Long wavelength photoinitiated free radical polymerization using conjugated thiophene derivatives in the presence of onium salts
Selim
Beyazit
,
Binnur
Aydogan
,
Ipek
Osken
,
Turan
Ozturk
and
Yusuf
Yagci
*
Istanbul Technical University, Department of Chemistry, Maslak, Istanbul, 34469, Turkey. E-mail: yusuf@itu.edu.tr; Fax: +90-212-2856386; Tel: +90-212-2853241
First published on 1st March 2011
Abstract
Long wavelength free radical photopolymerization of (meth)acrylic monomers using a conjugated thiophene derivative, namely 3,5-diphenyldithieno[3,2-b:2′,3′-d]-thiophene (DDT) and one of the following compounds, namely diphenyliodonium hexafluorophosphate (Ph2I+PF6−), triphenylsulfonium hexafluorophosphate (Ph3S+PF6−) and N-ethoxy-2-methylpyridinium hexafluorophosphate (EMP+PF6−), as photosensitizer and coinitiator, respectively, was described. Polymerization occurred only when DDT, in the presence of the onium salts, was irradiated at wavelengths where the salts have no absorption. A feasible initiation mechanism involves intermolecular electron transfer from photoexcited thiophene compound to the onium salt. The relative initiation efficiencies of the salts were investigated and evaluated in terms of their redox potentials.
Introduction
In recent years, photoinitiated free radical and cationic polymerizations have received revitalized interest as they congregate a wide range of economic and ecological anticipations.1–9Onium-type salts such as iodonium,10sulfonium11 and alkoxy pyridinium12 salts are known to be efficient photoinitiators for cationic polymerization due to their thermal stability, solubility in most of the cationically polymerizable monomers, and competence in generating reactive species upon photolysis.13 Because of the fact that besides Brønsted acids, radical species are also produced, these salts can also be used as photoinitiators for free radical, and concurrent free radical and cationic polymerizations.14,15 The performance of these photoinitiators particularly at long wavelengths can be improved by three main strategies: (i) oxidation of free radicals,16–18 (ii) electron transfer between a photoexcited molecule and an onium salt,19–28 and (iii) excitation of charge transfer complexes14,29 of certain onium salts. The spectral sensitivity of photoinitiated cationic polymerizations may be extended30–37 to the near UV and visible range by using appropriate free radical sources and aromatic sensitizers.
More recently, we have reported38 the use of a polynuclear thiophene derivative, namely 3,5-diphenyldithieno[3,2-b:2′,3′-d]-thiophene39,40 (DDT), as an electron-transfer photosensitizer for diaryliodonium salt cationic photoinitiators. DDT provides good sensitivity in the near UV region (350–400 nm). Several types of cationically polymerizable monofunctional monomers, such as cyclohexene oxide, n-butyl vinyl ether, styrene and N-vinylcarbazole, and bifunctional monomers, such as 3,4-epoxycyclohexyl-3′,4′-epoxycyclohexene carboxylate, were readily polymerized in bulk or dichloromethane solutions upon irradiation at 350 nm in the presence of DDT and diphenyliodonium hexafluorophosphate. A mechanism based on electron transfer between excited DDT and iodonium ion was proposed (Scheme 1). A wide range of polynuclear aromatic compounds are known to undergo similar electron transfer reactions.30–33,41,42Polymerizations were initiated either by DDT radical cations (reaction 4, Scheme 1) or the protons formed viahydrogen abstraction (reaction 5, Scheme 1) or coupling reactions (reaction 6, Scheme 1).
![Photoinitiated cationic polymerization by using 3,5-diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) and diphenyliodonium salt.](/image/article/2011/PY/c1py00019e/c1py00019e-s1.gif) | ||
| Scheme 1 Photoinitiated cationic polymerization by using 3,5-diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) and diphenyliodonium salt. | ||
By virtue of the thiophene-type radical cation formation and the involvement of the thiophene radical in coupling reactions in the electropolymerization, the described photoinduced process has the potential to initiate also free radical polymerization. It seemed, therefore, appropriate to expand the electron transfer reactions between photoexcited thiophene derivatives and onium salts to free radical polymerizing systems. This paper reports on the photoinitiated free radical polymerization at long wavelengths using highly conjugated thiophene derivative, DDT in the presence of onium salts.
Experimental part
Materials
3,5-Diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) was synthesized as described in literature.43Diphenyliodonium hexafluorophosphate (Ph2I+PF6−, 98%, Alfa Aesar) was used as received. Triphenylsulfonium hexafluorophosphate (Ph3S+PF6−)44 and N-ethoxy-2-methylpyridinium hexafluorophosphate (EMP+PF6−)17,45 were prepared according to the previously described procedures. Methyl acrylate (MA, 99%, Aldrich), butyl acrylate (BA, ≥99%, Aldrich), methyl methacrylate (MMA, 99%, Aldrich), trimethylolpropane triacrylate (TMPTA, 95%, Aldrich) and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 99%, Aldrich) were used as received. Dichloromethane (CH2Cl2, 99.8%, Baker) was extracted first with sulfuric acid, then with 5% NaOH solutions. After washing with water, CH2Cl2 was dried over anhydrous CaCl2 and CaH2 and finally distilled with a fractionation column.Photopolymerization
A typical photopolymerization procedure for methyl acrylate (MA) was as follows. MA (1 mL, 0.011 mol), Ph2I+PF6− (0.1 mL from 0.012 mol L−1 stock solution in CH2Cl2) and DDT (0.1 mL from 0.006 mol L−1 stock solution in CH2Cl2) were put in a Pyrex tube and filled with dry nitrogen prior to irradiation from a Polilight PL400 Forensic Plus light source at λ = 350 nm. At the end of irradiation, polymer was precipitated in 10-fold excess methanol and dried in vacuum. Conversions for all samples were determined gravimetrically.Radical scavenging
TEMPO (1.5 mg, 0.0096 mmol), Ph2I+PF6− (2.6 mg, 0.0063 mmol), DDT (1 mg, 0.0021 mmol) and CH2Cl2 (1 mL) were put in a Pyrex tube and filled with dry nitrogen. The solution was irradiated at λ = 350 nm for 30 min by using a Polilight PL400 Forensic Plus light source. At the end of irradiation, the solvent was removed by rotary evaporator and the obtained solid was characterized by 1H NMR.Instrumentation
1H NMR measurements were recorded in CDCl3 with Si(CH3)4 as an internal standard, using a Bruker AC250 (250.133 MHz) instrument. UV spectra were recorded on a Shimadzu UV-1601 spectrometer. Molecular weights were determined by gel permeation chromatography (GPC) instrument, Viscotek GPCmax Autosampler system, consisting of a pump, three ViscoGEL GPC columns (G2000HHR, G3000HHR and G4000HHR), a Viscotek UV detector and a Viscotek differential refractive index (RI) detector with a THF flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with PS standards having narrow molecular weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. A Rofin Polilight PL400 Forensic Plus light source, emitting light at several wavelengths (350, 415, 430, 450, LP530, 490, 505, 515, 530, 550, 560, LP560, 570, 590, 620, 650, white, half white and blank) with specific bandwidths, was used for the irradiation of the samples.Photocalorimetry (Photo-DSC)
The photo-differential scanning calorimetry (Photo-DSC) measurements were carried out by means of a modified Perkin-Elmer DiamondDSC equipped with a Polilight PL400 Forensic Plus light source at 350 nm. A uniform UV light intensity is delivered across the DSCcell to the sample and reference pans. The intensity of the light was measured as 67 mW cm−2 by a UV radiometer covering broad UV range. The mass of the samples was 3 mg and the measurements were carried out in an isothermal mode at 30 °C under a nitrogen flow of 20 mL min−1. The reaction heat liberated in the polymerization was directly proportional to the number of acrylate double bonds reacted in the system. By integrating the area under the exothermic peak, the conversion of the acrylate or methacrylategroups (C) or the extent of the reaction was determined according to eqn (1):| C = ΔHt/ΔHtheory0 | (1) |
Results and discussions
3,5-Diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) absorbs the light at 350–450 nm wavelength region where the onium salts are transparent and photoexcited DDT is rapidly quenched by onium salts (Fig. 1). Thus, photoinitiated polymerization of several (meth)acrylate monomers was performed at 350 nm by using DDT in the presence of diphenyliodonium salt. Polymerizations were conducted in dichloromethane solutions since onium salts are not soluble in the monomers employed. Typical results are presented in Table 1. They give clear indication for the sensitization effect of the thiophene compound for the polymerization as no polymerization occurred in the absence of DDT since iodonium salt is transparent at the irradiation wavelength. However, at very high iodonium concentrations some but little polymerization occurred due to the pronounced tail absorption of the salt. As can also be seen that among the monomers tested, methyl acrylate (MA) seemed to undergo polymerization more readily due to the less sterical hindrance and higher propagation rate constant. The polymerization was totally inhibited by the addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as the radical scavenger indicating that the free radical mechanism is responsible for the initiation. Possible effects of oxygen in the polymerization was also examined. Expectedly, the polymerization was totally inhibited when air was present in the system (Table 1, Run 5). For comparison, MA was polymerized using benzophenone/N,N-dimethylaniline as conventionally used photoinitiator for various applications. As can be seen, benzophenone initiates polymerization more efficiently than the described system. However, our system provides flexibility to apply to both radical and cationic formulations without any additional component.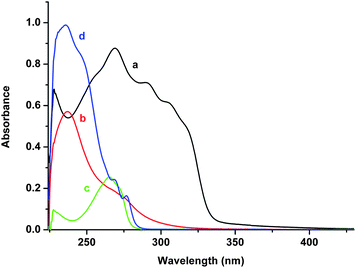 | ||
| Fig. 1 UV spectra of (a) DDT (5 × 10−5 mol L−1), (b) Ph2I+PF6− (5 × 10−5 mol L−1), (c) EMP+PF6− (5 × 10−5 mol L−1) and (d) Ph3S+PF6− (5 × 10−5 mol L−1) in CH2Cl2. | ||
| Run | Monomer/mol L−1 | Photosensitizer/mol L−1 | Onium salt/mol L−1 | Time/min | Conversion (%) | M n/g mol−1 | M w/Mn |
|---|---|---|---|---|---|---|---|
| a The experiment was performed in the presence of a radical scavengerTEMPO (2.5 × 10−3 mol L−1). b Oxygen saturated. c Benzophenone/N,N-dimethylaniline system was used instead of DDT/Ph2+PF6−. | |||||||
| 1 | MA(0.0093) | 2.5 × 10−4 | 0.5 × 10−3 | 5 | 10.1 | 323![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
1.87 |
| 2 | MA(0.0093) | 5 × 10−4 | 1 × 10−3 | 5 | 14.0 | 253300 |
1.45 |
| 3 | MA(0.0093)a | 5 × 10−4 | 1 × 10−3 | 5 | — | — | — |
| 4 | MA(0.0093) | — | 1 × 10−3 | 5 | — | — | — |
| 5 | MA(0.0093)b | 5 × 10−4 | 1 × 10−3 | 5 | — | — | — |
| 6 | MA(0.0093)c | 5 × 10−4 | 1 × 10−3 | 5 | 23.0 | 260800 |
1.92 |
| 7 | BA(0.0058) | 5 × 10−4 | 1 × 10−3 | 20 | 17.6 | 179600 |
2.03 |
| 8 | BA(0.0058) | — | 1 × 10−3 | 20 | — | — | — |
| 9 | MMA(0.0078) | 5 × 10−4 | 1 × 10−3 | 20 | 2.7 | 28000 |
1.54 |
| 10 | MMA(0.0078) | — | 1 × 10−3 | 20 | — | — | — |
Table 2 gives data for a series of polymerization of MA utilizing DDT and different onium salts. Although all onium salts are effective in initiating the polymerization, diphenyliodonium salt seemed to be the most efficient oxidizing agent in the process. Expectedly, the other onium salts, sulfonium and N-alkoxy pyridinium salts, exhibit lower efficiency due to their less favorable redox potentials compared to the iodonium salt.
| Onium salt | Time/min | Conversion (%) | M n/g mol−1 | M w/Mn | Free-energy changesa | |
|---|---|---|---|---|---|---|
| ΔGS/kcal mol−1 | ΔGT/kcal mol−1 | |||||
| a For diphenyliodonium salt Ered = −0.20 V vs.SCE,20 for triphenylsulfonium salt Ered = −1.20 V vs.SCE20 and for N-ethoxy-2-methylpyridinium salt Ered = −0.70 V vs.SCE.31 | ||||||
| Ph2I + PF6 − | 5 | 14.0 | 253300 |
1.45 | −57.8 | −38.8 |
| Ph3S+PF6− | 30 | 9.4 | 546800 |
3.50 | −34.7 | −15.7 |
| EMP + PF6 − | 30 | 5.7 | 422800 |
2.95 | −46.3 | −27.3 |
According to the Rehm–Weller equation (eqn (2)) electron transfer from the excited sensitizer to onium salt is feasible if the change in free energy (ΔG) is negative. Based on the oxidation potential (Eox) and active excitation energy (E*) of the photosensitizer (PS) and the reduction potential (Ered) of the initiator (PI), the free energy change (ΔG) for the photoinduced electron transfer process was estimated.34
| ΔG = Eox(PS) − Ered(PI) − E*(PS) | (2) |
Table 2 also summarizes the ΔGS and ΔGT values of the free energy changes for the electron transfer from the singlet and triplet excited states, respectively, of DDT to the ground-state cationic salts. Eox for DDT determined by cyclic voltammetry is 0.88 V,43 whereas the singlet excitation energy ES* and the triplet excitation energy ET* have been found to be 82.6 kcal mol−1 and 63.7 kcal mol−1, respectively. Scheme 2 shows the mechanism for free radical polymerization of MA based on electron transfer concerning the reaction of excited DDT with onium salt as the example of iodonium salt. As in the case of corresponding cationic polymerization (see Scheme 1), initially DDT radical cations are formed. Polymerization is expected to occur at the α-position of thiophene first by reaction of DDT radical cations with MA and then deprotonation.
![Photoinitiated free radical polymerization of acrylates by using 3,5-diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) and diphenyliodonium salt.](/image/article/2011/PY/c1py00019e/c1py00019e-s2.gif) | ||
| Scheme 2 Photoinitiated free radical polymerization of acrylates by using 3,5-diphenyldithieno[3,2-b:2′,3′-d]thiophene (DDT) and diphenyliodonium salt. | ||
Similar mechanism was accounted for the photoinitiated free radical polymerization through thiophene radical cations formed on gold nanoparticles. Neckers and coworkers47 elegantly showed that the charge-separated states of the gold nanoparticles attached to the bithiophenegroup generated upon exposure to UV light are active intermediates that can cause polymerization of an acrylic monomer. In addition to the efficient photoinitiation, the process leads to the uniform distribution of gold nanoparticles in the polymer matrix and the polymer nanocomposite with a unique texture was obtained by the one step photochemical process.
To investigate further if the thiophene based radical cations are the reactive intermediates playing a crucial role in the polymerization process and to determine the initiating radicals, the experiments using an excess of a stable radical, such as TEMPO, in the absence of an added monomer was performed. Indeed, the 1H NMR spectrum of the product (Fig. 2) formed from the photolysis of DDT and iodonium salt in the presence of TEMPO in methylene chloride presents a weak resonance of the aromatic proton at 4.1 ppm indicative of the thiophene radical cation coupling product of the following structure (Scheme 3).
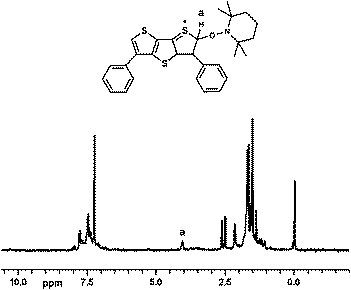 | ||
| Fig. 2 1H NMR spectrum of the product formed from the photolysis of DDT and iodonium salt in the presence of TEMPO in CDCl3. | ||
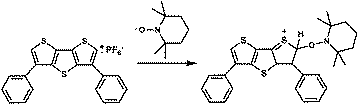 | ||
| Scheme 3 Structure of the coupling product formed from the photolysis of DDT and diphenyliodonium salt in the presence of TEMPO. | ||
The efficiency of the photoinitiating system in the photocuring of formulations containing multifunctional monomers such as trimethylolpropane triacrylate (TMPTA) was also studied. In Fig. 3, Photo-DSC profiles referring to the polymerization of TMPTA in the presence and absence of DDT under 350 nm are shown. Fig. 4 displays plots of the conversion versus irradiation time derived from Fig. 3. It can be seen that rapid polymerization takes place with DDT in conjunction with diphenyliodonium salt.
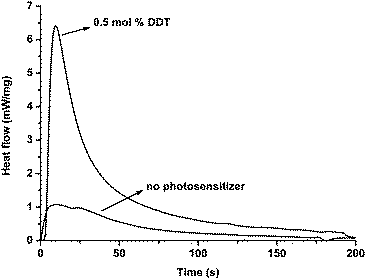 | ||
| Fig. 3 Heat flow vs. time for the polymerization of TMPTA initiated by 1 mol% Ph2I+PF6− in the absence and presence of 0.5 mol% DDT at 30 °C (350 nm, light intensity = 67 mW cm−2). | ||
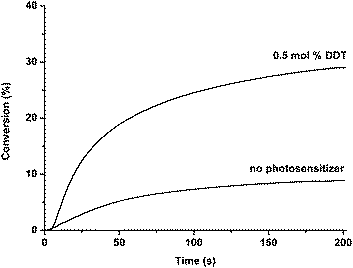 | ||
| Fig. 4 Conversion vs. time for the polymerization of TMPTA initiated by 1 mol% Ph2I+PF6− in the absence and presence of 0.5 mol% DDT at 30 °C (350 nm, light intensity = 67 mW cm−2). | ||
Conclusions
Preliminary experimental results fully support the idea that suitable thiophene derivatives in combination with oxidizing cations may be used to promote not only cationic polymerization but also free radical polymerization of (meth)acrylates. Mechanistic details remain to be evaluated, an important feature is the generation of radical cations by electron transfer from the excited state of the thiophene derivative to the onium salt; leading to formation of radical species with obvious implications to UV curing applications at the near UV region. Detailed studies are now in progress and it is clear that other types of highly conjugated compounds may react similarly given appropriate matching of redox potentials for electron transfer to the onium salt.Acknowledgements
The authors would like to thank Istanbul Technical University for financial support. One of the authors (Y.Y.) thanks the Turkish Academy of Sciences for partial financial support.References
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- R. S. Davidson, J. Photochem. Photobiol., A, 1993, 73, 81–96 CrossRef CAS.
- J. P. Fouassier, in Photoinitiation, Photopolymerization and Photocuring: Fundamentals and Applications, Hanser Publishers, Munich, 1995 Search PubMed.
- N. Moszner, U. K. Fischer, B. Ganster, R. Liska and V. Rheinberger, Dent. Mater., 2008, 24, 901–907 CrossRef CAS.
- N. Moszner, I. Lamparth, J. Angermann, U. K. Fischer, F. Zeuner, T. Bock, R. Liska and V. Rheinberger, Beilstein J. Org. Chem., 2010, 6, 26 Search PubMed.
- N. Moszner, F. Zeuner, I. Lamparth and U. K. Fischer, Macromol. Mater. Eng., 2009, 294, 877–886 CrossRef CAS.
- S. P. Pappas, in UV Curing: Science and Technology, Technology Marketing Corporation, Stamford, 1985 Search PubMed.
- J. Pavlinec and N. Moszner, J. Appl. Polym. Sci., 2009, 113, 3137–3145 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, Macromolecules, 1977, 10, 1307–1315 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 977–999 CrossRef CAS.
- Y. Yagci, A. Kornowski and W. Schnabel, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1987–1991 CrossRef CAS.
- F. Kasapoglu and Y. Yagci, Macromol. Rapid Commun., 2002, 23, 567–570 CrossRef CAS.
- G. Hizal, S. E. Emiroglu and Y. Yagci, Polym. Int., 1998, 47, 391–392 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 759–764 CrossRef CAS.
- Y. Yagci and A. Ledwith, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1911–1918 CrossRef CAS.
- F. A. M. Abdulrasoul, A. Ledwith and Y. Yagci, Polymer, 1978, 19, 1219–1222 CrossRef CAS.
- Y. Yagci and W. Schnabel, Makromol. Chem., Macromol. Symp., 1992, 60, 133–143 Search PubMed.
- J. V. Crivello and M. Sangermano, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 343–356 CrossRef CAS.
- J. V. Crivello and U. Bulut, Macromol. Symp., 2006, 240, 1–11 CrossRef CAS.
- J. V. Crivello and U. Bulut, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5217–5231 CrossRef CAS.
- Y. Yagci, I. Lukac and W. Schnabel, Polymer, 1993, 34, 1130–1133 CrossRef CAS.
- D. Dossow, Q. Q. Zhu, G. Hizal, Y. Yagci and W. Schnabel, Polymer, 1996, 37, 2821–2826 CrossRef CAS.
- J. V. Crivello and J. L. Lee, Macromolecules, 1981, 14, 1141–1147 CrossRef CAS.
- Y. H. Chen, T. Yamamura and K. Igarashi, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 90–100 CrossRef CAS.
- E. W. Nelson, T. P. Carter and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 247–256 CrossRef CAS.
- E. W. Nelson, T. P. Carter and A. B. Scranton, Macromolecules, 1994, 27, 1013–1019 CrossRef CAS.
- M. R. Rodrigues and M. G. Neumann, Macromol. Chem. Phys., 2001, 202, 2776–2782 CrossRef CAS.
- G. Hizal, Y. Yagci and W. Schnabel, Polymer, 1994, 35, 2428–2431 CrossRef CAS.
- S. Denizligil, R. Resul, Y. Yagci, C. McArdle and J. P. Fouassier, Macromol. Chem. Phys., 1996, 197, 1233–1240 CrossRef CAS.
- Z. Gomurashvili, Y. J. Hua and J. V. Crivello, Macromol. Chem. Phys., 2001, 202, 2133–2141 CrossRef CAS.
- Z. Gomurashvili and J. V. Crivello, Macromolecules, 2002, 35, 2962–2969 CrossRef CAS.
- Z. Gomurashvili and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1187–1197 CrossRef CAS.
- J. V. Crivello and F. Jiang, Chem. Mater., 2002, 14, 4858–4866 CrossRef CAS.
- Y. Y. Durmaz, N. Moszner and Y. Yagci, Macromolecules, 2008, 41, 6714–6718 CrossRef CAS.
- A. Bottcher, K. Hasebe, G. Hizal, Y. Yagci, P. Stellberg and W. Schnabel, Polymer, 1991, 32, 2289–2293 CrossRef.
- U. Bulut, G. E. Gunbas and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 209–213 CrossRef CAS.
- B. Aydogan, A. S. Gundogan, T. Ozturk and Y. Yagci, Macromolecules, 2008, 41, 3468–3471 CrossRef CAS.
- E. Ertas and T. Ozturk, Tetrahedron Lett., 2004, 45, 3405–3407 CrossRef CAS.
- T. Ozturk, E. Ertas and O. Mert, Tetrahedron, 2005, 61, 11055–11077 CrossRef CAS.
- B. Aydogan, A. S. Gundogan, T. Ozturk and Y. Yagci, Chem. Commun., 2009, 6300–6302 RSC.
- B. Aydogan, G. E. Gunbas, A. Durmus, L. Toppare and Y. Yagei, Macromolecules, 2010, 43, 101–106 CrossRef CAS.
- O. Mert, E. Sahin, E. Ertas, T. Ozturk, E. A. Aydin and L. Toppare, J. Electroanal. Chem., 2006, 591, 53–58 CrossRef CAS.
- J. V. Crivello and J. H. W. Lam, J. Org. Chem., 1978, 43, 3055–3058 CrossRef CAS.
- A. Ledwith, Polymer, 1978, 19, 1217–1219 CrossRef CAS.
- E. Andrzejewska and M. Andrzejewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 665–673 CrossRef CAS.
- K. C. Anyaogu, X. C. Cai and D. C. Neckers, Macromolecules, 2008, 41, 9000–9003 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |