Lex. The 3,6-disulfo-galactose-modified glycopolymer was also shown to induce a certain degree of L-selectin down-regulation on the surface of leukocytes, also with a lower degree of shedding compared with the glycopolymer bearing 3′,6-disulfo Lex.11
Previous studies have also indicated opportunities for scaffolds with defined ligand densities in the development of receptor cluster-specific therapeutics and tools. Sulfated tyrosine was conjugated to poly[N-(2-hydroxyethyl)acrylamide] backbones together with oligosaccharide ligands sLex or sLea, at various ratios and ligand densities. These macromolecules were tested as inhibitors of L-selectin binding. The best inhibition of L-selectin binding was achieved by the polymer containing average densities of 20% sLea and 5% of sTyr, indicating that variations in average ligand density are important for the binding,15 although in the reported studies, the uniform presentation of ligands was not possible.
Therefore we employed here, as in our previous studies, alanine-rich sequences with an α-helical conformation, and controlled number and placement of glutamic acid residues; these approaches permitted control of valencies and spacing of ligands. Given that L-selectin natural ligands, such as mucin-like glycoproteins, have rigid backbones for multivalent display of oligosaccharides,54,55polypeptides with α-helical conformation may be better suited for such studies. These sequences, with Rg values roughly estimated to be in the order of 25 nm,56 are sufficiently large to address L-selectin clusters on the surface of leukocytes, given that the size of the L-selectin lectin domain, as estimated from its crystal structure, is approximately 22 Å.57
These scaffolds also provide a means to evaluate the impact of ligand spacing and number on the L-selectin shedding event. The approximate spacings between ligands on the 17-H-X and 35-H-6 scaffolds, which are 17 and 35 Å, are controlled by the spacing between glutamic acid residues in the sequence and calculated based on an ideal α-helical chain conformation. Although the polypeptide backbones are not 100% helical (there are helical to random coil dynamic transitions within short peptide domains that decrease their overall helicities), even with a complete loss of the helical conformation, the distance between the ligands on the 17-H-X and 35-H-6 scaffolds will be approximately 37.8 and 53.5 Å, based on root-mean-square (rms) calculations for a random-flight chain model with a characteristic ratio Cn = 9.0.58 Therefore, the spacing between two adjacent ligands could range from approximately 17–35 Å and 35–50 Å for SulfoCap 17-H-X and SulfoCap 35-H-X, respectively.
Architecture effects on induced L-selectin shedding
Our L-selectin shedding results show that the monovalent ligand N-(ε-aminocaproyl)-3,6-disulfo-β-D-galactosylamine did not induce L-selectin shedding, consistent with previously reported studies.21 While the unmodified polypeptides also induced no shedding, the SulfoCap 17-H-3 and SulfoCap 17-H-6 induced L-selectin shedding at much lower saccharide concentration than the monovalent saccharide. These results are consistent with the generally accepted mechanism that the formation of receptor clusters20 causes the L-selectin cytoplasmic tail to release CaM and the extracellular domain to undergo a conformational change that renders it susceptible to cleavage by sheddase.2,59 The L-selectin shedding induced by the SulfoCap 17-H-3 and SulfoCap 17-H-6, both equipped with an N-linked 3,6-disulfo-galactosylamine, compares favorably with results previously reported by Kiessling and coworkers, in which L-selectin shedding from Jurkat cells was induced by a norbornene-based glycopolymer (n = 15) carrying an O-linked 3′,6-disulfo Lex ligand.11 The efficiency of L-selectin shedding of the SulfoCap 17-H-3 and SulfoCap 17-H-6 is comparable with that of the poly(norbornene)-based glycopolymers, although the polypeptide scaffolds are equipped with a weaker ligand.In contrast, however, the 35-H-6 and 35-RC-6 scaffolds induced no detectable shedding, even at high saccharide concentrations. These differences in shedding suggest that there may exist an optimal ligand density for L-selectin shedding induced by these scaffolds, and that simple presentation of multiple copies of a ligand is an insufficient condition for shedding (Fig. 8). The binding of SulfoCap 17-H-X, which presents ligands at distances (17–35 Å) that directly commensurate with the known size of the L-selectin (22 Å57) may permit organization of the L-selectin at distances (Fig. 8B) that initiate conformational changes that result in shedding. On the other hand, the binding of SulfoCap 35-H-6 (with similar chain length as SulfoCap 17-H-6) may not bring the L-selectins in sufficiently close proximity to induce this conformational change and trigger shedding (Fig. 8C). Thus, it is likely that effective L-selectin clusters are not formed upon binding by multivalent ligands with an approximate 35–50 Å interligand distance. The fact that competitive binding studies show that the SulfoCap 35-H-6 multivalent ligands are capable of inhibition of L-selectin binding indicates that the spacing between ligands represents a parameter that may be tuned to modulate L-selectin clustering and induced shedding.
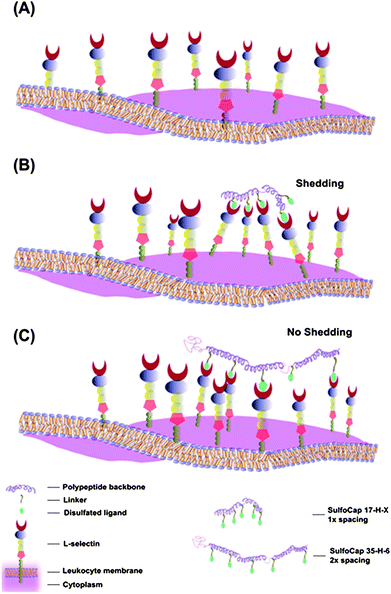 | ||
| Fig. 8 Schematic of L-selectin distribution and cluster formation on leukocyte surfaces with different multivalent ligands. (A) Evenly distributed L-selectin without clustering; (B) L-selectin clustering induced by multivalent ligands with closer spacing (SulfoCap 17-H-3 and SulfoCap 17-H-6); (C) L-selectin clustering induced by multivalent ligands with larger spacing (SulfoCap 35-H-6). | ||
The fact that the SulfoCap 17-H-3 is more efficient at inducing shedding than the SulfoCap 17-H-6 suggests that there may also be an optimal number of receptors in a cluster. The 15-mer norbornene polymer employed by the Kiessling laboratories, sufficiently long to induce shedding, has multiple conformations that yield a chain length ranging from 30–75 Å.60,61 Given that the lectin domain of L-selectin has a diameter of approximately 22 Å,57 a 15-mer norbornene-based polymer could theoretically bind up to 3 copies of L-selectin. The SulfoCap17-H-3, with 6 ligands, may allow a greater number of L-selectin to cluster, resulting in increased signaling. Interestingly, the SulfoCap 17-H-6, although it presents a greater number of ligands at the same inter-ligand spacing as SulfoCap 17-H-3, showed higher EC100 values, and less efficiently induced shedding, than SulfoCap 17-H-3. These observations suggest that the number of ligands of SulfoCap 17-H-3 is sufficient to induce effective L-selectin shedding. Indeed, clustering of a limited number of protein receptors is known to be a crucial mediator for inducing signal transduction mostly through varying the protein kinase and phosphatase activities,20,62 such as the integrin clustering,63 G-protein-coupled receptors,64 and T cell receptors.65
Although our current studies have not thoroughly probed the role of chain conformation in the shedding event, the fact that neither the SulfoCap 35-H-6 nor the SulfoCap 35-RC-6 induced shedding suggests that increased flexibility of the polymer chain does not permit reorganization of L-selectins to overcome a sub-optimal ligand density. In fact, the increased chain flexibility of the random coil scaffolds may make it difficult for L-selectins to maintain an effective cluster density and arrangement, based on both the smaller dimensions of the random coil polypeptides and their increased backbone flexibility; a random coil scaffold may thus be expected to be a poorer inducer of L-selectin shedding than a helical scaffold. Future evaluation of SulfoCap 17-RC-X samples and other random coil ligands with 17 Å spacing are under investigation to confirm the relative roles of ligand spacing and backbone conformation in the manipulation of L-selectin shedding.
Taken altogether, the comparison of these, and previously reported, results suggests that the polypeptide-based multivalent scaffolds are efficient scaffolds for the induction of L-selectin shedding, and that for polypeptides of a given size, that the density of ligands is a tunable parameter for manipulating L-selectin clustering and shedding. Further assays of leukocyte rolling and L-selectin binding are underway to verify these suppositions. The control over spacing and chain conformation afforded by the polypeptide scaffolds may be exploited in increasing the efficiency of multivalent binding or in the mapping cell surface receptors.
Conclusions
Polymers offer significant advantages as multivalent backbones to probe cell surface receptor arrangement and cell signaling, providing useful cell biology tools and potential therapeutic approaches. In this work, a series of polypeptide-based multivalent ligands containing a weak-affinity ligand—3,6-disulfo galactopyranoside—and equipped with a well-controlled architecture were synthesized and the architecture effects on L-selectin shedding were studied by L-selectin shedding assay and ELISA. The results showed clearly that L-selectin shedding was induced by the multivalent ligands of select architecture, but not by monovalent ligands or polypeptide backbones. Inter-ligand distance was suggested to be a key variable, as ligands with smaller spacing (17–35 Å), but not those with larger spacing (35–50 Å), induced L-selectin shedding. These data suggest that optimal ligand number and inter-receptor distance may be necessary for the induction of L-selectin shedding. Polypeptide-based multivalent ligands with random coil conformation and optimized spacing are under investigation to confirm the role of chain conformation and ligand spacing. Given the various important biological functions of L-selectin binding and shedding in the regulation of leukocyte tethering and rolling, lymphocyte homing, and metastasis, as well as the broad importance of cell receptor topology on cell function, these polypeptide-based multivalent ligands may have potential physiological and pathological applications in as cell biology tools, therapeutics, and in the regulation of cell functions or inflammatory responses.References
- W. I. Weis, M. E. Taylor and K. Drickamer, Immunol. Rev., 1998, 163, 19–34 CrossRef CAS.
- J. J. Grailer, M. Kodera and D. A. Steeber, J. Dermatol. Sci., 2009, 56, 141–147 CrossRef CAS.
- T. H. Rainer, Resuscitation, 2002, 52, 127–141 CrossRef CAS.
- R. P. McEver, Nat. Immunol., 2005, 6, 1067–1069 CrossRef CAS.
- A. Varki, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 7390–7397 CAS.
- D. Vestweber and J. E. Blanks, Phys. Rev., 1999, 79, 181–213 CAS.
- D. M. Smalley and K. Ley, J. Cell. Mol. Med., 2005, 9, 255–266 CrossRef CAS.
- R. Alon, S. Q. Chen, R. Fuhlbrigge, K. D. Puri and T. A. Springer, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11631–11636 CrossRef CAS.
- A. Hafezi-Moghadam, K. L. Thomas, A. J. Prorock, Y. Huo and K. Ley, J. Exp. Med., 2001, 193, 863–872 CrossRef CAS.
- B. Schleiffenbaum, O. Spertini and T. F. Tedder, J. Cell Biol., 1992, 119, 229–238 CrossRef CAS.
- P. Mowery, Z. Q. Yang, E. J. Gordon, O. Dwir, A. G. Spencer, R. Alon and L. L. Kiessling, Chem. Biol., 2004, 11, 725–732 CrossRef CAS.
- M. C. Phong, P. Gutwein, S. Kadel, K. Hexel, P. Altevogt, O. Linderkamp and B. Brenner, Biochem. Biophys. Res. Commun., 2003, 300, 563–569 CrossRef CAS.
- G. Preece, G. Murphy and A. Ager, J. Biol. Chem., 1996, 271, 11634–11640 CrossRef CAS.
- P. E. Mattila, C. E. Green, U. Schaff, S. I. Simon and B. Walcheck, Am. J. Physiol., 2005, 289, C323–C332 CrossRef CAS.
- S. Enders, G. Bernhard, A. Zakrzewicz and R. Tauber, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 1441–1449 CrossRef CAS.
- Y. Lee and N. S. Sampson, Curr. Opin. Struct. Biol., 2006, 16, 544–550 CrossRef CAS.
- Y. Lee and N. S. Sampson, ChemBioChem, 2009, 10, 929–937 CrossRef CAS.
- J. Geng, J. Lindqvist, G. Mantovani, G. Chen, C. T. Sayers, G. J. Clarkson and D. M. Haddleton, QSAR Comb. Sci., 2007, 26, 1220–1228 CrossRef CAS.
- M. Hetzer, G. J. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef CAS.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS.
- R. M. Owen, J. E. Gestwicki, T. Young and L. L. Kiessling, Org. Lett., 2002, 4, 2293–2296 CrossRef CAS.
- R. E. Connor and D. A. Tirrell, Polym. Rev., 2007, 47, 9–28 Search PubMed.
- N. Sharma, R. Furter, P. Kast and D. A. Tirrell, FEBS Lett., 2000, 467, 37–40 CrossRef CAS.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- I. Kwon, K. Kirshenbaum and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 7512–7513 CrossRef CAS.
- K. L. Kiick, Science, 2007, 317, 1182–1183 CrossRef CAS.
- Y. Wang and K. L. Kiick, J. Am. Chem. Soc., 2005, 127, 16392–16393 CrossRef CAS.
- S. Liu and K. L. Kiick, Macromolecules, 2008, 41, 764–772 CrossRef CAS.
- B. D. Polizzotti and K. L. Kiick, Biomacromolecules, 2006, 7, 483–490 CrossRef CAS.
- B. D. Polizzotti, R. Maheshwari, J. Vinkenborg and K. L. Kiick, Macromolecules, 2007, 40, 7103–7110 CrossRef CAS.
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- M. A. Frederick, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, K. Struhl, Short Protocols in Molecular Biology, Wiley John & Sons, New York, 1999 Search PubMed.
- S. Liu and K. L. Kiick, Macromolecules, 2008, 41, 764–772 CrossRef CAS.
- T. E. Creighton, Protein Folding, W.H. Freeman, 1992 Search PubMed.
- DuoSet® ELISA Development System Protocol.
- W. P. Bowen and J. C. Jerman, Trends Pharmacol. Sci., 1995, 16, 413–417 CrossRef CAS.
- B. Guilbert, N. J. Davis and S. L. Flitsch, Tetrahedron Lett., 1994, 35, 6563–6566 CrossRef CAS.
- W. J. Sanders, T. R. Katsumoto, C. R. Bertozzi, S. D. Rosen and L. L. Kiessling, Biochemistry, 1996, 35, 14862–14867 CrossRef CAS.
- C. Mitsuoka, N. KawakamiKimura, M. KasugaiSawada, N. Hiraiwa, K. Toda, H. Ishida, M. Kiso, A. Hasegawa and R. Kannagi, Biochem. Biophys. Res. Commun., 1997, 230, 546–551 CrossRef CAS.
- C. Galustian, A. M. Lawson, S. Komba, H. Ishida, M. Kiso and T. Feizi, Biochem. Biophys. Res. Commun., 1997, 240, 748–751 CrossRef CAS.
- R. S. Farmer, L. M. Argust, J. D. Sharp and K. L. Kiick, Macromolecules, 2006, 39, 162–170 CrossRef CAS.
- R. S. Farmer and K. L. Kiick, Biomacromolecules, 2005, 6, 1531–1539 CrossRef CAS.
- R. S. Farmer, A. Top, L. M. Argust, S. Liu and K. L. Kiick, Pharm. Res., 2008, 25, 700–708 CAS.
- J. S. Miller, R. J. Kennedy and D. S. Kemp, J. Am. Chem. Soc., 2002, 124, 945–962 CrossRef CAS.
- M. E. Holtzer and A. Holtzer, Biopolymers, 1992, 32, 1675–1677 CrossRef.
- J. M. Mo, M. E. Holtzer and A. Holtzer, Biopolymers, 1990, 30, 921–927 CrossRef CAS.
- N. Taddei, F. Chiti, T. Fiaschi, M. Bucciantini, C. Capanni, M. Stefani, L. Serrano, C. M. Dobson and G. Ramponi, J. Mol. Biol., 2000, 300, 633–647 CrossRef CAS.
- A. Top, K. L. Kiick and C. J. Roberts, Biomacromolecules, 2008, 9, 1595–1603 CrossRef CAS.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Curr. Opin. Chem. Biol., 2000, 4, 696–703 CrossRef CAS.
- Y. Suzuki, Y. Toda, T. Tamatani, T. Watanabe, T. Suzuki, T. Nakao, K. Murase, M. Kiso, A. Hasegawa, K. Tadanoaritomi, I. Ishizuka and M. Miyasaka, Biochem. Biophys. Res. Commun., 1993, 190, 426–434 CrossRef CAS.
- D. D. Manning, X. Hu, P. Beck and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 3161–3162 CrossRef CAS.
- S. Hemmerich, H. Leffler and S. D. Rosen, J. Biol. Chem., 1995, 270, 12035–12047 CrossRef CAS.
- R. Maheshwari, S. Liu, B. D. Polizzotti, Y. Wang and K. L. Kiick, Chemical Glycobiology, Oxford University Press, Washington, DC, 2008, p. 288 Search PubMed.
- S. Baumhueter, M. S. Singer, W. Henzel, S. Hemmerich, M. Renz, S. D. Rosen and L. A. Lasky, Science, 1993, 262, 436–438 CAS.
- N. Jentoft, Trends Biochem. Sci., 1990, 15, 291–294 CrossRef CAS.
- A. Top, PhD dissertation, University of Delaware, 2010 Search PubMed.
- P. Mehta, V. Oganesyan, S. Terzyan, T. Mather, R. P. McEver, Protein Data Bank 3CFW, 2008 Search PubMed.
- Protein Structure, ed. T. E. Creighton, 2nd edn, 1997 Search PubMed.
- A. Ivetic and A. J. Ridley, Biochem. Soc. Trans., 2004, 32, 1118–1121 CrossRef CAS.
- J. E. Gestwicki, L. E. Strong and L. L. Kiessling, Chem. Biol., 2000, 7, 583–591 CrossRef CAS.
- M. Kanai, K. H. Mortell and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 9931–9932 CrossRef CAS.
- F. D. Smith and J. D. Scott, Curr. Biol., 2002, 12, R32–R40 CrossRef CAS.
- L. J. Kornberg, H. S. Earp, C. E. Turner, C. Prockop and R. L. Juliano, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8392–8396 CrossRef CAS.
- S. Angers, A. Salahpour, E. Joly, S. Hilairet, D. Chelsky, M. Dennis and M. Bouvier, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3684–3689 CrossRef CAS.
- K. H. Lee, A. D. Holdorf, M. L. Dustin, A. C. Chan, P. M. Allen and A. S. Shaw, Science, 2002, 295, 1539–1542 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00063b |
| This journal is © The Royal Society of Chemistry 2011 |