Block copolymer conjugates prepared by sequentially grafting from proteinsvia RAFT†
Hongmei
Li
,
Ming
Li
,
Xiao
Yu
,
Abhijeet P.
Bapat
and
Brent S.
Sumerlin
*
Southern Methodist University, Department of Chemistry, 3215 Daniel Ave., Dallas, Texas 75275-0314, USA. E-mail: bsumerlin@smu.edu; Fax: +1-214-768-4089; Tel: +1-214-768-8802
First published on 1st March 2011
Abstract
Well-defined thermoresponsive block copolymer conjugates were prepared by two consecutive grafting-from polymerizationsvia reversible addition–fragmentation chain transfer (RAFT) polymerization. A RAFT agent containing an activated ester was conjugated to amines on lysozyme, and N-isopropylacrylamide was polymerized by grafting directly from the modified protein in aqueous buffer. Retention of the active thiocarbonylthio moieties on the ω end of the conjugated chains allowed chain extension viapolymerization of N,N-dimethylacrylamide to yield lysozyme–poly(N-isopropylacrylamide)-b-poly(N,N-dimethylacrylamide) (LYS–PNIPAM-b-PDMA) block copolymer conjugates. Analysis of the conjugates, as well as the cleaved homopolymers and block copolymers, indicated the grafting-from polymerizations were well controlled and that chain extension during block copolymerization was efficient. The temperature-responsive aqueous solution behavior of the resulting conjugates was investigated.
Introduction
Conjugation to synthetic polymers is a viable means to modify the solubility, activity, and potential utility of proteins.1–5 For applications in vivo, functionalization of a protein therapeutic with an appropriate high molecular weight polymer can substantially reduce immunogenicity and rates of renal clearance/proteolysis. Accordingly, many diverse routes of polymer–protein conjugation have been reported. Generally, these approaches can be classified as either grafting-to, which involves conjugation of a preformed polymer with a protein by reactive coupling, or grafting-from, which describes the polymerization of monomer from a protein capable of initiation.Many previous reports of polymer–protein conjugation have involved immobilization of poly(ethylene glycol) (PEG) or its derivatives. Recent advances in controlled radical polymerization (CRP) have facilitated access to other polymers suitable for preparing well-defined polymer–protein conjugates.6,7 For example, atom transfer radical polymerization (ATRP),8–14nitroxide-mediated polymerization (NMP),15,16 and reversible addition–fragmentation chain transfer (RAFT) polymerization17–24 have all been employed to prepare polymer conjugates of proteins or peptides.
In addition to allowing precise control of polymer molecular weight and end group functionality, one of the most valuable features of CRP is the ability to prepare block copolymers by sequential polymerization of two or more monomers.25,26Block copolymer bioconjugates may have particular benefits over conventional homopolymer conjugates for a variety of applications. For instance, Stayton and coworkers suggested that addition of an outer hydrophilic block to the end of a enzyme-conjugated responsive polymer could prevent stimuli-induced collapse of the responsive segment from leading to intermolecular aggregation, while still allowing modulation of the protein's activity.27 Low-to-moderate molecular weight block copolymer conjugates can be readily prepared by grafting-to biomolecules via reactive coupling.27,28 However, given that the efficiency of the grafting-to approach is likely to decrease with increasing molecular weight of the polymer to be conjugated, an attractive alternative involves the synthesis of high molecular weight block copolymer conjugates by sequentially polymerizing two monomers from a biomolecule labeled with an initiating species. However, only a few examples of block copolymer bioconjugates prepared exclusively by grafting-from methods have been reported. For instance, Wooley et al. successfully employed NMP and ATRP to graft block copolymers from resin-supported peptides15 and amino acids.29Proteins have also been modified by grafting-from viaCRP, though the majority of examples have been limited to the synthesis of homopolymer–protein conjugates.6,7,30,31 Methods that allow the synthesis of well-defined block copolymer–protein conjugates by two consecutive grafting-from polymerizations would be highly advantageous.
Herein, we describe the synthesis of block copolymer–protein conjugates by RAFT polymerization. By immobilizing the RAFT agent via its “R-group”,23 the first monomer was polymerized under mild aqueous conditions32 to yield homopolymer–protein conjugates with active thiocarbonylthio moieties on the distal end of the conjugated chains (Scheme 1). The resulting polymer–protein macro-chain transfer agent (macroCTA) was employed for a subsequent polymerization of a second monomer33 to yield thermoresponsive block copolymer–protein conjugates. In addition to demonstrating end group retention, this report unambiguously demonstrates the molecular weight control possible while grafting-from a proteinviaCRP.
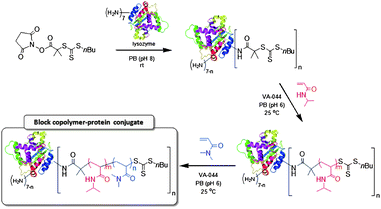 | ||
| Scheme 1 Synthesis of LYS–PNIPAM-b-PDMA conjugates by sequential RAFT polymerizations from CTA-modified LYS. | ||
Experimental
Materials
The N-hydroxysuccinimide functionalized trithiocarbonate chain transfer agent (NHS–CTA) was prepared as previously reported.342,2-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) was purchased from Wako Pure Chemical Industries, Ltd. and was recrystallized (×3) from methanol. N-Isopropylacrylamide (NIPAM, TCI America) was recrystallized from hexanes. N,N-Dimethylacrylamide (DMA, Fluka, 98%) was passed through basic alumina prior to use. Lysozyme (LYS, MP Biochemicals), methanol (EMD Chemicals), and deuterium oxide (D2O, Acros, 99.8% D) were used as received. Dialysis membranes were purchased from Pierce. Protein markers were purchased from Fisher Science. The Quick Start Bradford™ Protein Assay kit was purchased from Bio-Rad. All other chemicals were purchased from VWR and used without further purification, unless otherwise noted.Analyses
UV-Vis spectroscopy for trithiocarbonate quantification and turbidity measurements for characterization of responsive solution behavior were carried out on a Beckman Coulter DU Series 800 UV-Vis spectrophotometer equipped with a Peltier temperature controller. Lysozyme concentration was determined by comparison with a quantified lysozyme solution based on a relative protein standard curve made with a protein assay kit. 1H NMR spectroscopy was conducted with a JEOL Delta 500 spectrometer operating at 500 MHz. Conversion of NIPAM and DMA during polymerization was determined by 1H NMR spectroscopy. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed with a Bio-Rad electrophoresis system using 12% polyacrylamide gels at 160 V constant voltage for 60 min. Samples were dissolved in deionized (DI) water (18 μL, [LYS] = 2 mg mL−1), mixed with 6 μL sample buffers (containing 3% of β-mercaptoethanol), and heated at 95 °C for 5 min before loading. Staining was accomplished with Coomassie blue. Size exclusion chromatography (SEC) of the cleaved poly(N-isopropylacrylamide) PNIPAM or PNIPAM-b-poly(N,N-dimethylacrylamide) (PDMA) was conducted in DMF with 50 mM LiBr at 55 °C at a flow rate of 1.0 mL min−1 (Viscotek VE 2001 GPCmax; columns: guard + two Polymer Laboratories PolarGel-M mixed bed columns). The detector setup included a Viscotek VE 3580 refractive index detector operating at 660 nm and a Viscotek Model 270 Series Platform, consisting of a laser light scattering detector (operating at 3 mW, λ = 670 nm with detection angles of 7° and 90°) and a four capillary viscometer. Molecular weights were determined by the triple detection method.Preparation of lysozyme macroCTA (LYS–macroCTA)
LYS (0.20 g, 14 μmol) was dissolved in phosphate buffer (PB) solution (pH 8.0, 69.2 mL) in a 150 mL flask and stirred gently with a magnetic bar while purging with nitrogen for 20 min. A solution of NHS–CTA (48.0 mg, 137 μmol) dissolved in methanol (10.8 mL) was added drop wise. The resulting solution was stirred at 25 °C for 51 h. The reaction mixture was filtered with a 0.22 μm hydrophilic nylon membrane filter and subsequently dialyzed against DI water (3 × 20 L) for 15 h (molecular weight cut-off (MWCO) = 10 kDa). The resulting solution was lyophilized to yield the LYS–macroCTA.Determination of trithiocarbonate content in LYS–macroCTA and LYS–PNIPAM macroCTA
S-Butyl-S′-(α,α′-dimethyl-α′′-acetic acid)-trithiocarbonate 34 was dissolved in methanol to make a 1.76 mg mL−1 (6.97 × 10−3 M) concentrated solution, which was then diluted to 0.0176 mg mL−1 (6.97 × 10−5 M) with DI water to obtain a stock solution. The above prepared CTA solution was again diluted with DI water in six plastic cuvettes (1 mL, 1 cm path length) to contain 0, 20, 40, 60, 80, and 100% of the stock solution, respectively. The absorbance of each solution was measured at 310 nm to obtain a linear plot of CTA concentration versus absorbance. The CTA extinction coefficient was determined to be ε = 9686 M−1 cm−1, and this value was used to calculate the concentration of trithiocarbonate groups within the LYS–macroCTA and LYS–polymer conjugates.RAFT polymerization of NIPAM with LYS–macroCTA
A typical grafting-from RAFT polymerization of NIPAM from the LYS–macroCTA was conducted as follows. NIPAM (121 mg, 1.07 mmol), LYS–macroCTA (53.7 mg, 3.65 μmol protein, which contained 4.89 μmol of CTA functionality, as determined by UV-Vis), and PB (pH 6.0, 3.8 mL) were sealed in a 4 mL glass vial equipped with a magnetic stir bar. The solution was cooled in an ice bath and purged with nitrogen for 30 min. A nitrogen-purged solution of VA-044 (6.46 μmol) in PB (pH 6.0, 0.2 mL) was added by syringe, and the reaction vial was placed in a preheated reaction block at 25 °C. Samples were removed periodically by nitrogen-purged syringe to monitor monomer conversion by 1H NMR spectroscopy. The polymerization was quenched after 6.9 h (83% conversion) by opening the vial to air and cooling the reaction in an ice bath. The reaction mixture was dialyzed against DI water (3 × 20 L) for 15 h using a membrane of MWCO = 10 kDa. The resulting LYS–PNIPAM was isolated by lyophilization. For the block copolymerization study, CTA content per protein (2.1 trithiocarbonates per LYS) was determined by dividing the concentration of trithiocarbonates determined from the absorbance of the conjugate at 310 nm by the protein concentration determined by protein assay.LYS–PNIPAM purification by thermoprecipitation
While dialysis proved effective, thermoprecipitation was an alternative method for the removal of unreacted LYS from the LYS–PNIPAM product that resulted from the polymerization of NIPAM with the LYS–PNIPAM macroCTA. The crude LYS–PNIPAM solution (∼10 mg mL−1) was heated at 40 °C for 5 min, followed by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 rpm at 40 °C for 3 min. The supernatant containing unreacted LYS was decanted, and the precipitate was dissolved in DI water and lyophilized to isolate LYS–PNIPAM. While one round of thermoprecipitation successfully removed most of the unreacted LYS, the bioactivity of the purified conjugates was negatively affected. Control experiments with native LYS being exposed to the same heating procedure also resulted in significant loss of activity, suggesting the denaturation was due to the thermal treatment instead of the presence of the polymer.
200 rpm at 40 °C for 3 min. The supernatant containing unreacted LYS was decanted, and the precipitate was dissolved in DI water and lyophilized to isolate LYS–PNIPAM. While one round of thermoprecipitation successfully removed most of the unreacted LYS, the bioactivity of the purified conjugates was negatively affected. Control experiments with native LYS being exposed to the same heating procedure also resulted in significant loss of activity, suggesting the denaturation was due to the thermal treatment instead of the presence of the polymer.
Block copolymerization of DMA from the LYS–PNIPAM macroCTA
LYS–PNIPAM with Mn,PNIPAM = 11.2 kg mol−1 (LYS–PNIPAM) was used as macroCTA for the RAFT polymerization of DMA. DMA (88 μL, 0.85 mmol), LYS–PNIPAM (1.67 μmol in LYS, which contained 3.55 μmol of CTA functionality, as determined by UV-Vis), and PB (pH 6.0, 3.6 mL) were sealed in a 4 mL glass vial equipped with a magnetic stir bar. The resulting solution was purged with nitrogen for 30 min in an ice bath. A nitrogen-purged solution of VA-044 (2.23 mg, 6.89 μmol) in PB (pH 6.0, 0.2 mL) was added by syringe, and the reaction vial was placed in a preheated reaction block at 25 °C. Samples were removed periodically by nitrogen-purged syringe to monitor monomer conversion by 1H NMR spectroscopy. The polymerization was quenched after 6.5 h (79% conversion) by opening the vial to air and cooling the reaction in an ice bath. The reaction mixture was dialyzed against DI water (3 × 20 L) for 15 h using a membrane of MWCO = 10 kDa. The resulting LYS–PNIPAM-b-PDMA was isolated by lyophilization.Cleavage of grafted polymer by protein decomposition
PNIPAM and PNIPAM-b-PDMA were cleaved from their respective LYS conjugates by protein decomposition in the presence of Tergazyme. A typical procedure was as follows. LYS–PNIPAM (∼0.17 μmol) was dissolved in 360 μL of PB buffer (0.1 M, pH 7.5). An aliquot of a Tergazyme stock solution (40 μL, 1 mg mL−1 in PB buffer) was added, and the solutions were stirred at room temperature for 2 days. For the reactions involving the cleavage of PNIPAM from the LYS–PNIPAM conjugates, the reaction solution was then saturated with NaCl, and PNIPAM was extracted with an equal volume of ethyl acetate. The resulting organic phase was divided equally into two scintillation vials and the solvent was evaporated. During the cleavage of PNIPAM-b-PDMA from LYS–PNIPAM-b-PDMA, the conjugate/Tergazyme solution was dialyzed (MWCO = 10 kDa) for 24 h, separated into two scintillation vials, and lyophilized. For both of the methods described above, DMF was added to one of the vials for SEC analysis and DI water was added to the other vial for SDS-PAGE analysis. The SEC results are shown in the main text. No protein band was observed during SDS-PAGE, indicating the successful decomposition of LYS (Fig. S3†). As a control experiment, a PNIPAM homopolymer and PNIPAM-b-PDMA block copolymer were treated with Tergazyme under the identical conditions as described above. No noticeable difference was observed in the SEC traces and NMR spectra of the polymer before and after incubation with Tergazyme (ESI†).Cloud point measurements of LYS–PNIPAM and LYS–PNIPAM-b-PDMA
After incubating aqueous solutions (0.11 mM) of the conjugates at 15 °C to ensure complete dissolution, turbidity measurements were monitored by recording solution absorbance at 600 nm. The temperature was gradually raised from 24 to 48 °C by increments of 1 °C with 5 min equilibration time at each increment. The cloud point was defined as the temperature at 10% of the maximum absorbance.Results and discussion
A RAFT chain transfer agent containing an N-hydroxysuccinimide activated ester was reacted at room temperature with LYS in phosphate buffer at pH 8.0 (Scheme 1). After removal of the unreacted CTA by filtration and extensive dialysis, the trithiocarbonate content of the resulting LYS–macroCTA was determined by UV-Vis spectroscopy using the thiocarbonylthio extinction coefficient at 310 nm (Fig. S1, ESI†). NIPAM was polymerized in PB (pH 6.0) at 25 °C in the presence of the LYS–macroCTA ([NIPAM]/[LYS–macroCTA]/[initiator] = 219/1/1.3). A relatively high concentration of initiator was required because of the long half-life of VA-044 at 25 °C.7SDS-PAGE indicated the molecular weight of the LYS–PNIPAM conjugates increased with conversion (Fig. 1A). A small amount of unreacted LYS and LYS dimer (∼28 kDa) was present throughout the polymerization but was significantly reduced by subsequent dialysis (Fig. 2A, lane 1). Alternatively, the temperature-responsive nature of PNIPAM allowed purification by selective precipitation of the LYS–PNIPAM from aqueous solution (conc. = 10 mg mL−1) upon heating to 40 °C for 5 min.7,16 While LYS contains seven primary amines, careful control of pH during reactions with activated esters leads to amine protonation and reduced nucleophilicity, allowing the degree of functionalization to be limited.35–37UV-Vis analysis of the isolated homopolymer conjugates indicated that an average of 2.1 polymer chains were conjugated to each LYS.![Results from the aqueous homopolymerization of NIPAM with a LYS–macroCTA. (A) SDS-PAGE results as a function of time (Lanes 1–6: t = 0, 1.9, 2.6, 3.5, 4.5, and 6.9 h, respectively). (B) Mn and Mw/Mnvs. monomer conversion for cleaved PNIPAM. (C) SEC traces of cleaved PNIPAM [numbers correspond to lane labels in (A)].](/image/article/2011/PY/c1py00031d/c1py00031d-f1.gif) | ||
| Fig. 1 Results from the aqueous homopolymerization of NIPAM with a LYS–macroCTA. (A) SDS-PAGE results as a function of time (Lanes 1–6: t = 0, 1.9, 2.6, 3.5, 4.5, and 6.9 h, respectively). (B) Mn and Mw/Mnvs. monomer conversion for cleaved PNIPAM. (C) SEC traces of cleaved PNIPAM [numbers correspond to lane labels in (A)]. | ||
![(A) SDS-PAGE from the polymerization of DMA with a LYS–PNIPAM macroCTA (Lanes 1–5: t = 0, 1.7, 2.6, 3.8, and 6.5 h, respectively). (B) SEC traces of cleaved PNIPAM-b-PDMA as a function of time [numbers correspond to lane labels in (A)]](/image/article/2011/PY/c1py00031d/c1py00031d-f2.gif) | ||
| Fig. 2 (A) SDS-PAGE from the polymerization of DMA with a LYS–PNIPAM macroCTA (Lanes 1–5: t = 0, 1.7, 2.6, 3.8, and 6.5 h, respectively). (B) SEC traces of cleaved PNIPAM-b-PDMA as a function of time [numbers correspond to lane labels in (A)] | ||
To gain further insight into the controlled nature of the polymerization, conjugate samples taken at various points during the polymerization were treated with Tergazyme, which led to protein decomposition and retention of the PNIPAM chains, as determined by SDS-PAGE and NMR spectroscopy, respectively. After dialysis to remove low molecular weight (poly)peptide fragments, SEC analysis verified that the number average molecular weight (Mn) of the “cleaved” polymer increased linearly with conversion and was in good agreement with theoretical values, as expected for the RAFT process (Fig. 1B). Additionally, the molecular weight distributions of the cleaved chains were narrow and unimodal, with polydispersity indices (Mw/Mn) remaining less than 1.32 over the conversion range considered (Table 1).
| Entrya | Timeb/h | Conv.c (%) | Cleaved polymer | ||
|---|---|---|---|---|---|
| M n,theory d/kg mol−1 | M n e/kg mol−1 | M w/Mne | |||
| a Conjugate from which each polymer was cleaved. b Polymerization time. c Determined by NMR spectroscopy. d Theoretical Mn e Determined by SEC. | |||||
| LYS–macroCTA | 0 | 0 | — | — | — |
| LYS–PNIPAM | 1.9 | 20 | 5.0 | 7.5 | 1.31 |
| LYS–PNIPAM | 2.6 | 31 | 7.7 | 8.9 | 1.32 |
| LYS–PNIPAM | 3.5 | 47 | 11.6 | 13.9 | 1.32 |
| LYS–PNIPAM | 4.5 | 68 | 16.8 | 16.9 | 1.32 |
| LYS–PNIPAM | 6.9 | 83 | 20.5 | 19.6 | 1.31 |
| LYS–PNIPAM macroCTA | 0 | 0 | 11.2 | 11.2 | 1.30 |
| LYS–PNIPAM-b-PDMA | 1.7 | 26 | 17.4 | 18.6 | 1.33 |
| LYS–PNIPAM-b-PDMA | 2.6 | 46 | 22.1 | 23.8 | 1.35 |
| LYS–PNIPAM-b-PDMA | 3.8 | 59 | 25.2 | 29.2 | 1.21 |
| LYS–PNIPAM-b-PDMA | 6.5 | 79 | 30.0 | 35.7 | 1.31 |
While we previously demonstrated grafting-from a protein by RAFT is a viable means to prepare homopolymer–protein conjugates,23 the possibility of CTA degradation by hydrolysis, aminolysis, etc. suggests retention of the end group should be unambiguously confirmed. In this case, end group retention after the polymerization of NIPAM with the LYS–macroCTA was demonstrated by chain extension with DMA. LYS–PNIPAM with a molecular weight of 25.5 kg mol−1 (14.3 kg mol−1 LYS + 11.2 kg mol−1 PNIPAM) was employed as a macroCTA for the RAFT polymerization of DMA in PB (pH 6.0) at 25 °C ([DMA]/[LYS–PNIPAM macroCTA]/[VA-044] = 240/1/1.9). SDS-PAGE analysis indicated an increase in the apparent molecular weight of the conjugate over the course of the block copolymerization (Fig. 2A). SEC analysis of residual PNIPAM-b-PDMA obtained by LYS decomposition demonstrated a clear increase in molecular weight of the cleaved chains over the course of the block copolymerization (Fig. 2B). A small high molecular weight shoulder was detected, potentially arising from bimolecular coupling during polymerization or incomplete protein decomposition, though the results indicate Mw/Mn remained less than 1.35 throughout the polymerization (Table 1).
Preliminary thermoresponsive solution behavior of the homopolymer and block copolymer conjugates was also investigated. Turbidity measurements indicated the cloud point of the conjugates increased from 34–36 °C for LYS–PNIPAM to 38–40 °C for LYS–PNIPAM-b-PDMA (Fig. 3). Therefore, while the increased hydrophilicity of the PDMA block increased the cloud point of the conjugates, it did not prevent intermolecular aggregation directed by the internal thermoresponsive PNIPAM segment. In fact at a concentration of 0.11 mM, precipitation and sedimentation of the block copolymer conjugates were observed at high temperatures, as evidenced by the apparent decrease in turbidity at high temperatures (>43 °C) (Fig. 3B).
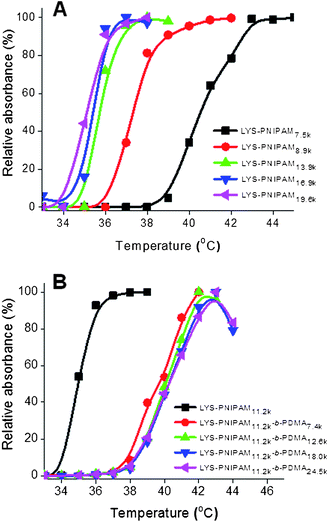 | ||
| Fig. 3 Turbidity measurements for the cloud point determination of (A) LYS–PNIPAM and (B) LYS–PNIPAM-b-PDMA conjugates. | ||
Conclusions
Grafting-from viaRAFT polymerization is possible when the initial CTA is conjugated to the proteinvia its R-group. Conveniently, this method leads to the retained thiocarbonylthio end group being readily available for subsequent chain extension during block copolymerization. However, given that a high degree of end group retention is a fundamental criterion of living polymerization and that there are many unfavorable fates the end groups may meet in an aqueous polymerization with a highly functional protein (e.g., hydrolysis, aminolysis), the significance of these results goes beyond demonstrating that this method is a viable means to prepare block copolymer–protein conjugates. Indeed, these findings indicate that grafting-from by aqueous RAFT polymerization proceeds with considerable control that rivals that observed during more typical RAFT polymerizations conducted with low molecular weight CTAs. Moreover, as compared to the alternative grafting-to approach, the reduced steric limitations of this method may substantially facilitate access to a wide variety of block copolymer bioconjugates with high molecular weight synthetic components.Acknowledgements
A portion of this work was supported by the National Science Foundation (DMR-0846792) and an Alfred P. Sloan Research Fellowship (BSS).References
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916–1922 RSC.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka, C.-W. Chang and H. D. Maynard, Biomacromolecules, 2009, 10, 2207–2212 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263–6266 CrossRef CAS.
- J. P. Magnusson, S. Bersani, S. Salmaso, C. Alexander and P. Caliceti, Bioconjugate Chem., 2010, 21, 671–678 CrossRef CAS.
- M. L. Becker, J. Liu and K. L. Wooley, Chem. Commun., 2003, 180–181 RSC.
- M. L. Becker, J. Liu and K. L. Wooley, Biomacromolecules, 2004, 6, 220–228.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854–859 RSC.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650–7656 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860–3863 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- F. D. Jochum, P. J. Roth, D. Kessler and P. Theato, Biomacromolecules, 2010, 11, 2432–2439 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477–2479 RSC.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Müller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736–2741 CrossRef CAS.
- C. L. Duvall, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Mol. Pharmacol., 2010, 7, 468–476 CrossRef CAS.
- S. Venkataraman and K. L. Wooley, Macromolecules, 2006, 39, 9661–9664 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697–4699 RSC.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- M. Li, H. Li, P. De and B. S. Sumerlin, Macromol. Rapid Commun., 2011, 32, 354–359 CrossRef CAS.
- H. Li, A. P. Bapat, M. Li and B. S. Sumerlin, Polym. Chem., 2011, 2, 323–327 RSC.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, J. Am. Chem. Soc., 2009, 131, 13908–13909 CrossRef CAS.
- L. Tao, J. Liu, J. Xu and T. P. Davis, Org. Biomol. Chem., 2009, 7, 3481–3485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1py00031d |
| This journal is © The Royal Society of Chemistry 2011 |