Synthesis of polyurethane-g-poly(ethylene glycol) copolymers by macroiniferter and their protein resistance†
Guoying
Zhou
a,
Chunfeng
Ma
ab and
Guangzhao
Zhang
*ab
aHefei National Laboratory for Physical Sciences at Microscale, Department of Chemical Physics, University of Science and Technology of China, Hefei, China 230026
bFaculty of Materials Science and Engineering, South China University of Technology, Guangzhou, China 510640
First published on 15th April 2011
Abstract
We have synthesized polyurethane (PU)-g-poly(ethylene glycol) (PEG) copolymers for protein resistance. A macroiniferter consisting of polyurethane (PU) with tetraphenylethane groups is synthesized by the reaction of 1,1,2,2-tetraphenyl-1,2-ethanediol and isocyanate-terminated prepolymer with poly(tetramethylene glycol) (PTMG) segments. Such a macroiniferter initiates the polymerization of poly(ethylene glycol) methyl ether methacrylate to yield the target copolymer. Fourier transform infrared spectroscopy (FTIR), proton nuclear magnetic resonance spectroscopy (1H NMR) and thermal analysis confirm the structure of the copolymer. Thermogravimetric analysis (TGA) also shows that the introduction of PEG side chains slightly decreases the thermal stability of the polymer. By use of quartz crystal microbalance with dissipation (QCM-D) and surface plasmon resonance (SPR), we have investigated the adsorption of fibrinogen, bovine serum albumin (BSA) and lysozyme on a PU-g-PEG surface in real time. It shows that the PU-g-PEG surface can resist protein adsorption depending on the PEG content.
Introduction
Protein adsorption is an important issue in many bio-related fields including biomaterials, biomedical devices and anti-biofouling.1–5 Once proteins are adsorbed on a surface of any system, they profoundly modify the surface properties. Sometimes this has negative effect on the surface functions. For example, it can degrade the sensitivity of diagnostic devices,6 cause a foreign-body inflammatory response for implanted biomaterials,7 and induce undesirable biofouling on ship hulls.8 To resist or reduce protein adsorption, a number of materials such as dextran,9heparin,10 polyoxazolines,11poly(ethylene glycol) (PEG) or oligo(ethylene glycol) (OEG),12,13 and polymers based on sulfobetaine14 or phosphorylcholine15 have been developed. Particularly, PEG-based materials exhibit excellent nonfouling capability and biocompatibility.16,17Polyurethanes (PUs) are widely used for their unique properties such as good adhesion and biocompatibility, but they cannot resist protein adsorption.18–21 The incorporation of PEG into PUs is expected to produce materials with a combination of the properties of PU and protein resistance. Accordingly, a PEG-modified PU surface has been prepared by oxygen plasma treatment of PU followed by surface-initiated atom transfer radical polymerization of oligo(ethylene glycol) methacrylate (OEGMA).22,23 Alternatively, hydroxyl or amine terminated-PEG chains have been chemically grafted on PU surfaces.24,25 However, such grafting on a large scale is hard to control, which prevents the materials from use in applications. We have prepared polyurethanes with PEG soft segments which exhibit excellent protein resistance.26 Yet, the polymers have undesirable swelling when PEG content is high. In this work, by use of a polyurethane macroiniferter with 1,1,2,2-tetraphenyl-1,2-ethane groups,27–32 we have prepared a copolymer with PU and PEG as the main and side chains via a facile procedure which is a combination of condensation reaction and radical polymerization. Our aim is to develop a polymeric material with good protein resistance and adhesion to surfaces.
Experimental section
Materials
Poly(tetramethylene glycol) (PTMG) (Mw = 2000 g mol−1) (PTMG2000) (Aladdin) was dried under reduced pressure for 2 h at 110 °C prior to use. 1,1,2,2-tetraphenyl-1,2-ethanediol (TPE) (Yangcun) was recrystallized from glacial acetic acid three times. Methoxy polyethylene glycol (Mw = 2000 g mol−1) (MPEG2000) was kindly provided by BASF and dried over an anhydrous toluene by azeotropic distillation. 4,4′-Diphenylmethane diisocyanate (MDI) (Alfa), dibutyltindilaurate (DBTDL) (Alfa), methacryloyl chloride (Aladdin) were all used as received. Triethylamine was refluxed with acetic anhydride, dried with CaH2 and then distilled. Tetrahydrofuran (THF) and methylene chloride (CH2Cl2) were refluxed over CaH2 and distilled prior to use. N,N′-dimethylformamide (DMF) was dried over anhydrous magnesium sulfate (MgSO4) and distilled at reduced pressure. Fibrinogen (fraction I from human plasma, Mw = 340 kDa, pI = 5.5) from Merck Chemicals, lysozyme from chicken egg white (Mw = 14.7 kDa, pI = 11.1) from Sangon and BSA (Mw = 68 kDa, pI = 4.8) from Hualvyuan Bio-technology company were used as received. Physiological phosphate buffered saline (PBS, 0.14 M, pH 7.4) was prepared by dissolving NaCl, KCl, Na2HPO4, KH2PO4 in Milli-Q water (Millipore, resistivity = 18.2 MΩ·cm, 25 °C). Each protein solution (1.0 mg mL−1) was prepared by dissolving the protein in PBS buffer. Other reagents were used as received.The synthesis of PU-g-PEG copolymers is illustrated in Scheme 1.
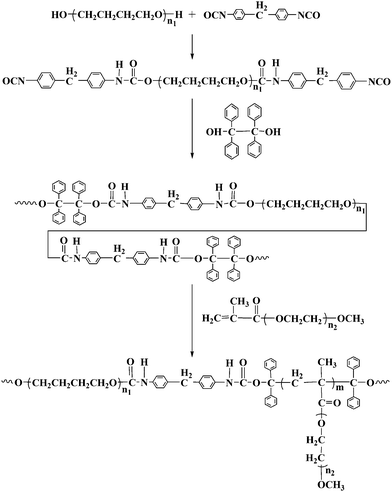 | ||
| Scheme 1 Synthesis of PU-g-PEG copolymersvia a PU macroiniferter. | ||
Synthesis of PU macroiniferter
As shown in Scheme 1, isocyanate end capped prepolymer was synthesized via reaction of PTMG2000 with MDI, which further reacted with TPE as chain extender to yield the PU macroiniferter (PUI). Specifically, PTMG2000 (30 g, 0.015 mol) and MDI (7.56 g, 0.03 mol) were added into a 500 mL flask and stirred at 60 °C under nitrogen atmosphere. When diisocyanate content decreased to the half of the initial value, the reaction mixture was cooled to 30 °C. Subsequently, TPE (5.55 g, 0.015 mol) and DBTDL (0.1 wt% based on the initial isocyanate content) were added and reacted for 24 h at 30 °C. At the end of the reaction, 5.0 mL of methanol was added and stirred for another 15 min. The product was precipitated in methanol-water mixture (v/v: 2/1), filtered, washed thoroughly with methanol to remove the unreacted TPE and dried in vacuum. The product was characterized by FTIR and 1H NMR. FTIR (KBr): 3300 cm−1 (NH), 2950 cm−1 (CH3), 1730 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1100 cm−1 (C–O–C). 1H NMR (400 MHz, CDCl3) δ: 1.61 ppm (CH2CH2CH2CH2O), 3.40 ppm (CH2CH2CH2CH2O), 3.80 ppm (C6H4CH2C6H4), 4.15 ppm (NHCOOCH2), 7.10–7.40 ppm (C6H5).
O), 1100 cm−1 (C–O–C). 1H NMR (400 MHz, CDCl3) δ: 1.61 ppm (CH2CH2CH2CH2O), 3.40 ppm (CH2CH2CH2CH2O), 3.80 ppm (C6H4CH2C6H4), 4.15 ppm (NHCOOCH2), 7.10–7.40 ppm (C6H5).
Synthesis of poly(ethylene glycol) methyl ether methacrylate (PEGMA)
PEGMA was synthesized following a procedure reported before.33 Typically, MPEG2000 (40 g, 0.02 mol), CH2Cl2 (200 ml) and triethylamine (23.1 mL, 0.16 mol) were added into a 250 mL three-neck flask equipped with a magnetic stirrer. Methacryloyl chloride (15.4 mL, 0.16 mol) was slowly dropped into the mixture at 0 °C. The mixture was further stirred at room temperature for 24 h. 6.0 mL methanol was slowly added at 0 °C to react with the excess of methacryloyl chloride. The reaction solution was filtered and passed through an alkaline alumina column to remove the triethylammonium chloride. The product was precipitated into cold diethyl ether, filtered and dried under vacuum. 1H NMR (400 MHz, CDCl3) δ: 3.35 ppm (CH3O(CH2CH2O)n), 3.64 ppm (CH3O(CH2CH2O)n), 4.3 ppm (COOCH2CH2), 5.56 and 6.13 ppm (CH2C(CH3)), 1.96 ppm (CH2C (CH3)).
Synthesis of PU-g-PEG copolymers
A known amount of PUI, PEGMA, and DMF were added into a Pyrex vial. After three freeze-pump-thaw cycles, the vial was sealed under vacuum and then reacted at 80 °C for 24 h. The reaction was arrested by dipping in liquid N2. The product was precipitated in cold methanol and washed thoroughly with methanol to remove the unreacted PEGMA monomer. Centrifugal separation was used to separate PU-g-PEG copolymer when the solution became an emulsion at a high PEGMA content. The copolymer is designed as PU-g-PEGx, where x is the weight percentage of PEG determined by 1H NMR. The number (Mn) and weight-average (Mw) molecular weights and polydispersity index (Mw/Mn) were determined by gel permeation chromatography (GPC). All characterization data are summarized in Table 1.Characterizations
FTIR spectra of the polymers were recorded on a Bruker VECTOR-22 IR spectrometer using the KBr disk method. The spectra were collected at 64 scans with a spectral resolution of 4 cm−1.All 1H NMR spectra were recorded on a Bruker AV400 NMR spectrometer (resonance frequency of 400 MHz for 1H) operated in the Fourier transform mode with CDCl3 as the solvent.
GPC measurements were conducted at 35 °C on a Waters 1515 by using a series of monodisperse polystyrenes as the standard and THF as the fluent with a flow rate of 1.0 mL min−1.
Themogravimetric analysis (TGA) measurements were performed under nitrogen atmosphere using a TA SDT Q600 instrument at a heating rate of 10 °C min−1 over the range from room temperature to 800 °C. Differential scanning calorimetry (DSC) was performed on a TA Instruments Q2000 differential scanning calorimeter under a nitrogen flow of 50 mL min−1. After the sample was quickly heated to 120 °C and equilibrated at the temperature for 5 min, it was cooled to −90 °C at a rate of 10 °C min−1. Finally, it was re-heated to 120 °C at the same rate. All DSC traces are from the second heat to minimize effects of thermal history.
QCM-D and the AT-cut quartz crystal with a fundamental resonant frequency of 5 MHz were from Q-sense AB.34 The quartz crystal was mounted in a fluid cell with one side exposed to the solution. The effects of surface roughness were minimized by using highly polished crystals with a root-mean-square roughness less than 3 nm.35 The details for QCM-D can be found elsewhere.36,37 In short, the mass of a thin layer on quartz crystal relates to the decrease in the resonant frequency of the crystal, whereas the dissipation factor is to the viscoelastic properties of additional layer. In the present study, the changes in the frequency (Δf) and dissipation (ΔD) give information about the protein adsorption and structural change of the protein layer. All the presented data are from the 3rd overtone (n = 3). Δf and ΔD values from the fundamental were discarded because they were usually noise due to insufficient energy trapping.38 Data from 5th and 7th overtones were not presented since they were similar to those from the 3rd overtone. The experiments were performed with PBS as the reference at 25 °C. Each film for QCM-D experiments was prepared by spin-casting of a polymer solution in THF (0.5 wt%) on a quartz crystal with a spin coater (CHEMAT, KW-400) at 4000 rpm in air. The film together with the crystal was heated at 40 °C for 24 h in an oven before use. Note that further increasing the PEG content leads to the film to detaching from the QCM-D crystal surface in aqueous solution. Thus, we only studied the samples with PEG content less than 53 wt%.
SPR measurements were carried out on Biacore X at 25 °C. The gold-coated glass plate was attached to a glass prism (n ≈ 1.65) with a silicone opto-interface between them so that they matched in refractive index.39,40 The response was linearly related to the mass of added layer with 1000 RU ≈ 1 ng mm−2, where RU was the response unit.41 Light from a near-infrared light-emitting diode was focused through the prism on to the sensor surface in a wedge-shaped beam to give a fixed range of incident light angle. Light reflected from the sensor was monitored by a linear array of light-sensitive diodes with a resolution of ∼0.1°. The protein solution was delivered to the surface at a flow rate of 10 μL min−1. The amount of protein adsorbed designated as the change in RUs (ΔRU) was determined by subtracting the RU value for the reference (PBS) from that for the protein solution. The film for SPR experiments was also prepared by spin-casting like that for QCM-D experiments.
Results and discussion
Fig. 1 shows the FTIR spectra of TPE, PUI and PU-g-PEG17 copolymer. We can observe a broad band at ∼3550 cm−1 for TPE, which is attributed to OH. For PUI, the band at ∼3300 cm−1 is attributed to the urethane amide bond. The absence of an OH band in TPE indicates the completion reaction. In the spectrum of PU-g-PEG copolymer, the intensity regarding C–O–C at ∼1100 cm−1 increases, indicating the incorporation of PEGMA into PU backbone.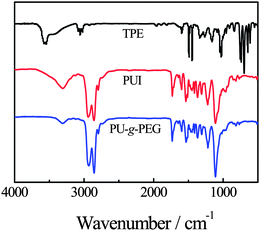 | ||
| Fig. 1 FTIR spectra of TPE, PUI and PU-g-PEG. | ||
Fig. 2 shows the 1H NMR spectrum of PEGMA, PUI and PU-g-PEG copolymer. The new peak (δ = 3.64 ppm) in PU-g-PEG is attributed to CH2O in PEGMA monomer, so that PEGMA is incorporated to PU. In addition, the absence of double bond (δ = 5.56 ppm and δ = 6.13 ppm) in PEGMA monomer further indicates incorporation.
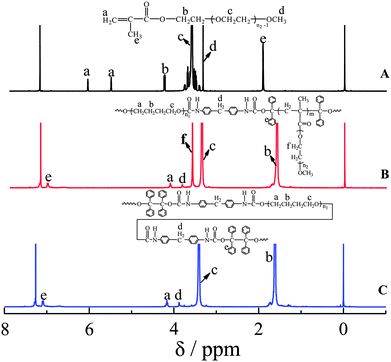 | ||
| Fig. 2 1H NMR spectra of PEGMA (A), PU-g-PEG (B) and PUI (C). | ||
Fig. 3 shows typical TGA curves of PUI and PU-g-PEG copolymers. PUI exhibits two degradation stages located at ∼280–360 °C and ∼360–490 °C, respectively. The former is attributed to the degradation of hard segment as a consequence of the relatively low thermal stability of the urethane groups, whereas the latter is related to the decomposition of PTMG soft segments.42,43 In contrast, only one stage located at ∼280–480 °C is observed for each PU-g-PEG copolymer. The decomposition of the hard segments almost disappears. This is understandable because the weight percent of urethane groups in the copolymer is low, so that the signal for their decomposition is masked. The derivative of thermogravimetric analysis (DTGA) in the inset shows the temperature (Td) regarding the maximum decomposition rate for PU-g-PEG7, PU-g-PEG17, PU-g-PEG32 and PU-g-PEG53 are about 415, 413, 408 and 405 °C, respectively. Clearly, Td decreases with PEG content. This is because the decomposition temperature (∼400 °C) of PEG side chains is close but some lower than that of PTMG.44,45 What we observed is their combination whose Td shifts to that of PEG as PEG content increases. Anyhow, the introduction of PEG side chains only slightly decreases the thermal stability of the polymer.
 | ||
| Fig. 3 TGA and DTGA (the inset) curves of PUI and PU-g-PEG copolymers. | ||
Fig. 4 shows the DSC thermograms of PU-g-PEG copolymers together with PUI. We can observe a single endothermic peak at ∼20 °C for PUI due to the melting of crystallized PTMG segments.46 In contrast, all PU-g-PEG copolymers show two melting endothermic peaks at ∼20 and ∼50 °C, which are attributed to the melting of the crystallized PTMG and PEG segments, respectively.46,47 As PEG content increases, the crystallinity of PTMG decreases because its weight percent decreases. Meanwhile, the melting temperature of PEG segments increases and shifts to that of pure PEG. The DSC results agree with those of TGA, further indicating the structure of the copolymer.
 | ||
| Fig. 4 DSC curves of PUI and PU-g-PEGs with different PEG content. | ||
We have examined the protein resistance of PU-g-PEG copolymer. Fig. 5 shows the frequency shift (Δf) and energy dissipation shift (ΔD) in real time in QCM-D measurement of the adsorption of fibrinogen onto different polymer surfaces. It is known that Δf decreases but ΔD increases as added mass on the sensor surface increases.37 For PUI surface, after fibrinogen solution is added, Δf decreases and ΔD increases sharply, implying that the protein molecules are adsorbed on the surface. Then, Δf and ΔD gradually level off, indicating the saturation of adsorption. Upon rinsing with PBS, Δf and ΔD still show obvious changes in comparison with those of PBS reference, indicating that fibrinogen is strongly absorbed on the PUI surface. Note that the changes in Δf and ΔD for each PU-g-PEG surface are much smaller than those for the PUI surface, clearly indicating that PU-g-PEG copolymers have protein resistance. In addition, the protein adsorption on PU-g-PEG surfaces gradually decreases with increasing PEG content. In particular, for PU-g-PEG53, Δf and ΔD return to the baseline upon rinsing with PBS. Namely, the protein adsorption is almost zero when PEG content is high enough.
 | ||
| Fig. 5 Time dependence of frequency shift (Δf) and dissipation shift (ΔD) for the adsorption of fibrinogen on PUI and PU-g-PEG surfaces at 25 °C. | ||
Fig. 6 shows a rapid increase in SPR signal after fibrinogen solution is added due to the change in refractive index caused by the adsorbed protein. For the PUI surface, the SPR response (ΔRU) after rinsing is ∼2500 RU, clearly indicating the adsorption of fibrinogen on PUI surface. ΔRU values after rinsing are ∼1100, 400, 170 and 50 RU for PU-g-PEG7, PU-g-PEG17, PU-g-PEG32 and PU-g-PEG53, respectively. Clearly, the protein adsorption decreases with PEG content, that is, the protein resistance increases with PEG content. For PU-g-PEG53, ΔRU is only ∼50 RU or ∼0.05 ng mm−2 after rinsing, indicating a very low adsorption of fibrinogen. Note that unlike the QCM-D results, the adsorption detected by SPR is not equal to zero. This is because SPR is more sensitive than QCM-D.48
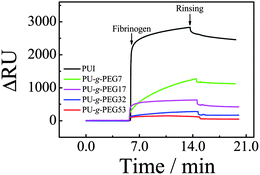 | ||
| Fig. 6 SPR sensorgrams for fibrinogen adsorption on PUI and PU-g-PEG surfaces at 25 °C. | ||
Fig. 7 shows the frequency shift (Δf) and dissipation shift (ΔD) in real time for the adsorption of BSA on PUI and PU-g-PEG surfaces. BSA is a very abundant protein in blood with size smaller than that of fibrinogen. Similar to fibrinogen, BSA is easily adsorbed on PUI surface, reflecting in the large changes in Δf and ΔD after rinsing. For PU-g-PEG surfaces, BSA adsorption gradually decreases from PU-g-PEG7 to PU-g-PEG53, indicating protein resistance increases as PEG content increases. Finally, undetectable BSA adsorption is observed for PU-g-PEG53. Clearly, PU-g-PEG surfaces can resist BSA adsorption depending on the PEG content. Fig. 8 also shows that BSA is adsorbed on PUI surface, reflecting in the large ΔRU. However, ΔRU for PU-g-PEG surfaces decreases as PEG content increases, further indicating that the protein adsorption decreases. A very small ΔRU is observed for PU-g-PEG53 surface, indicating its best protein resistance here.
 | ||
| Fig. 7 Time dependence of frequency shift (Δf) and dissipation shift (ΔD) for the adsorption of BSA on PUI and PU-g-PEG surfaces at 25 °C. | ||
 | ||
| Fig. 8 SPR sensorgrams for the adsorption of BSA on PUI and PU-g-PEG surfaces at 25 °C. | ||
Fig. 9 shows the frequency shift (Δf) and dissipation shift (ΔD) in real time for the adsorption of lysozyme on PUI and PU-g-PEG surfaces. Lysozyme is the smallest in size among the three proteins. It is positively charged under the experimental conditions (PBS, 0.14 M, pH 7.4). For PUI, Δf decreases and ΔD increases markedly, indicating the adsorption of lysozyme. For PU-g-PEG copolymers, the changes in Δf and ΔD become smaller as PEG content increases, indicating that the protein resistance increases. SPR measurements in Fig. 10 give the same information. However, unlike the cases about fibrinogen and BSA, either QCM-D or SPR measurements demonstrate some adsorption of lysozyme even for the surface constructed by PU-g-PEG53. This is consistent with the results reported before.49 As suggested by Halperin,50 small proteins can penetrate the space between grafted chains, but proteins with a large enough size cannot. Thus, some lysozyme molecules should be located at the space between PEG side chains, leading to a small adsorption. Anyhow, the above QCM-D and SPR results reveal that PU-g-PEG copolymers can resist protein adsorption depending on the PEG content. The above experiments clearly demonstrate that the protein resistance of PU-g-PEG comes from PEG segments because PUI without PEG segments can adsorb proteins. The mechanism for protein resistance of PEG is not well understood so far. It could be explained in terms of the water barrier theory. Namely, water molecules form a tight layer around PEG chains viahydrogen bonds, which can create a physical and energetic barrier to prevent protein adsorption.26,51
 | ||
| Fig. 9 Time dependence of frequency shift (Δf) and dissipation shift (ΔD) for the adsorption of lysozyme on PUI and PU-g-PEG surfaces at 25 °C. | ||
 | ||
| Fig. 10 SPR sensorgrams for the adsorption of lysozyme on PUI and PU-g-PEG surfaces at 25 °C. | ||
Conclusion
We have synthesized polyurethane-g-poly(ethylene glycol) (PU-g-PEG) copolymers by using a macroiniferter. The copolymer structures were confirmed by FTIR, 1H NMR and thermal analysis. TGA also indicates that the presence of PEG side chains slightly decreases the thermal stability of the polymer. QCM-D and SPR measurements demonstrate that the PU-g-PEG copolymers have protein resistance which can be tuned by varying PEG content.Acknowledgements
The financial support of the National Distinguished Young Investigator Fund (20725414), the Science and Technology Cooperation Project of China and Australia (51011120051) and Ministry of Science and Technology of China (2007CB936401) is acknowledged.References
- J. H. Lee, Y. M. Ju and D. M. Kim, Biomaterials, 2000, 21, 683–691 CrossRef CAS.
- D. F. Williams, Biomaterials, 2008, 29, 2941–2953 CrossRef CAS.
- C. Werner, M. F. Maitz and C. Sperling, J. Mater. Chem., 2007, 17, 3376–3384 RSC.
- W. Senaratne, L. Andruzzi and C. K. Ober, Biomacromolecules, 2005, 6, 2427–2448 CrossRef CAS.
- R. L. Townsin, Biofouling, 2003, 19, 9–15 CrossRef.
- N. Wisniewski and M. Reichert, Colloids Surf., B, 2000, 18, 197–219 CrossRef CAS.
- P. Thomsen and C. Gretzer, Curr. Opin. Solid State Mater. Sci., 2001, 5, 163–176 CrossRef CAS.
- L. D. Chambers, K. R. Stokes, F. C. Walsh and R. J. K. Wood, Surf. Coat. Technol., 2006, 201, 3642–3652 CrossRef CAS.
- A. De Sousa Delgado, M. Léonard and E. Dellacherie, Langmuir, 2001, 17, 4386–4391 CrossRef CAS.
- M. Johnell, R. Larsson and A. Siegbahn, Biomaterials, 2005, 26, 1731–1739 CrossRef CAS.
- R. Konradi, B. Pidhatika, A. Müehlebach and M. Textor, Langmuir, 2008, 24, 613–616 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS.
- C. Roberts, C. S. Chen, M. Mrksich, V. Martichonok, D. E. Ingber and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 6548–6555 CrossRef CAS.
- J. Ladd, Z. Zhang, S. F. Chen, J. C. Hower and S. Y. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS.
- S. F. Chen, J. Zheng, L. Y. Li and S. Y. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS.
- N. A. Alcantar, E. S. Aydil and J. N. Israelachvili, J. Biomed. Mater. Res., 2000, 51, 343–351 CrossRef CAS.
- R. L. C. Wang, H. J. Kreuzer and M. Grunze, J. Phys. Chem. B, 1997, 101, 9767–9773 CrossRef CAS.
- D. S. Vara, G. Punshon, K. M. Sales, H. J. Salacinski, S. Dijk, R. A. Brown, G. Hamilton and A. M. Seifalian, Biomaterials, 2005, 26, 3987–3993 CrossRef CAS.
- Z. F. Liu, X. Wu, X. Y. Yang, D. P. Liu, C. Jun, R. M. Sun, X. P. Liu and F. X. Li, Biomacromolecules, 2005, 6, 1713–1721 CrossRef CAS.
- R. L. Oréfice, E. Ayres, M. M. Pereira and H. S. Mansur, Macromolecules, 2005, 38, 4058–4060 CrossRef CAS.
- D. Fournier and F. D. Prez, Macromolecules, 2008, 41, 4622–4630 CrossRef CAS.
- Z. L. Jin, W. Feng, S. P. Zhu, H. Sheardown and J. L. Brash, J. Biomed. Mater. Res., Part A, 2009, 91A, 1189–1201 CrossRef CAS.
- Z. L. Jin, W. Feng, K. Beisser, S. P. Zhu, H. Sheardown and J. L. Brash, Colloids Surf., B, 2009, 70, 53–59 CrossRef CAS.
- J. G. Archambault and J. L. Brash, Colloids Surf., B, 2004, 33, 111–120 CrossRef CAS.
- J. G. Archambault and J. L. Brash, Colloids Surf., B, 2004, 39, 9–16 CrossRef CAS.
- C. F. Ma, Y. Hou, S. Liu and G. Z. Zhang, Langmuir, 2009, 25, 9467–9472 CrossRef CAS.
- G. C. Jiang, X. L. Tuo, D. R. Wang and Q. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3248–3256 CrossRef CAS.
- G. N. Mahesh, A. Sivaraman, K. Tharanikkarasu and G. Radhakrishnan, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1237–1244 CrossRef CAS.
- S. Sundar, K. Tharanikkarasu, A. Dhathathreyan and G. Radhakrishnan, Colloid Polym. Sci., 2002, 280, 915–921 CrossRef CAS.
- D. Braun and K. H. Becker, Angew. Makromol. Chem., 1969, 6, 186–189 CrossRef CAS.
- K. Tharanikkarasu and G. Radhakrishnan, J. Appl. Polym. Sci., 1997, 66, 1551–1560 CrossRef CAS.
- T. Otsu and M. Yoshida, Makromol. Chem. Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- Y. G. Li, P. J. Shi, Y. S. Zhou and C. Y. Pan, Polym. Int., 2004, 53, 349–354 CrossRef CAS.
- M. Rodahl, F. Höök, A. Krozer, B. Kasemo and P. Brzezinski, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- L. Daikhin and M. Urbakh, Faraday Discuss., 1997, 107, 27–38 RSC.
- G. Z. Sauerbrey, Z. Phys., 1959, 155, 206–212 Search PubMed.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- V. E. Bottom, Introduction to Quartz Crystal Unit Design, Van Nostrand Reinhold Co., New York, 1982 Search PubMed.
- E. Kretschmann and H. Raether, Z. Naturforsch., A, 1968, 23, 2135–2136 CAS.
- B. Liedberg, C. Nylander and I. Lunstrom, Sens. Actuators, 1983, 4, 299–304 CrossRef CAS.
- E. Stenberg, B. Persson, H. Poss and C. Urbaniczky, J. Colloid Interface Sci., 1991, 143, 513–526 CrossRef CAS.
- J. M. Cervantes-Uc, J. I. Moo Espinosa, J. V. Cauich-Rodríguez, A. Ávila-Ortega, H. Vázquez-Torres, A. Marcos-Fernández and J. San Román, Polym. Degrad. Stab., 2009, 94, 1666–1677 CrossRef CAS.
- Z. S. Petrović, Z. Zavargo, J. H. Flyn and W. J. Macknight, J. Appl. Polym. Sci., 1994, 51, 1087–1095 CrossRef CAS.
- F. Y. Wang, C. C. M. Ma and W. J. Wu, J. Mater. Sci., 2001, 36, 943–947 CrossRef CAS.
- N. Kizilyar, L. Toppare, A. Önen and Y. Yağci, Polym. Bull., 1998, 40, 639–645 CrossRef CAS.
- C. B. Hu, R. S. Ward and N. S. Schneider, J. Appl. Polym. Sci., 1982, 27, 2167–2177 CrossRef CAS.
- X. J. Loh, K. B. C. Sng and J. Li, Biomaterials, 2008, 29, 3185–3194 CrossRef CAS.
- Q. Wang, J. F. Wang, P. H. Geil and G. W. Padua, Biomacromolecules, 2004, 5, 1356–1361 CrossRef CAS.
- W. Feng, S. P. Zhu, K. Ishihara and J. L. Brash, Langmuir, 2005, 21, 5980–5987 CrossRef CAS.
- A. Halperin, Langmuir, 1999, 15, 2525–2533 CrossRef CAS.
- S. F. Chen, L. Y. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00016k |
| This journal is © The Royal Society of Chemistry 2011 |