Tetra-alkoxy substituted PPV derivatives: a new class of highly soluble liquid crystalline conjugated polymers†
Joke
Vandenbergh
a,
Ineke
Van Severen
a,
Laurence
Lutsen
b,
Peter
Adriaensens
a,
Henk J.
Bolink
c,
Thomas J.
Cleij
*a and
Dirk
Vanderzande
ab
aHasselt University, Institute for Materials Research (IMO), Agoralaan, Building D, B-3590, Diepenbeek, Belgium. E-mail: thomas.cleij@uhasselt.be; Fax: +32-11-268301; Tel: +32-11-268310
bIMEC, Division IMOMEC, Wetenschapspark 1, B-3590, Diepenbeek, Belgium
cInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, P.O. Box 22085, 46071, Valencia, Spain
First published on 28th March 2011
Abstract
Two first examples of highly soluble tetra-alkoxy substituted PPV derivatives, poly(2,3,5,6-tetrahexyloxy-1,4-phenylene vinylene) (TH-PPV) and poly[2,3,5,6-tetra(2′-ethyl-hexyloxy)-1,4-phenylene vinylene] TEH-PPV, are presented. Both polymers have been fully characterized and the solubility and UV-Vis absorption characteristics have been studied in various organic solvents. Due to the symmetric nature of the repeating unit, TH-PPV and TEH-PPV have an inherently regioregular nature, which is unusual for PPV-type polymers. Observations from polarized light microscopy and differential scanning calorimetry indicate that TH-PPV exhibits thermally induced order resulting in a liquid crystalline mesophase. In contrast, due to steric hindrance of the branched alkyl side chains, TEH-PPV is rather disordered. As a first demonstration of the charge transporting properties of TH-PPV, a simple solution processed electroluminescent device has been prepared. The electroluminescence spectrum shows a detailed structure similar to that observed from the solid-state photoluminescence spectrum and shows one of the highest energy emissions observed for PPVs. It is speculated that the enhanced order in TH-PPV will benefit the usage of this new class of polymers in optoelectronic applications, in which the improved charge carrier mobility is desirable.
Introduction
Poly(p-phenylene vinylene) (PPV) and related polymers exhibit numerous attractive optical and electronic properties, which make them interesting candidates for applications such as organic light emitting diodes (OLEDs), organic field effect transistors (OFETs), (bio)sensors and photovoltaic cells.1–5 For the successful integration of PPV-type polymers in devices, it is often of considerable importance to obtain a well defined ordered morphology of the active layer.6–8 Potentially, this can be achieved by using polymeric materials, which exhibit ordering phenomena. However, the 2,5-substituted structure of commonly used PPV-derivatives, i.e.OC1C10–PPV and MEH–PPV, has been originally designed to minimize aggregation phenomena, which usually result in a lowering of the luminescent quantum yields in thin films. For example, while successfully optimizing the electroluminescence, only moderate charge carrier mobilities have been reported for these polymers.7 Hence, considerable interest exists in the development of novel PPV-type polymers, which display enhanced stacking and ordering phenomena. To this end, various approaches have been presented in the literature. Examples include the introduction of linear side chains,7–9 the introduction of regioregularity6,10 as well as the introduction of chiral substituents.11,12 Although indeed notable improvements have been observed for most of these modified PPV-derivatives, the quest for even further improved PPV-derivatives continues. Especially novel PPV-derivatives, which combine ordering phenomena and straightforward accessibility, are of considerable interest.Remarkably, all of the previously mentioned approaches towards novel PPV-type materials focus on 2,5-dialkoxy substituted PPV-derivatives. This is a result of the synthetic accessibility of this class of materials for the past decades.13 In contrast, thus far interest in the synthesis and optoelectronic properties of tetra-alkoxy substituted PPV derivatives has been rather limited. This is surprising, since they have an inherently regioregular nature and potentially have a favorable geometry for intra- and intermolecular ordering phenomena. Whereas soluble trialkoxyalkyl substituted PPV derivatives have been reported,14 tetra-alkoxy PPV derivatives remain mostly elusive. Hitherto, only one tetra-alkoxy PPV-derivative has been reported in the literature, i.e. poly(2,3,5,6-tetramethoxy-1,4-phenylene vinylene) (TM-PPV).15–17 This polymer contains the readily accessible tetramethoxybenzene structure. Furthermore, a number of copolymers containing this structural unit have been prepared.15,18,19 Both TM-PPV and the copolymers were found to have unusual optical and electronic properties. However, the obtained conjugated polymers were in all cases completely insoluble, which impedes not only a full characterization, but also hampers their purification and processability towards applications. Hence, our attention focused on the development of a novel and versatile route towards soluble tetra-alkoxy substituted PPV-derivatives.
Here we present such a route as well as the synthesis and full characterization of the first two examples of highly soluble tetra-alkoxy substituted PPV-derivatives, i.e. poly(2,3,5,6-tetrahexyloxy-1,4-phenylene vinylene) (TH-PPV) and poly[2,3,5,6-tetra(2′-ethyl-hexyloxy)-1,4-phenylene vinylene] (TEH-PPV). In addition, the solubility and UV-Vis absorption characteristics of TH-PPV and TEH-PPV in various solvents with different polarities have been studied. Observations from polarized light microscopy (PLM) and differential scanning calorimetry (DSC) indicate that TH-PPV exhibits thermally induced order, resulting in an ordered liquid crystalline mesophase. This confirms our assumption that the tetra-alkoxy substitution pattern indeed can give rise to desirable ordering phenomena. However, due to steric hindrance of the branched alkyl side chains, the second polymer, i.e. TEH-PPV, has a comparatively lower molecular weight and remains rather disordered. Finally, the first data on the application and optoelectronic characterization of TH-PPV in a P-LED device will be presented.
Experimental section
General
NMR spectra were recorded with a Varian Inova Spectrometer (1H-NMR 300 MHz, 13C-NMR 75 MHz). Analytical Size Exclusion Chromatography (SEC) (eluent THF, flow rate 1.0 mL min−1) was performed using a Spectra series P100 pump equipped with a pre-column and two mixed-B columns (Polymer Labs) and a Refractive Index (RI) detector (Shodex) at 40 °C. Molecular weight distributions are given relative to polystyrene standards. GC-MS data were obtained with a Varian TSQ 3400 Gas Chromatograph and a TSQ 700 Finnigan Mat mass spectrometer. Direct insertion probe pyrolysis mass spectrometry (DIP-MS) analysis was carried out on a Finnigan TSQ 70 (EI; mass range 35–500; heating rate 10 °C min−1; electron energy 70 eV). TGA analysis was carried out on a TA Instruments 951 (heating rate 20 °C min−1, Ar atmosphere). FT-IR spectra were collected with a Perkin Elmer Spectrum One spectrometer. Fluorescence spectra were collected on a Perkin Elmer LS-5B luminescence spectrometer. UV-Vis measurements were performed on a Cary 500 UV-Vis-NIR spectrophotometer (scan rate 600 nm min−1). Elemental analysis was performed with a Flash EA 1112 Series CHNS-O analyzer. DSC measurements were carried out on a DSC 910-2000 analyzer (temperature program −100 °C to 200 °C; heating rate 10 °C min−1, N2 atmosphere). The second heating curves were evaluated. Polarized Light Microscopy (PLM) observations were made with a Nikon 144040 polarization microscope equipped with a Linkam temperature-controlled stage. Thin films were drop-cast from CHCl3 solutions on glass substrates, made apolar by treatment with trimethylsilyl chloride. The thin-film quantum yield measurements were performed in a nitrogen environment and determined on a thin film (100 nm) using the quantum yield measurement system from Hamamatsu, model C9920-01, equipped with an integrated sphere.Synthesis
All chemicals were purchased from Aldrich or Acros and used without further purification. 1,4-Dioxane was distilled from sodium/benzophenone.1,2,4,5-Tetrahexyloxybenzene 1 (Scheme 1)
1,2,4,5-Tetrahydroxybenzene was generated quantitatively by in situhydrogenation of 2,5-dihydroxy-1,4-benzoquinone (5.2 g, 37 mmol) in dry DMF (1220 mL) at atmospheric pressure (pH2 = 1 atm) with PtO2 (40 mg) as a catalyst according to an adapted literature procedure.20,21 Subsequently 1-bromohexane (27.0 g, 163.0 mmol) and cesium carbonate (72.4 g, 222.3 mmol) were added. The reaction mixture was stirred for 6 days at 65 °C. After cooling to room temperature DMF was removed under reduced pressure and CH2Cl2 (600 mL) was added. The reaction mixture was washed with H2O (3 × 100 mL), after which the organic extracts were dried over MgSO4 and concentrated under reduced pressure to yield crude 1 as a brown solid, which was recrystallized twice from MeOH giving 13.9 g (78% yield) white crystalline 1.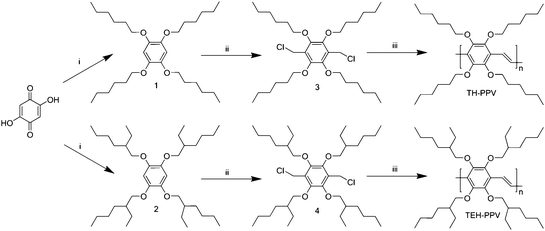 | ||
| Scheme 1 Synthesis of 1, 2, 3, 4, TH-PPV and TEH-PPV. (i): PtO2, H2(g) followed by Cs2CO3, C6H13–Br or CH3(CH2)3CH(C2H5)CH2Br; (ii): p-CH2O, Ac2O, HCl; (iii): KtBuO, 1,4-dioxane. | ||
1H NMR (300 MHz, CDCl3, δ): 6.55 (s, 2H, Ar H), 3.91 (t, J = 5.7 Hz, 8H, OCH2), 1.74 (m, 8H, CH2), 1.43 (m, 8H, CH2), 1.30 (m, 16H, CH2), 0.89 (t, J = 7.1 Hz, 12H, CH3); 13C NMR (75 MHz, CDCl3, δ): 143.4, 105.4, 70.4, 31.5, 29.4, 25.6, 22.5, 13.9; EIMS (m/z (%)): 478 [M+], 394, 309, 225, 141.
1,2,4,5-Tetra(2′-ethyl-hexyloxy)benzene 2
Using a similar procedure as described for 1, first 1,2,4,5-tetrahydroxybenzene was generated quantitatively by the in situhydrogenation of 2,5-dihydroxy-1,4-benzoquinone (5.2 g, 37 mmol). Subsequently 1-bromo-2-ethylhexane (31.5 g, 163 mmol) and cesium carbonate (72.4 g, 222.3 mmol) were added, giving after reaction workup and column chromatography (SiO2, eluent hexane/ethyl acetate 9/1) 12 g (55% yield) clear yellow oil 2.1H NMR (300 MHz, CDCl3, δ): 6.55 (s, 2H, Ar H), 3.80 (d, J = 5.7 Hz, 8H, OCH2), 1.70 (m, 4H, CH), 1.43 (m, 16H, CH2), 1.30 (m, 16H, CH2), 0.91 (t, J = 7.3 Hz, 24H, CH3); 13C NMR (75 MHz, CDCl3, δ): 143.5, 107.5, 53.5, 40.3, 30.5, 29.5, 24.0, 23.5, 14.5, 11.5; EIMS (m/z (%)): 590 [M+], 477, 363, 250, 141.
1,4-Bis-chloromethyl-2,3,5,6-tetrahexyloxybenzene 3
To a stirred mixture of 1 (5.0 g, 10.4 mmol) and p-formaldehyde (3.1 g, 104 mmol) at 0 °C, concentrated HCl (9.6 mL, 114.8 mmol) was added dropwise under N2 atmosphere. Subsequently, acetic anhydride (19.6 mL, 208.8 mmol) was added. After addition was complete, the resulting solution was stirred at 95 °C for 6 days after which it was cooled down and poured into water (100 mL). The reaction mixture was extracted with CH2Cl2 (3 × 100 mL) and dried over MgSO4. Evaporation of the solvent gave the crude product. The pure product was obtained by column chromatography (SiO2, eluent hexane/ethyl acetate 95/5) as a white solid (4.1 g, 68% yield).1H NMR (300 MHz, CDCl3, δ): 4.65 (s, 4H, CH2Cl), 4.02 (t, J = 6.6 Hz, 8H, OCH2), 1.78 (m, 8H, CH2), 1.48 (m, 8H, CH2), 1.30 (m, 16H, CH2), 0.89 (t, J = 7.1 Hz, 12H, CH3); 13C NMR (75 MHz, CDCl3, δ): 147.0, 126.9, 73.8, 35.6, 31.5, 30.1, 25.5, 22.5, 13.9; EIMS (m/z (%)): 574 [M+], 539 [M+ − Cl], 492 [M+ − 2Cl–CH2], 456, 370.
1,4-Bis-chloromethyl-2,3,5,6-tetra(2′-ethyl-hexyloxy)benzene 4
Using a similar procedure as described for 3, compound 4 was prepared from 2 (12 g, 20.3 mmol), p-formaldehyde (6.1 g, 203 mmol), concentrated HCl (18.3 mL, 223 mmol) and acetic anhydride (38.4 mL, 406 mmol). After column chromatography (SiO2, eluent hexane/ethyl acetate 99/1), 4 was obtained as a clear colorless oil (4.6 g, 33% yield).1H NMR (300 MHz, CDCl3, δ): 4.65 (s, 4H, CH2Cl), 3.90 (d, J = 6.6 Hz, 8H, OCH2), 1.80 (m, 4H, CH), 1.50 (m, 8H, CH2), 1.42 (m, 8H, CH2), 1.30 (m, 16H, CH2), 0.89 (t, J = 7.3 Hz, 24H, CH3); 13C NMR (75 MHz, CDCl3, δ): 147.5, 127.5, 53.5, 40.3, 36.0, 30.5, 29.5, 24.0, 23.5, 14.5, 11.5; EIMS (m/z (%)): 687 [M+], 574, 462, 350, 238, 202.
Poly(2,3,5,6-tetrahexyloxy-1,4-phenylene vinylene) (TH-PPV)
1.5 g (3.1 mmol) of 2 was dissolved in dry 1,4-dioxane (200 mL) under N2 atmosphere. After heating to 96 °C, a solution of KtBuO (0.91 g, 8.1 mmol) in dry 1,4-dioxane (9 mL) was added dropwise while stirring. During this addition, the solution turned from colorless to white. After 5 min, a second solution of KtBuO (0.70 g, 6.2 mmol) in dry 1,4-dioxane (7 mL) was added at which the solution turned yellow. The polymerization was allowed to proceed for 3 h at 98 °C after which the total volume was reduced to 20 mL by evaporation and the solution was precipitated dropwise in cold MeOH (200 mL). The resulting polymer was filtered off, washed with MeOH and dried under reduced pressure at room temperature giving TH-PPV as a yellow solid (702 mg, 55% yield).1H NMR (300 MHz, CDCl3, δ): 7.8 (br d, 2H, CHCH), 3.9 (br t, 8H, OCH2), 1.7 (br m, 8H, CH2), 1.4 (br m, 8H, CH2), 1.3 (br m, 16H, CH2), 0.8 (br t, 12H, CH3); 13C NMR (75 MHz, CDCl3, δ): 147.3, 126.2, 125.4, 73.3, 31.7, 30.3, 25.7, 22.5, 13.9; FT-IR (NaCl): 2955, 2859, 1445, 1378, 1301, 1266, 1121, 726 cm−1; UV-Vis (CHCl3) λmax = 430 nm and (thin film) λmax = 442 nm; SEC (THF) Mw = 4 × 105 g mol−1 (PD = Mw/Mn = 3.1) (73.4%) + 5 × 103 g mol−1 (PD = Mw/Mn = 1.3) (26.6%); Anal. calcd for (C32H54O4)n: C 76.5, H 10.8, O 12.7; found C 76.7, H 11.4, O 11.9%.
Poly[2,3,5,6-tetra(2′-ethyl-hexyloxy)-1,4-phenylene vinylene] (TEH-PPV)
Using a similar procedure as described for TH-PPV, TEH-PPV was prepared from 0.5 g (0.73 mmol) of 4 and KtBuO in two batches (total amount 0.382 g, 3.34 mmol). After polymerization and purification TEH-PPV was isolated as a yellow sticky oil (260 mg, 58% yield).1H NMR (300 MHz, CDCl3, δ): 7.8 (br d, 2H, CHCH), 3.7 (br d, 8H, OCH2), 1.7 (br m, 4H, CH), 1.4 (br m, 16H, CH2), 1.3 (br m, 16H, CH2), 0.8 (br t, 24H, CH3); 13C NMR (75 MHz, CDCl3, δ): 147.3, 127.2, 125.4, 53.5, 40.2, 30.7, 29.3, 23.7, 13.9, 11.1; FT-IR (NaCl): 2960, 2929, 2873, 2858, 1445, 1378, 1295, 1260, 1117, 1046, 730 cm−1; UV-Vis (CHCl3): λmax = 370 nm and (thin film) λmax = 372 nm; SEC (THF) Mw = 2.2 × 104 g mol−1 (PD = Mw/Mn = 2.5) (64.9%) + 1 × 103 g mol−1 (PD = Mw/Mn = 1.1) (35.1%); Anal. calcd for (C40H70O4)n: C 78.1, H 11.5, O 10.4; found C 77.2, H 11.7, O 11.1%.
Device preparation and characterization
Poly(3,4-ethylenedioxythiophene):polystyrenesulfonate (PEDOT:PSS) was purchased from HC-Starck and solvents used were obtained from Aldrich. Indium tin oxide (ITO)-coated glass plates obtained from Naranjo substrates were patterned using conventional photolithography. The substrates were extensively cleaned using sonification in subsequently water-soap, water and 2-propanol baths. After drying, the substrates were placed in a UV-ozone cleaner for 20 minutes. The electroluminescent devices were prepared as follows. Transparent thin films of TH-PPV were obtained by spinning from toluene solutions using concentrations of 20 mg mL−1 at 2000 rpm for 40 seconds, resulting in a 110 nm thick film. Prior to the deposition of the emitting layer a 100 nm layer of PEDOT:PSS was deposited to increase the device preparation yield. The thickness of the films was determined using an Ambios XP1 profilometer. After spinning the organic layers, the samples were transferred to an inert atmosphere glove box (<0.1 ppm O2 and H2O, MBraun) and dried on a hot plate at 80 °C for 1 hour. Barium and silver metal electrodes (5 and 80 nm, respectively) were thermally evaporated using a shadow mask under a vacuum (<1 × 10−6 mbar) using an Edwards Auto500 evaporator integrated into an inert atmosphere glove box. Current density and luminance versus voltage were measured using a Keithley 2400 sourcemeter and a photodiode coupled to a Keithley 6485 pico-amperometer using a Minolta LS100 to calibrate the photocurrent. External quantum efficiencies (EQEs) were determined using an integrated sphere coupled to an UDT instruments S370 Optometer.Results and discussion
Monomer and polymer synthesis
Key to the preparation of tetra-alkoxy substituted PPV-type polymers is a straightforward method to obtain the corresponding benzene derivatives in good yield. Previously, for the studies on TM-PPV the corresponding benzene derivative was prepared from benzoquinone in three steps.22 However, this procedure gave unsatisfactory yields when attempted with larger alkoxy substituents. Fortunately, an elegant method has been presented in the literature to prepare benzenes with 2,3,5,6-substitution patterns.20,21 Using this method, the synthesis of 1,2,4,5-tetrahexyloxybenzene 1 and 1,2,4,5-tetra(2′-ethyl-hexyloxy)benzene 2 has been accomplished in a satisfactory yield (78% and 55%, respectively) via a Williamson ether synthesis of 1,2,4,5-tetrahydroxybenzene obtained in situ by hydrogenation of 2,5-dihydroxybenzene-1,4-benzoquinone (Scheme 1). Subsequently, 1 and 2 have been chloromethylated according to an adapted literature procedure23 using concentrated HCl and formaldehyde in acetic anhydride to give 3 and 4. Whereas for 2,5-subsituted benzenes this reaction usually proceeds within hours at circa 60 °C, the preparation of 3 and 4 requires more elevated temperatures (95 °C) and extended reaction times (6 days) to achieve a reasonable yield (68% and 33%, respectively). At lower temperatures and shorter reaction times only mono-chloromethylated compounds could be identified. The reduced reaction rates presumably are a result of steric hindrance of the neighboring alkyl chains. It should be noted that the yields of 2 and 4 are significantly lower than the yields observed for 1 and 3. This difference is a result of a difference in steric hindrance between the linear alkyl substituents present in 1 and 3 and the more sterically demanding branched alkyl substituents present in 2 and 4.Monomers 3 and 4 have been polymerized according to the Gilch procedure (dehydrohalogenation polymerization), which is one of the common methods to prepare PPV-type polymers.24,25 TH-PPV has been obtained in a yield of 55% as a yellow solid. In contrast, TEH-PPV was obtained as a yellow oil, albeit in a similar yield (58%). Both TH-PPV and TEH-PPV have been fully characterized using different analytical techniques. NMR-spectroscopy confirms that, as expected, these tetra-substituted PPV-derivatives have an inherently regioregular nature. Analytical SEC measurements of TH-PPV display a bimodal character, with both high molecular weight (Mw = 4 × 105 g mol−1; 73.4%) and low molecular weight polymers (Mw = 5 × 103 g mol−1; 26.6%) being present in the mixture. The same observation can be made for TEH-PPV, although the observed molecular weights are significantly lower, i.e. high molecular weight Mw = 2.2 × 104 g mol−1 (64.9%) and low molecular weight Mw = 1 × 103 g mol−1 (35.1%). To our knowledge, this is the first time that such a bimodal character of the molecular weight distribution is observed for a Gilch polymerization. Previously reported mechanistic studies have demonstrated that the main propagation mechanism for the Gilch route is a self-initiating radical polymerization, usually leading to a very high molecular weight material.26–32 However, the simultaneous occurrence of an anionic mechanism cannot be fully excluded. In other precursor routes, a competition between a radical and an anionic mechanism has already been observed.33–35 This competition leads to a bimodal molecular weight distribution in which typically the high molecular weight material is associated with the self-initiating radical mechanism and the low molecular weight material is produced via an anionic mechanism. Hence, the low molecular weight material formed during the Gilch polymerization of TH-PPV and TEH-PPV may well originate from an anionic polymerization mechanism. Apparently, tetra-substitution and thus increased steric hindrance results in an increased competition between the normal radical mechanism and the anionic mechanism. In view of this, it can be expected that the molecular weight distributions can be improved by further optimization of the polymerization conditions.
Optical characterization
The solubility and UV-Vis absorption characteristics of TH-PPV and TEH-PPV in various solvents with different polarities have been studied. The polymers are readily soluble in a variety of solvents ranging from apolar solvents, such as n-hexane and toluene, to solvents of medium polarity, such as chloroform and dichloromethane. It is noteworthy that TH-PPV and TEH-PPV are even soluble in cyclohexane, a very apolar solvent in which most other available PPV derivatives are insoluble. The UV-Vis absorption spectrum of TH-PPV and TEH-PPV exhibits typical absorption bands associated with the π–π* transition, both in thin film and solution (Fig. 1). To study the influence of the bimodal weight distribution on the optical properties, the high and low molecular weight fractions of the TH-PPV were separated using preparative size exclusion chromatography on Bio-Beads® (S-X1) with toluene as the solvent. Subsequently, the optical characterization of TH-PPV was executed both before and after separation and identical results were obtained. The solution UV-Vis absorption maximum λmax of TH-PPV is virtually independent of the solvent used and is positioned at 429 nm. The λmax of TH-PPV in a thin film is slightly red-shifted and positioned at 442 nm. Both values are somewhat red-shifted in comparison to the previously reported TM-PPV derivatives.17,18 This is probably a result of the enhanced purity of TH-PPV due to its improved solubility, which allows for a careful purification of all intermediates as well as the conjugated polymer itself. In contrast, for TEH-PPV, both in solution and in a thin film, a broad absorption band is found between 300 and 400 nm, which is composed of several overlapping smaller bands. Apparently, the backbone of TEH-PPV is considerably distorted as compared to TH-PPV, which results in a distribution of polymer segments with a strongly reduced effective conjugated length. This is not surprising since all observed λmax values for all tetra-substituted polymers are significantly blue shifted as compared to OC1C10–PPV, which exhibits a λmax in the 480–520 nm range, with the exact position being dependent on the preparation procedure.36–38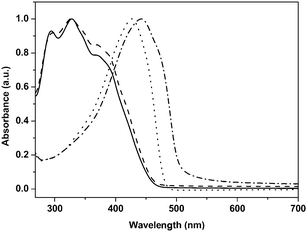 | ||
| Fig. 1 UV-Vis absorption spectra of TH-PPV (solution dotted; thin film dash-dotted) and TEH-PPV (solution solid; thin film dashed). | ||
The blue shifts of the absorption maxima can be attributed to the steric hindrance between the side chains of both TH-PPV and TEH-PPV. This causes a more “twisted” conformation of the polymer backbone, which would directly affect the band gap and thus the λmax. The presence of this effect was previously predicted in a quantum-chemical investigation on the conformations and electronic properties of a variety of methoxy-substituted PPV-derivatives.39 According to these studies, strong steric repulsions between adjacent sites in the phenylene ring lead to rotation of the substituents around the C(sp2)–O bonds and consequently the overlap between the π-type p-orbital of oxygen and the π molecular orbital of the polymer decreases. This effect leads to a wider band gap for tetra-methoxy substituted PPV derivatives, as compared to their dialkoxy-substituted counterparts. However, it should be noted that by using other precursor routes, a different λmax may be found for these polymers, since it is well known that the λmax of PPV-type polymers is dependent on the polymerization route utilized.38,40 TH-PPV and TEH-PPV have also been studied using fluorescence spectroscopy (Fig. 2). In a thin film, TH-PPV exhibits a distinct fluorescence emission at λem = 495 nm with a shoulder around 530 nm (excitation wavelength λex = 442 nm). In dilute solution, a similar λem = 488 nm and shoulder around 519 nm are found (λex = 430 nm). Whereas the shape is similar to OC1C10–PPV, also in this case the maxima are significantly blue shifted, as was observed for the absorption characteristics. This corroborates the hypothesis of a wider band gap due to stronger distortion of the effective conjugation length in the polymer chain. For TEH-PPV, in thin film the fluorescence emission maximum is positioned at λem = 462 nm with a shoulder around 490 nm (excitation wavelength λex = 370 nm). In dilute solution, a similar λem = 460 nm is found (λex = 370 nm). This confirms the reduced conjugation length of TEH-PPV as compared to TH-PPV. In view of this observation, further characterization and analysis, as described in the following sections, have focused on TH-PPV.
 | ||
| Fig. 2 Photoluminescence excitation (PLE) and emission (PL) spectra of thin films of TH-PPV (PLE for emission at 495 nm dash-dotted; PL with excitation at 442 nm dotted) and TEH-PPV (PLE for emission at 460 nm dashed; PL with excitation at 370 nm solid). | ||
Thermal stability
The impact of tetra-substitution on the thermal stability of the PPV-derivative is of considerable interest for application of this type of materials in optoelectronics. Thermogravimetric analysis (TGA) indicates that for TH-PPV polymer degradation commences above 250 °C, with the derivative of weight loss to the temperature exhibiting one maximum at 378 °C. A similar observation has been made with direct insertion probe pyrolysis mass spectrometry (DIP-MS). The total reconstructed ion chromatogram shows an increasing current starting around 250 °C with a peak at 293 °C related to polymer degradation. It should be noted that both TGA and DIP-MS give information on the thermal stability of the polymer, but do not monitor the actual degradation of the conjugated system itself. Therefore, the thermal stability of specifically the conjugated system has been specifically probed using a third technique, i.e. in situUV-Vis spectroscopy. Heating the TH-PPV to temperatures up to 220 °C results in a significant thermochromic shift. Upon cooling, the room temperature UV-Vis spectrum is fully recovered, confirming the reversibility of this process. However, heating at temperatures of 240 °C and higher results in an irreversible blue-shift of λmax, indicating that the degradation of the conjugated system starts between 220 °C and 240 °C. This thermal stability is in a similar order of magnitude as was observed for OC1C10–PPV and indicates that the thermal stability of our novel tetra-substituted material is sufficient for optoelectronic applications.38Phase behavior
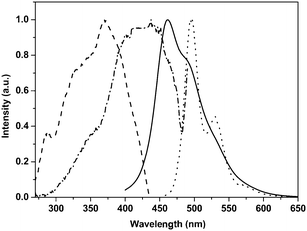
The behavior of TH-PPV upon thermal treatment has been studied in more detail with DSC and PLM. For the PLM studies, TH-PPV has been drop-cast from a CHCl3 solution onto a trimethylsilyl chloride treated glass substrate. The obtained film is only weakly birefringent under cross-polarization. However, upon heating to temperatures above 100 °C, followed by cooling, nematic-like optical textures become more apparent (Fig. 3). A similar observation has been previously made for 2,5-subsituted PPV-derivatives with linear alkoxy side chains.9 During the subsequent thermal cycling between room temperature and 200 °C at a heating/cooling rate of 10 °C min−1, little variation in texture is observed. The birefringence weakens above 200 °C, due to isotropization, but is fully recovered upon subsequent cooling. Above 220 °C degradation phenomena are observed, which are in line with the observations on the thermal stability of the conjugated system (vide supra). | ||
| Fig. 3 PLM of a thin film of TH-PPV at RT after thermal treatment (Magnification: 60 P, cross-polarization). | ||
Mesophase formation is further evident from DSC measurements (Fig. 4). The lower curve corresponds to a heating process and shows double “melting endotherms” (Tm = 51 °C and 89 °C). These endotherms are associated with order–disorder transitions of the side chains. The subsequent cooling process displays a distinct “crystallization” peak at Tc = 72 °C. In contrast, DSC measurements on TEH-PPV do not indicate the presence of any thermal transitions at temperatures as high as 200 °C. Apparently, the branched side chains hamper the formation of an organized phase. Because the monomer of TH-PPV does not display liquid crystalline behavior, it can be assumed that the observed mesophase originates from the rod-like nature of the main chain of TH-PPV. The formation of a mesophase in TH-PPV is a direct indication for the enhanced structural order of this material. Such an enhanced structural order is a desirable property, since often concomitantly an increase in charge carrier mobility is observed. Furthermore, it can be anticipated that by altering the length of the alkoxy side chains, the mesogenic properties of the polymer (layer spacing, packing of the side chains) and as a result, the optoelectronic properties can be controlled and tuned.
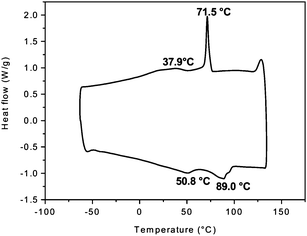 | ||
| Fig. 4 DSC curve of the heating (bottom) and cooling (top) of TH-PPV between −60 and 135 °C. The second heating/cooling cycle is shown. | ||
Device characterization
Many PPV-type polymers have been studied on their performance in organic light-emitting diodes (OLEDs) due to their very good PL efficiencies and ambipolar charge transport. With TH-PPV now being readily available as a tetra-substituted representative with a high purity, this compound was chosen for further device studies. To verify the performance of the TH-PPV in an OLED, a simple two layer solution processed OLED was prepared (cf. Experimental section for details). Due to the soft character of the TH-PPV, the determination of the active layer could not be directly obtained with a profilometer. As an alternative, first a 50 nm small molecular layer of (1,3,5-tris(2-N-phenylbenzimidazolyl) benzene (TPBI) was evaporated on top of the TH-PPV layer and the total thickness of the double layer was determined. Subsequently, by subtracting the layer thickness of the TPBI layer, the thickness of the TH-PPV layer was obtained. The layer thickness of the TH-PPV layer (110 nm) was slightly more than optimal, but was needed to obtain working devices. It can be assumed that the low Tg of the TH-PPV allows for some penetration of the metal cathode into the active layer, which results in the formation of weak spots that eventually lead to the formation of short circuits.The current density and the luminance versus applied voltage are depicted in Fig. 5. The current density rises at voltages slightly higher than what would be expected for conventional PPV based OLEDs. A part of this is due to the thicker films. Upon injection, diode behavior is clearly observed as the current density rises rapidly. It rises continuously with increasing voltage levels. The luminance turns on at similar voltages indicating that the charge injection is balanced. The luminance, however, does level off at moderate values around 100 cd m−2. This indicates that there is a dominant charge carrier being transported through the TH-PPV as the current density appears to be space charge limited. Further analysis is required to determine the nature of the majority carrier and how its mobility compares with other PPVs. This is of interest since on the one hand the TH-PPV is regioregular, which should increase the solid state ordering and hence increase the carrier mobility. However, on the other hand the conjugation is limited by the out of plane distortion due to steric hindrance, which is expected to decrease the carrier mobility.41 The electroluminescence (EL) observed from the above described device is almost identical to the luminance spectrum obtained by photoexcitation (PL) (Fig. 6). Both are very similar to the photoluminescence spectra obtained from the TH-PPV in solution. The EL spectrum starts at slightly lower wavelength when compared to the PL spectrum, which may be due to the slightly increased self-absorption for the photoexcitation spectrum.
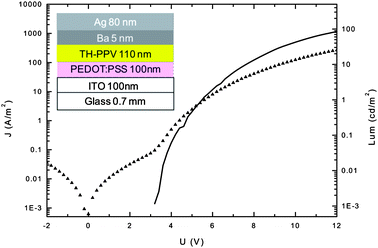 | ||
| Fig. 5 Current density (triangles) and luminance (solid) for a ITO/PEDOT:PSS/TH-PPV/Ba/Ag device. | ||
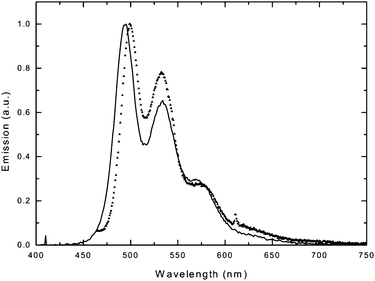 | ||
| Fig. 6 Photoluminescence (triangles) and electroluminescence (solid) spectra obtained from a ITO/PEDOT:PSS/TH-PPV/Ba/Ag device by photoexcitation at 400 nm or electroexcitation at 7 volts, respectively. | ||
Conclusions
The synthesis and full characterization of the first two soluble tetra-alkoxy substituted PPV derivatives, poly(2,3,5,6-tetrahexyloxy-1,4-phenylene vinylene) TH-PPV and poly[2,3,5,6-tetra(2′-ethyl-hexyloxy)-1,4-phenylene vinylene] TEH-PPV, is reported. The monomer synthesis is straightforward and feasible in high yields. The polymerization is accomplished using the well-known dehydrohalogenation route. The resulting polymers are highly soluble in various organic solvents ranging from solvents at the apolar side of the polarity scale, such as cyclohexane, to the more common organic solvents, such as dichloromethane. Furthermore, the conjugated structure has a thermal stability until well above 200 °C. TH-PPV exhibits desirable ordering phenomena as well as a liquid crystalline mesophase. In contrast, due to steric hindrance of the branched alkyl side chains, TEH-PPV is rather disordered. As a first demonstration of the charge transporting properties of TH-PPV, a simple solution processed electroluminescent device has been prepared, which functions well. With the described route, soluble tetra-alkoxy substituted PPV-derivatives are now readily available for further study and application.Acknowledgements
The authors thank H. Penxten for the PL and PLE measurements and Sonsoles Garcia Santamaria for the careful device preparation. The authors gratefully acknowledge the Fund for Scientific Research (FWO) for granting a PhD fellowship (I.V.S. and J.V.) and for the funding of the project G.0091.07, the Institute for the promotion of Innovation by Science and Technology in Flanders (IWT, SBO-project “Polyspec” IWT-060843), the Spanish Ministry of Science and Innovation (MICINN) and CSD2007-00010) and the Generalitat Valenciana. Furthermore, this work is a part of the European Science Foundation EUROCORES Program SONS II (CRP no. 05-SONS-FP-021 “SOHYDs”) that is supported by funds from the FWO (G.068506), and by MICINN (MAT2006-28185-E) and the EC Sixth Framework Program, under Contract no. ERAS-CT-2003-980409.References
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- S. Scheinert and G. Paasch, Phys. Status Solidi A, 2004, 201, 1263 CrossRef CAS.
- D. Tyler McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS.
- H. Hoppe, M. Niggemann, C. Winder, J. Kraut, R. Hiesgen, A. Hinsch, D. Meissner and N. S. Sariciftci, Adv. Funct. Mater., 2004, 14, 1005 CrossRef CAS.
- A. J. Mozer, P. Denk, M. C. Scharber, H. Neugebauer, N. S. Sariciftci, P. Wagner, L. Lutsen and D. Vanderzande, J. Phys. Chem. B, 2004, 108, 5235 CrossRef CAS.
- A. J. J. M. van Breemen, P. T. Herwig, C. H. T. Chlon, J. Sweelssen, H. F. M. Schoo, E. M. Benito, D. M. de Leeuw, C. Tanase, J. Wildeman and P. W. M. Blom, Adv. Funct. Mater., 2005, 872 CrossRef CAS.
- L. Breban, L. Lutsen, G. Vanhoyland, J. D'Haen, J. Manca and D. Vanderzande, Thin Solid Films, 2006, 511–512, 695 CrossRef CAS.
- S. H. Chen, A. C. Su, S. R. Han, S. A. Chen and Y. Z. Lee, Macromolecules, 2004, 37, 181 CrossRef CAS.
- K. Tajima, Y. Suzuki and K. Hashimoto, J. Phys. Chem. C, 2008, 112, 8507 CrossRef CAS.
- E. Peeters, A. Delmotte, R. A. J. Janssen and E. W. Meijer, Adv. Mater., 1997, 9, 493 CrossRef CAS.
- M. Kemerink, J. K. J. van Duren, P. Jonkheijm, W. F. Pasveer, P. M. Koenraad, R. A. J. Janssen, H. W. M. Salemink and J. H. Wolters, Nano Lett., 2003, 3, 1191 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474 CrossRef CAS.
- J.-H. Lee and D.-H. Hwang, Synth. Met., 2008, 158, 273 CrossRef CAS.
- J.-I. Jin, C.-K. Park and H.-K. Shim, Macromolecules, 1993, 26, 1799 CrossRef CAS.
- O. Levi, G. Perepelitsa, D. Davidov, S. Shalom, I. Benjamin, R. Neumann, A. J. Agranat and Y. Avny, J. Appl. Phys., 2000, 88, 1236 CrossRef CAS.
- A. Donval, D. Josse, G. Kranzelbinder, R. Hierle, E. Toussaere, J. Zyss, G. Perpelitsa, O. Levi, D. Davidov, I. Bar-Nahum and R. Nemann, Synth. Met., 2001, 124, 59 CrossRef CAS.
- J.-I. Jin, H.-J. Kang and H.-K. Shim, Bull. Korean Chem. Soc., 1990, 11, 415 CAS.
- S.-J. Chung, D. W. Lee, D.-K. Oh, C. E. Lee and J.-I. Jin, Acta Polym., 1999, 50, 298 CrossRef CAS.
- E. M. D. Keegstra, B.-H. Huisman, E. M. Paardekooper, F. J. Hoogesteger, J. W. Zwikker, L. W. Jenneskens, H. Kooijman, A. Schouten, N. Veldman and A. L. Spek, J. Chem. Soc., Perkin Trans. 1, 1996, 229 RSC.
- J. J. H. Schlotter, I. J. A. Mertens, A. M. A. van Wageningen, F. P. J. Mulders, J. W. Zwikker, H.-J. Buschmann and L. W. Jenneskens, Tetrahedron Lett., 1994, 35, 7255 CrossRef CAS.
- F. Benington, R. D. Morin and L. C. Clark, J. Org. Chem., 1955, 20, 102 CrossRef CAS.
- I. Van Severen, F. Motmans, L. Lutsen, T. J. Cleij and D. Vanderzande, Polymer, 2005, 46, 5466 CrossRef CAS.
- H. G. Gilch and W. L. Wheelwright, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 1337 CrossRef CAS.
- H. Becker, H. Spreitzer, K. Ibrom and W. Kreuder, Macromolecules, 1999, 32, 4925 CrossRef CAS.
- T. Schwalm, J. Wiesecke, S. Immel and M. Rehahn, Macromolecules, 2007, 40, 8842 CrossRef CAS.
- J. Wiesecke and M. Rehahn, Angew. Chem., Int. Ed., 2003, 42, 567 CrossRef CAS.
- J. Wiesecke and M. Rehahn, Macromol. Rapid Commun., 2007, 28, 78 CrossRef CAS.
- J. Wiesecke and M. Rehahn, Macromol. Rapid Commun., 2007, 28, 188 CrossRef CAS.
- T. Schwalm and M. Rehahn, Macromolecules, 2007, 40, 33921.
- L. Hontis, V. Vrindts, L. Lutsen, D. Vanderzande and J. Gelan, Polymer, 2001, 42, 5793 CrossRef CAS.
- L. Hontis, V. Vrindts, D. Vanderzande and L. Lutsen, Macromolecules, 2003, 36, 3035 CrossRef CAS.
- L. Hontis, M. Van Der Borght, D. Vanderzande and J. Gelan, Polymer, 1999, 40, 6615 CrossRef CAS.
- P. Adriaensens, M. Van Der Borght, L. Hontis, A. Issaris, A. van Breemen, M. de Kok, D. Vanderzande and J. Gelan, Polymer, 2000, 41, 7003 CrossRef CAS.
- D. J. M. Vanderzande, L. Hontis, A. Palmaerts, D. Van Den Berghe, J. Wouters, L. Lutsen and T. Cleij, Proc. SPIE, 2005, 5937, 116.
- L. Lutsen, P. Adriaensens, H. Becker, A. J. Van Breemen, D. Vanderzande and J. Gelan, Macromolecules, 1999, 32, 6517 CrossRef CAS.
- E. Kesters, D. Vanderzande, L. Lutsen, H. Penxten and R. Carleer, Macromolecules, 2005, 38, 1141 CrossRef CAS.
- A. Henckens, I. Duyssens, L. Lutsen, D. Vanderzande and T. J. Cleij, Polymer, 2006, 47, 123 CrossRef CAS.
- S. Y. Hong, Bull. Korean Chem. Soc., 1999, 20, 42 CAS.
- H. Roex, P. Adriaensens, D. Vanderzande and J. Gelan, Macromolecules, 2003, 36, 5613 CrossRef CAS.
- P. W. M. Blom and M. C. J. M. Vissenberg, Mater. Sci. Eng., R, 2000, 27, 53 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of monomers 3 and 4 and 1H and 13C NMR spectra of TH-PPV and TEH-PPV. See DOI: 10.1039/c1py00027f |
| This journal is © The Royal Society of Chemistry 2011 |