Benzotriazole containing conjugated polymers for multipurpose organic electronic applications
Abidin
Balan†
a,
Derya
Baran‡
a and
Levent
Toppare
*abc
aDepartment of Chemistry, Middle East Technical University, 06531, Ankara, Turkey
bDepartment of Biotechnology, Middle East Technical University, 06531, Ankara, Turkey
cDepartment of Polymer Science and Technology, Middle East Technical University, 06531, Ankara, Turkey
First published on 24th February 2011
Abstract
Benzotriazole (BTz) containing polymers have recently emerged in organic electronic applications and they are increasingly attracting a great deal of attention. These polymers are reviewed from a general perspective in terms of their potential use in three main fields, electrochromics (ECs), organic solar cells (OSCs) and organic light emitting diodes (OLEDs). In order to have a better insight into the properties of these polymers, they were compared with similar polymers. Good solubility, optical and electronic properties and synthetic availability make them multipurpose materials. They combine many desired properties in polymers and their use in different device applications is examined in detail.
 Abidin Balan | Abidin Balan received his BSc in chemistry from Bilkent University in 2007. He then joined the group of Professor Levent Toppare to pursue an MSc in chemistry. In his MSc studies, he focused on the synthesis of the Donor–Acceptor type polymers, mainly benzotriazole derivatives. He worked in the group of Prof. N. S. Sariciftci as a short term visitor during his MSc. He has published many papers on electrochromic and photovoltaic properties of conjugated polymers. Recently, he has started his PhD at Eindhoven University of Technology, NL under the supervision of Prof. Rint Sijbesma. His research interests include supramolecular polymers and mechanochemical catalysis. |
 Derya Baran | Derya Baran began her BSc studies in 2004 and joined the group of Prof. Levent Toppare in 2007 as a junior student. After her graduation in 2008, she pursued her MSc studies in the same group and during her studies she worked in Linz Institute for Organic Solar Cells (LIOS) with Prof. N. S. Sariciftci. Her MSc thesis was based on multipurpose applications of benzotriazole bearing conjugated polymers. Recently, she has started her PhD studies in the group of Prof. Christoph J. Brabec at Friedrich-Alexander University Erlangen-Nürnberg, DE. Her research interests include the characterizations of conjugated polymers for electrochromic devices and organic solar cells. |
 Levent Toppare | Levent Toppare is a professor of chemistry at the Department of Chemistry, Department of Polymer Science and Technology and Department of Biotechnology in Middle East Technical University with expertise in electrochemistry. He is also a member of Center for Solar Energy Research and Applications (GÜNAM). His research interests include conducting polymers for over 25 years. His group has recently been involved in electrochromism, electrochromic devices and photovoltaic cells. Toppare obtained his PhD (1982) degree from METU. He has published over 300 scientific papers. He is the recipient of British Council, Fulbright and Alexander von Humboldt Scholarships. |
Introduction
Conjugated polymers (CPs) have been a fundamental subject over the past couple of decades and they have been used as active materials in numerous industrial applications.1 Many different types and derivatives of conducting polymers have been successfully prepared and their potential as advanced materials has been investigated since synthetic availability for small structural modifications on the polymer backbone allows tuning of the band gap and hence their optical and electronic properties.2 In order to obtain polymers with desired properties recent research interest has mainly focused on donor–acceptor (DA) type materials where alternating electron rich and electron deficient groups are present on the backbone.3The DA type CPs generally exhibit lower band gaps and wider bandwidths than either of the corresponding parent homo-polymers. This statement is true for the polymers which satisfy necessary intramolecular interactions. The basic idea here is the contributions of donor and acceptor heterocyclics to HOMO and LUMO energy levels of the polymer.4 Mainly, the polymer concerned is expected to have an ionization potential (IP) close to the donor and an electron affinity (EA) close to the acceptor units.5 Salzner et al. theoretically proved that the D–A match is the keystone towards lower band gap polymers.6 It has been shown that in DA type polymers band gap can be decreased only if there is a high enough interaction between D and A moieties which is a consequence of low difference in HOMO–LUMO energy levels of these different groups. However, DA theory is applied not only to obtain low band gap polymers, but also to produce polymers with optical, mechanical and electronic properties which otherwise cannot be achieved by their corresponding homopolymers.
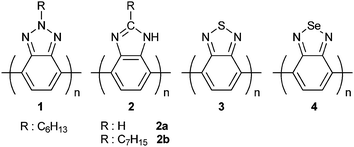
Benzotriazole (BTz) is a nitrogen containing heterocyclic benzazole derivative which exhibits high electron transporting ability due to the electron withdrawing imine (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) on its backbone. Katritzky et al. reported the extensive use of BTz as a precursor in several types of reactions.7–9 However, conjugated polymers containing BTz units in the main chain were investigated in the comprehensive reports of Yamamoto et al. They elaborated on n-type π-conjugated polymers; poly(benzotriazole) (p(BTz)) (1), poly(benzimidazole) (p(BIm)) (2), poly(benzothiadiazole) (p(BTd)) (3) and poly(benzoselenadiazole) (p(BSe)) (4) where all are potential acceptor units (Scheme 1).10–15 It has been showed that their electrochemical and optical characteristics are slightly different since they possess different atoms in the 2-positions (N, C, S, Se) in their isoelectronic structures. Among them 4 was shown to be the best electron accepting unit which can be explained by the large polarizability and the electrochemical amphotericity of selenium atoms.11,16 All these aromatic heterocycles have electronic characters affected by the polarizability differences between the atoms at 2-positions. Although a different substituent on the 2-position affects the electron density of the entire system in 1 and 2 and can change the order; present examples suggest that the electron accepting ability of these benzazoles increases from 1 to 4. Thus, 1, a moderate electron acceptorpolymer can be classified as the worst electron accepting homopolymer among its homologues2–4. However the properties achieved from the current examples of copolymers of BTz make BTz bearing CPs interesting for optoelectronic applications. Additionally, BTzpolymers are synthetically easy to achieve and due to their possible functionalization site they are easily modified, thus combining a huge synthetic diversity with great optoelectronic properties. BTz containing polymers with different DA combinations and numerous different repeating units utilized in three major fields of organic electronics, ECDs, OPVs and OLEDs, are examined in detail.
N–) on its backbone. Katritzky et al. reported the extensive use of BTz as a precursor in several types of reactions.7–9 However, conjugated polymers containing BTz units in the main chain were investigated in the comprehensive reports of Yamamoto et al. They elaborated on n-type π-conjugated polymers; poly(benzotriazole) (p(BTz)) (1), poly(benzimidazole) (p(BIm)) (2), poly(benzothiadiazole) (p(BTd)) (3) and poly(benzoselenadiazole) (p(BSe)) (4) where all are potential acceptor units (Scheme 1).10–15 It has been showed that their electrochemical and optical characteristics are slightly different since they possess different atoms in the 2-positions (N, C, S, Se) in their isoelectronic structures. Among them 4 was shown to be the best electron accepting unit which can be explained by the large polarizability and the electrochemical amphotericity of selenium atoms.11,16 All these aromatic heterocycles have electronic characters affected by the polarizability differences between the atoms at 2-positions. Although a different substituent on the 2-position affects the electron density of the entire system in 1 and 2 and can change the order; present examples suggest that the electron accepting ability of these benzazoles increases from 1 to 4. Thus, 1, a moderate electron acceptorpolymer can be classified as the worst electron accepting homopolymer among its homologues2–4. However the properties achieved from the current examples of copolymers of BTz make BTz bearing CPs interesting for optoelectronic applications. Additionally, BTzpolymers are synthetically easy to achieve and due to their possible functionalization site they are easily modified, thus combining a huge synthetic diversity with great optoelectronic properties. BTz containing polymers with different DA combinations and numerous different repeating units utilized in three major fields of organic electronics, ECDs, OPVs and OLEDs, are examined in detail.
 | ||
| Scheme 1 | ||
Electrochromics (ECs)
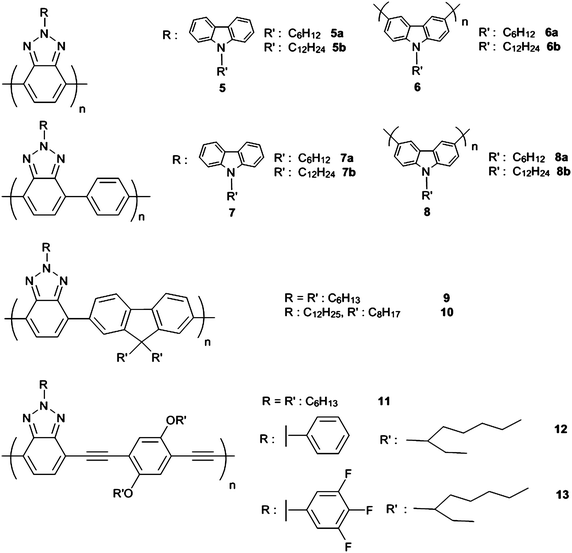
During the search for soluble, fast switching, high optical contrast and stable CPs for electrochromic devices, neutral state blue, red or green polymers with redox achievable transmissive states were aimed for. However, obtaining all these properties in a unique polymer was only possible with benzotriazole containing CPs which is the subject of this section.To the best of our knowledge, the first CPs containing BTz in the main chain (5, 9 and 11) were synthesized by Tanimoto and Yamamoto in 2004.13Hexyl substitution in 5 was not enough to have good solubility in common organic solvents. On the other hand all other polymers in the series 5–13 (Scheme 2) with longer pendant alkyl chains showed excellent solubility. Charge transfer between carbazole (CBz) pendant groups and BTz chain was proven for polymers5–8. 5a revealed a maximum absorption in the visible region at a shorter wavelength than its longer alkyl chain substituted analogue 5b. This is probably due to having different molecular weights in the soluble part of the polymers. However, longer wavelength absorption in 5a compared to 1 showed possible contribution of a charge transfer between CBz and BTz backbone.12 Additionally, photoluminescence (PL) experiments showed that the intensity of CBz emission is weaker in 5a than 5b. This suggests that photoenergy transfer from CBz pendant group to BTz main chain is more effective in shorter spacer case. Electrochemical polymerization of CBz units as in 6 and 8 yielded insoluble CPs which hindered further characterization.12 Hoger et al. reported the synthesis of orange-red polymers12 and 13 as the building blocks for low bandgap poly(arylene-ethynylene)s with 2.35 and 2.32 eV band gaps respectively.17 Red shifted visible absorption (40–50 nm) compared to the alkyl substituted 1113 can be attributed to the increase in π-stacking due to phenyl pendant group. Those polymers were not characterized in detail in terms of their optoelectronic properties however; they would be great candidates for OSCs and/or OLEDs.
 | ||
| Scheme 2 | ||
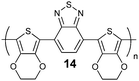
The first electro-polymerizable BTz containing heterocycle in the main chain was synthesized by our group in 2008.18BTz was coupled with ethylenedioxythiophene (EDOT) which is a well known electron donor, to give a linearly polymerizable, symmetric monomer structure like other benzazole derivatives (14–18) (Table 1). The polymer, 19 showed that the presence of BTz unit in the polymer backbone exceptionally enhanced the electrochromic properties of the parent polymer, PEDOT, in terms of optical contrast, switching time and coloration efficiency (Table 2). Additionally this polymer revealed an ambipolar character due to electron deficient BTz contribution with high stability (6% decrease in total charge after 4000 cycles), whereas PEDOT is not n-type dopable at all.19 Monomer of compound 19, 4,7-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)-2-dodecyl-2H-benzo [1,2,3] triazole (BEBT), was used as the co-monomer against benzoquinoxaline and pyrrole bearing ones and it has been shown that copolymerization with BEBT increases the optical and kinetic properties of the resulting polymer even more efficiently than simple copolymerization with Bi-EDOT.20,21
| Polymer | Colors | E m,a (V) | E p,a E p,c (V) | λ max (nm) | E g (eV) | %ΔTb | ST (s) | |
|---|---|---|---|---|---|---|---|---|
| a Optical band gaps calculated from the onset of the π–π* transition in the visible region of the neutral polymer. b Percent transmittance difference between neutral and fully oxidized states of the polymer film. c Switching time of the polymers in the visible region between neutral and fully oxidized states. 14 Adapted with permission from ref. 22, Copyright 2007 The Royal Society of Chemistry; 15 Adapted with permission from ref. 23, Copyright 2008 Wiley-VCH Verlag GmbH & Co. KGaA,Weinheim, Germany; 16, 17, 18 Adapted with permission from ref. 29, Copyright 2010 Elsevier B. V. | ||||||||
|

|
0.95 | −0.06 | 428 | 1.19 | 23.0%–755 nm | 0.4 | |
| −0.26 | 755 | 72.0%–1500 nm | ||||||

|

|
0.85 | −0.09 | 343 | 1.13 | 27.3%–448 nm | 2.1 | |
| −1.24 | 448 | 10.9%–796 nm | ||||||
| 796 | ||||||||
|

|

|
0.82 | 0.56 | 580 | 1.75 | 44.5%–580 nm | 0.4 |
| 0.38 | 75.3%–1800 nm | |||||||

|

|
0.80 | 0.51 | 560 | 1.69 | 44.5%–560 nm | 0.4 | |
| 0.33 | 71.4%–1800 nm | |||||||
|

|
0.96 | 1.06 | 513 | 1.77 | 10.0%–513 nm | 2.5 | |
| 0.40 | 36.0%–1370 nm | |||||||
| Polymer | Optical Contrast (%ΔT) | Switching Time (s) | Coloration Efficiency (cm2/C) | n-Doping |
|---|---|---|---|---|
| 19 | 53 | 1.1 | 211 | ✓ |
| PEDOT | 44 | 2.2 | 183 | ✗ |
Benzazole-EDOT bearing DA type ECPs, 14 and 15, revealed a green color in their neutral states accompanied by highly transmissive oxidized states.22,23 They showed two π–π* transitions which was claimed as essential to obtain green colored polymers.24–27 These are generally attributed to two transitions: (1) between the valence band and antibonding orbital, and (2) between the valence band and the narrow conduction band.28 Lower band gap and red shifted absorption in 15 (compared to 14) suggest that the D–A match is superior in 15. However, in terms of electrochromism, 14 showed better properties such as switching time and high optical contrast in the NIR region, besides being the first green to transmissive electrochromic polymer in the literature.22 Compared to others (14–18) 19 showed a high energy absorption with a negligibly weak intensity which indicates that the polymer have a dominant donor character rather than DA as was the case for BIm bearing polymers.29BIm and EDOT containing polymers16–18 are great examples of the fact that the substituent is affecting the entire electronic and optical properties in such polymers, i.e. fine color tuning can be achieved with different substituent groups.
Enhancing the electrochromic properties of PEDOT in 19 opened a new gate for electrochromic polymers (Fig. 1). Addition of benzotriazole might also yield soluble conjugated polymers due to the alkyl substitution on N1 position of BTz unit (designated as R in Fig. 1). In this manner, thiophene was also coupled to BTz in order to improve its electrochromic properties. Although solution processable red to highly transmissive polymers have been synthesized recently,30 poly(3,4-ethylenedioxypyrrole) (PEDOP) was the only example for red to transmissive switching ECP, although it lacks solution processability.31 Thus, upon addition of BTz units to the PTh chain a red to transmissive switching solution processable ECP could have been realized. However, this attempt in polymer20 showed unexpected properties such as switching between all primary colors of RGB,32,33 solution processability and photoluminescence.34 It is noteworthy that this was the first example to show all these properties in a single polymer. After this outstanding invention, curiosity to see the effect of other donor groups on color and kinetic properties of BTz bearing polymers was arisen, leading to the production of polymers21–26. Selenophene, for instance, has a lower oxidation potential and higher electron donating character than those of thiophene.35 Low oxidation potential increases the quality of electrochemically produced polymer film by minimizing overoxidation. Selenophene coupled polymer22 was shown to have red-purple neutral and highly transmissive oxidized states.36 Even though thiophene and selenophene seem very similar in their natures, 22 did not show multicolored oxidation states due to its low energy polaronic states (≥800 nm). Visible absorption of polymer chain was diminished upon oxidation and transition state colors between neutral and oxidized forms could not be observed.
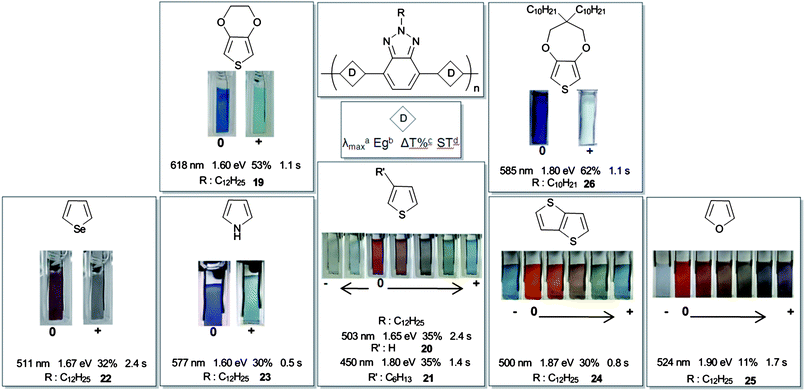 | ||
| Fig. 1 Chemical structures of representative BTz containing conjugated polymers, their colors in neutral, various oxidized and reduced states and electrochromic properties of the polymer films. a Absorption maximum of the neutral state polymer films in visible region. b Optical band gap. c Percent transmittance in the visible region. d Switching time in the visible region. 19 Adapted with permission from ref. 18, Copyright 2008 American Chemical Society; 20 Adapted with permission from ref. 34, Copyright 2009 The Royal Society of Chemistry; 21 Adapted with permission from ref. 146, Copyright 2010 American Chemical Society; 22 Adapted with permission from ref. 36, Copyright 2009 Elsevier B. V.; 23 Adapted with permission from ref. 41, Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany; 24, 25 Adapted with permission from ref. 45, Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. | ||
Diaz et al. reported electrochemical properties of polypyrrole which is an electron rich and easy to polymerize heterocycle both chemically and electrochemically.37,38,39 Thin polypyrrole film is yellow/green in the neutral and blue/violet in the doped state and it has 2.7 eV Eg.40 The most important drawback with the pyrrole containing CPs is their insolubility. However, pyrrole derivative 23 was shown to be both p and n type dopable and soluble41 which are unusual properties for a pyrrole containing polymer.4 Although, DA type benzoquinoxalinepolymers bearing a pyrrole unit showed green to transmissive electrochromic switching,42,43 thin film of 23 switched between blue and highly transmissive states in a low working potential range (between 0.2 V and 0.4 V).
Thienothiophene (TTh) is an electron rich aromatic heterocycle with extended conjugation compared to its single ring counterpart, thiophene.44 Despite this fact, the DAD type monomer of 24 has the highest oxidation potential among all BTz containing monomers.45 This can be attributed to the high resonance stabilization energy of the fused Th rings in TTh. It resulted in a low lying HOMO causing the extraction of electron difficult from the system.46 As its thiophene containing homologue20, 24 also showed multicolored electrochromic properties. Stepwise oxidation of 24 allowed the observation of several different colored oxidation states. Moreover, it surprisingly revealed the same color for p-doped and n-doped states which is not likely to be seen due to the energy difference between positively and negatively charged polymer chains.47
Usually, polymers for organic electronic applications are not biodegradable, and even worse, extensive use of them results in hazardous waste production.48 On the other hand, furan bearing conjugated polymers deserve a close scrutiny in terms of biodegradable polymer applications.49 Additionally, among aforementioned heterocyclics, only furan can be obtained from entirely renewable resources.50BTz and furan with all their good reputations were coupled in 25.45 In case of furan, electrochemical polymerization has always been difficult due to its high tendency to be overoxidized.51 Electrochemically produced polymer has low conductivity since overoxidation usually breaks the conjugation along the polymer backbone. However, reversible cycles for 25 in CV showed an increase in current density for polymerredox coupling during electrochemical polymerization. This indicates that the electroactive surface area on the electrode is increasing; hence a conducting polymer film is coated on the surface. This is related to the low potential requirement for monomer to initiate the electrochemical polymerization (1.15 V). Thin films of 25 also revealed multicolored electrochromism as its thiophene bearing analogue 20, with six different colored oxidation states. To the best of our knowledge, this was the maximum number of achievable different colors resulting from the oxidation of a single polymer. Additionally a highly transmissive n-doped state allows this polymer to be exploited in non-emissive electrochromic applications.
Although, 19 was introduced as the polymer which enhanced the electrochromic properties of EDOT, insolubility was the main drawback. Alkyl substitution on BTz unit was not sufficient to provide solubility to the electrochemically produced polymer. Later on, it has been shown that chemically produced polymer was soluble in common organic solvents with blue shifted absorption for neutral state. That might be due to shorter polymer chains compared to the electrochemically produced ones.52 On the other hand, Önal and Cihaner et al. reported the DA type polymer26; 3,3-didecyl-3,4-dihydro-2H-thieno[3,4-b]- [1,4]dioxepine coupled with BTz in the main chain which switches between deep blue and highly transmissive states.53 Additional alkyl chains on the donor group compared to 19 provided good solubility as well as further improvement in electrochromic properties which were also better than its BTd containing blue to transmissive switching counterpart.54
Polymers 27–30 in Fig. 2 illustrate the effects of substitution on optical and electrochemical properties for BTz bearing CPs. Benzyl substituted 27,55 analogous to the alkyl substituted 19, showed lower optical contrast. It should be noted here that long alkyl chains on the polymer backbone increase the optical contrast for the polymer which can be attributed to the separation between polymer chains.56 Increased separation facilitates the injection/ejection of the counter ion and provides higher optical contrast.57 In the case of 28 which has an isomeric structure to 27, benzyl group was substituted from N1-position of BTz instead of N2. This change in position of substitution decreased the effective conjugation length in 28 compared to 27, and entire properties of the polymer changed accordingly. Neutral state absorbance was blue shifted from 625 nm to 477 nm, thus colors of the polymer films in their neutral states were completely different. Polaronic state absorbance was also blue shifted to higher energy region which resulted in a colored oxidized state rather than transmissive. The difference between N2-substituted 29 and N1-substituted 30 was even more dramatic when thiophene was the donor group.58 Neither of the N1-substituted polymers showed tendency for n-type doping in the working potential range due to the change in the effective conjugation length of acceptor unit which hindered the negative charge carrier formation on polymer backbone.
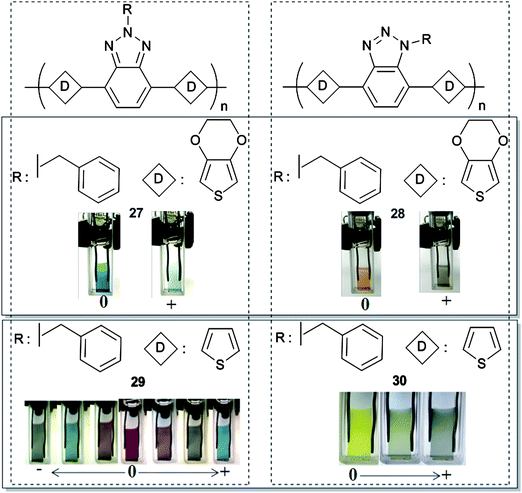 | ||
| Fig. 2 Chemical structures of D–A type BTzpolymers and their colors in neutral, oxidized and reduced states. 27, 28 Adapted with permission from ref. 55, Copyright 2010 Elsevier B. V.; 29, 30 Adapted with permission from ref. 58, Copyright 2010 Elsevier B. V. | ||
Towards solution processable multicolored electrochromic CPs, 31–34 with different benzotriazole bridged thiophene chains were investigated (Scheme 3). All three polymers, 31, 32 and 33, differing only in the length of alkyl chain, were red in their neutral states and blue in their oxidized states.59 However their spectral responses upon stepwise oxidation were different. Alkyl chains on the polymer backbone affected the doping rate due to aforementioned reasons and with increasing the length of the alkyl pendant group a greater number of different colors became detectable. The slowest switching time for 33 is also a possible reason for the observation of several different colored states. The most important properties for these polymers are their potential candidacy to be used as multicolor to transmissive switching ECPs since reduction of the neutral chains resulted in the depletion of optical absorbance in the visible region. Reported results showed that 33 has 27% optical contrast between red and transparent states and 12% optical contrast between blue and transparent states. It switches between these states within 1.7 and 2.3 s respectively. Additional substitution of the phenyl and thiophene units (compare to 20) increased the doping rates in 34 and 35, which in turn, led to the detection of fewer colors.60 The fraction of benzotriazole moieties per repeating unit on the 34 and 35 decreased compared to 20 and this allowed more accessible volume in the polymer films where dopant ions can easily be incorporated. Faster switching due to facilitated ion accumulation impeded the occurrence of two distinct transitions and observation of green colored state. On the other hand, they can be classified as fast switching ECPs accompanied with the multicolored to transmissive switching properties.
Poly-9,9-dialkylfluorenes (PFOs) and their copolymers with different groups are considered as some of the most promising materials to be used in organic electronics.61–64 Especially BTd-PFOcopolymers were exploited in highly efficient OPVs, OLEDs and OFETs.65–68Copolymerization of BTz derivatives with fluorene as in 10, 36 and 37, yielded soluble multicolored polymers with moderate band gaps compared to their parent polymers or BTd containing counterparts.69 Addition of trifluoroacetic acid (TFA) to the solutions of 36 and 37 in toluene also resulted in the observation of different colors. Since polymers regain their neutral state colors after the addition of triethylamine, these polymers can be used as solution processable pH responsive chromophores.
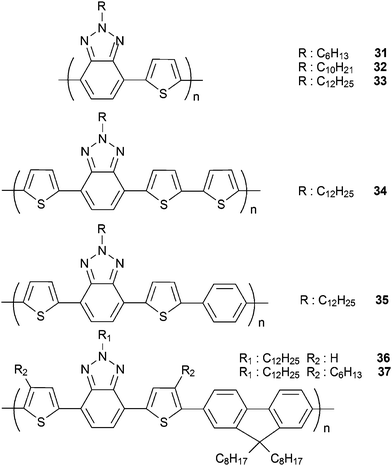 | ||
| Scheme 3 | ||
BTz bearing CPs were mostly multicolored polymers as explained so far. Multicolored polymers with a redox accessible transmissive state could be great tools for the realization of low cost and flexible devices in EC display technology.70 However, for possible smart windows applications of ECPs, black colored conjugated polymers integrated with a transmissive state should also be designed.71 They would provide easier processing through printing, spraying and coating methods and lower the cost compared to their inorganic counterparts.72 In addition, efficient light harvesting which is one of the most important parameters in organic solar cells could be achieved with these materials.73 It has been shown that black colored polymers with a transmissive oxidized state can be achieved by donor acceptor type BTd containing copolymers with extended neutral state absorption over the entire visible region.74,75 Recently BTz bearing random copolymers were also employed as black to transmissive switching ECPs. Electrochemical polymerization in the presence of two different monomers resulted in black colored soluble polymer38 (Scheme 4) which was deposited as a thin film on ITO.76
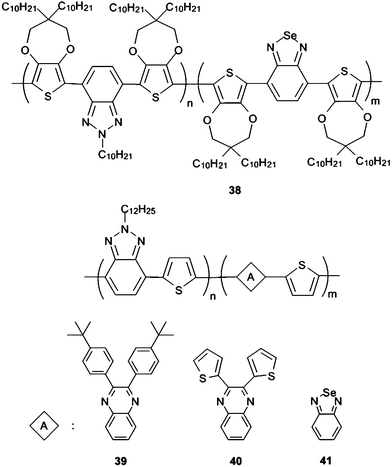 | ||
| Scheme 4 | ||
The main idea here is the combination of different oligomers absorbing in different regions of the spectrum to obtain full visible absorption. Benzoquinoxaline or BSe containing polymers were shown to be green in their neutral states.23,77 It is has been well established that neutral state green conjugated polymers reveal two distinct λmax maxima at around 400 and 700 nm.22 On the other hand, BTz and thiophene bearing polymers, for example, are red in their neutral states with λmax at ca. 500 nm. Thus, combination of benzoquinoxaline or BSe and BTz with Th as in 39–41 should allow different segments on the same polymer chain (Scheme 4) ending up with the absorption of all the visible light.78
Due to dominant absorbance at 600 nm, 39 was blue; whereas a strong and blue shifted absorption resulted in purple color in the neutral state for 41. However, since almost all visible light was absorbed by 40 it revealed black colored neutral state where the oxidation of polymer resulted in a highly transmissive oxidized state.
Organic solar cells (OSCs)
Polymer bulk heterojunction (BHJ) solar cells based on DA type conjugated homopolymers and composites as donor materials are aroused interest and offer pledge for the realization and improvement of a low cost, printable and flexible clean energy source.79–83 Over the past decade, classical polymer donor molecules, such as dialkoxy-substituted poly(para-phenylene)s (PPVs) and poly(3-hexylthiophene) (P3HT) have been investigated in detail regarding their photovoltaic applications.84,85 Therefore, in some sense, poly[2-methoxy-5-(2-ethylhexyloxy)-p-phenylenevinylene] (MEH-PPV), (poly[2-methoxy-5-(3,7-dimethyl- octyloxy)]-1,4-phenylenevinylene) (MDMO-PPV) and poly(3-hexylthiophene) (P3HT) (Scheme 5) can be classified as milestones and have been addressed in previous reviews.73,86–88 New avenues for the development of novel conjugated polymers as donor materials with improved efficiencies are required to be used in OPVs. In this part of the review, we summarize the most recent developments in conjugated polymers as active layers used either with [6,6]-phenyl-C61-butyric acid methyl ester(PC61BM) or [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) (Scheme 5) to construct highly efficient OPV devices. Several reviews on photovoltaic devices have summarized the device physics and use of conjugated polymers in organic solar cells (OSCs).89–91 Thus, we focused on the progress of the novel polymer donors starting from fluorenes, cabazoles and benzodithiophenes including benzotriazole (BTz) that have paved the pathway toward high efficiency photovoltaic devices which might be the promising candidates for the future applications of large area solar panels.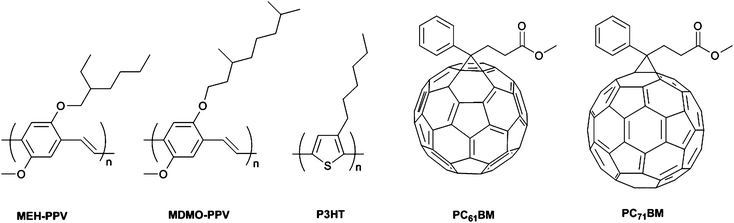 | ||
| Scheme 5 | ||
Polyfluorenes (PFO)s have been used in BHJ solar cells; however, these devices were mainly absorbing at short wavelengths due to their large band gaps, although a few exceptions exist.92–95 The first breakthrough in fluorene based polymer solar cells was pointed out by Andersson et al. in 2003.96 They synthesized poly[2,7-(9-2′-ethylhexyl)-9-hexylfluorene]-alt-5,5-(e′,7′-di-2-thienyl-2,1,3-benzo-thiadiazole] (42) (Scheme 6). This polymer has an extended absorption in the visible region to 650 nm and 2.5% efficiencies were achieved using an ITO/PEDOT:PSS/Polymer:PC61BM/LiF/Al device architecture. After their report, many groups highlighted the significance of low band gap fluorenecopolymers with benzothiadiazole and benzoselenadiazole (44–46) (Scheme 6).97–101 In 2005, alkyl-substituted fluorene with 4,7-diselenophen-2-yl-2,1,3-benzoselenadiazole was shown by Yang et al. and the polymer showed only 1% efficiency with 0.85 V Voc and 2.53 mAcm−2Jsc using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 polymer: PC61BM ratio.102 When the co-momoner was changed into 4,7-di-2-thienyl-2,1,3-benzoselenadiazole (43a) (Scheme 6) the absorption was shifted to near-infrared region and 0.91% PCE was recorded.103 The so-called “APFO-Green” series comprising PFO and various donor–acceptor segments have provided fresh air for OPVs by virtue of their high solubilities and low band gaps.104–107 With different alkyl chains or using strong electron acceptor units such as BTd,67BSe97 and thienopyrazine,108 several fluorene based copolymers have been synthesized and characterized in terms of their photovoltaic performances.68,109,110 Very recently, BTz was also involved in the PFO-based copolymers as given by Zhang et al.11147 (Scheme 6) was synthesized via Suzuki coupling and HOMO–LUMO energy levels and optical band gap values were calculated from CV measurements as −5.67, −3.43 and 2.24 eV, respectively. Fabricated devices with PC61BM and alcohol-soluble poly[(9,9-dioctyl-2,7-fluorene)-alt-(9,9-bis(3,2-(N,N-dimethylamino)propyl)-2,7-fluorene)] (PFN) as the cathode modification layer yielded a maximum of 1.3% PCE using 1:2 ratio. Its BTd bearing counterpart (APFO-3) 43b (Scheme 6) showed 2.7% PCE using dichlorobenzene as the solvent and PC71BM as the acceptor unit.112 Nevertheless, since low band gap PFOs have relatively low hole mobility, design of new materials was an urgent need for high efficiency solar cells.113–115 Poly(N-vinyl carbazole) (PVCz) was utilized due to its well-known excellent photoconductivity.116 In 2006, Müllen et al. have reported 0.6%117 and 0.8% efficient solar cells with PVCz derivatives as the start of these studies. However, with the growing demand for technology, important progress has been made in understanding the device structures and improvements in device efficiencies. Leclerc and his group reported a processable, high molecular weight and highly conjugated PVCz derivative to be used in efficient BHJ solar cells in 2007.11850 was synthesized via Suzuki coupling and its electrochromic and photovoltaic properties were reported. HOMO–LUMO energy levels were determined as −5.5 eV/−3.6 eV for the polymer which were reasonable electronic energy levels for OSCs (EHOMO level between −5.2 and −5.8 eV; and ELUMO level between −3.7 and −4.0 eV).119 3.6% efficient solar cells were fabricated using 50:PC61BM in 4:1 ratio with 0.89 V open circuit voltage (Voc), 6.52 mA cm−2 short circuit current (Jsc) and 63% fill factor (FF). Moreover, this study was improved using titanium oxide (TiOx) as the optical spacer by Park and colleagues in 2009.120 6.1% power conversion efficiency was obtained having 10.6 mA cm−2Jsc with BHJ composites comprising 50:PC71BM. Almost 100% internal quantum efficiencies were reported in the paper, implying that all photo-generated charge carriers were collected at the electrodes. The results were also confirmed by National Renewable Energy Laboratory (NREL). These studies were further investigated by replacing the BTdgroup with BTz on the polymer backbone. Very recently, 55 was synthesized from 4,7-dibromothieny-octyl-1,2,3-benzotriazole and a poly(2,7-carbazole) derivative via Suzuki cross-coupling polymerization.121 HOMO–LUMO energies and the optical band gap values were calculated as −5.70, −3.82 and 2.16 eV respectively. BTzgroup on the polymer chain lowered the HOMO–LUMO energy levels probably due to is strong electron accepting ability.122 The fabricated devices showed that 1:2; 55:PC61BM ratio has the highest performance and 77 nm thick solar cells yielded the best efficiency of 2.1%, 0.9 V Voc, 5.57 mA cm−2Jsc and 42% FF.
:3 polymer: PC61BM ratio.102 When the co-momoner was changed into 4,7-di-2-thienyl-2,1,3-benzoselenadiazole (43a) (Scheme 6) the absorption was shifted to near-infrared region and 0.91% PCE was recorded.103 The so-called “APFO-Green” series comprising PFO and various donor–acceptor segments have provided fresh air for OPVs by virtue of their high solubilities and low band gaps.104–107 With different alkyl chains or using strong electron acceptor units such as BTd,67BSe97 and thienopyrazine,108 several fluorene based copolymers have been synthesized and characterized in terms of their photovoltaic performances.68,109,110 Very recently, BTz was also involved in the PFO-based copolymers as given by Zhang et al.11147 (Scheme 6) was synthesized via Suzuki coupling and HOMO–LUMO energy levels and optical band gap values were calculated from CV measurements as −5.67, −3.43 and 2.24 eV, respectively. Fabricated devices with PC61BM and alcohol-soluble poly[(9,9-dioctyl-2,7-fluorene)-alt-(9,9-bis(3,2-(N,N-dimethylamino)propyl)-2,7-fluorene)] (PFN) as the cathode modification layer yielded a maximum of 1.3% PCE using 1:2 ratio. Its BTd bearing counterpart (APFO-3) 43b (Scheme 6) showed 2.7% PCE using dichlorobenzene as the solvent and PC71BM as the acceptor unit.112 Nevertheless, since low band gap PFOs have relatively low hole mobility, design of new materials was an urgent need for high efficiency solar cells.113–115 Poly(N-vinyl carbazole) (PVCz) was utilized due to its well-known excellent photoconductivity.116 In 2006, Müllen et al. have reported 0.6%117 and 0.8% efficient solar cells with PVCz derivatives as the start of these studies. However, with the growing demand for technology, important progress has been made in understanding the device structures and improvements in device efficiencies. Leclerc and his group reported a processable, high molecular weight and highly conjugated PVCz derivative to be used in efficient BHJ solar cells in 2007.11850 was synthesized via Suzuki coupling and its electrochromic and photovoltaic properties were reported. HOMO–LUMO energy levels were determined as −5.5 eV/−3.6 eV for the polymer which were reasonable electronic energy levels for OSCs (EHOMO level between −5.2 and −5.8 eV; and ELUMO level between −3.7 and −4.0 eV).119 3.6% efficient solar cells were fabricated using 50:PC61BM in 4:1 ratio with 0.89 V open circuit voltage (Voc), 6.52 mA cm−2 short circuit current (Jsc) and 63% fill factor (FF). Moreover, this study was improved using titanium oxide (TiOx) as the optical spacer by Park and colleagues in 2009.120 6.1% power conversion efficiency was obtained having 10.6 mA cm−2Jsc with BHJ composites comprising 50:PC71BM. Almost 100% internal quantum efficiencies were reported in the paper, implying that all photo-generated charge carriers were collected at the electrodes. The results were also confirmed by National Renewable Energy Laboratory (NREL). These studies were further investigated by replacing the BTdgroup with BTz on the polymer backbone. Very recently, 55 was synthesized from 4,7-dibromothieny-octyl-1,2,3-benzotriazole and a poly(2,7-carbazole) derivative via Suzuki cross-coupling polymerization.121 HOMO–LUMO energies and the optical band gap values were calculated as −5.70, −3.82 and 2.16 eV respectively. BTzgroup on the polymer chain lowered the HOMO–LUMO energy levels probably due to is strong electron accepting ability.122 The fabricated devices showed that 1:2; 55:PC61BM ratio has the highest performance and 77 nm thick solar cells yielded the best efficiency of 2.1%, 0.9 V Voc, 5.57 mA cm−2Jsc and 42% FF.
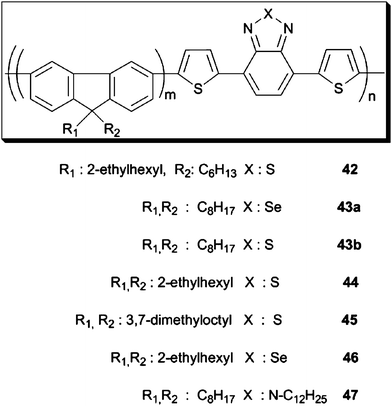 | ||
| Scheme 6 | ||
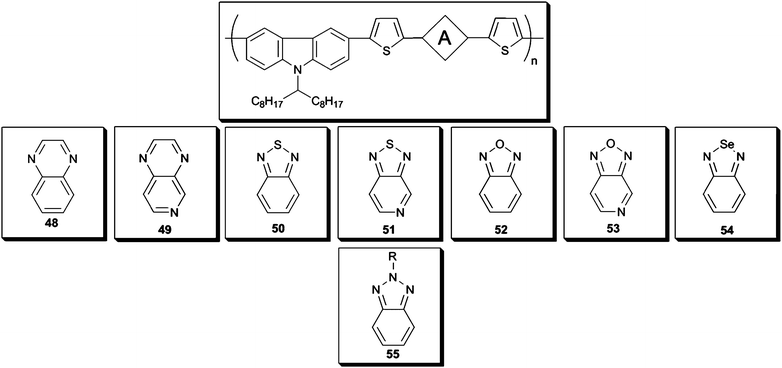 | ||
| Scheme 7 | ||
To have an insight into the chemical structure and ideal donor polymer for the BHJ solar cells, many acceptor units were inserted into the CBz containing polymer backbones. Numerous donor molecules bearing PVCz as the comonomer were designed with either benzazoles or pyridine derivatives (48–53) (Scheme 7).123 HOMO–LUMO energy levels were determined more precisely for the polymers48–53 due to the dual electrochemical properties of the polymers. It has been seen that altering the electron deficient part of the polymer chain did not change the polymeroxidation potential significantly. On the other hand, LUMO levels responded differently. The LUMO energy levels were strongly affected by the electron withdrawing center on the polymer chain and stronger electron-deficient units resulted in low lying LUMO levels. For comparison purposes a BTz and CBz derivative 55 (Scheme 7) was also synthesized via Suzuki coupling by Zhang et al.111 The polymer was used as the donor material together with PC61BM. Devices comprising either PFN as cathode modification layer showed obvious improvements of PV performance.124,125 Its BSehomologue54 (Scheme 7) was also synthesized recently by Zhao et al.126 Incorporation of BSe unit into the polymer backbone resulted in a lower optical band gap (1.73 eV) compared to its BT (1.88 eV) and BTz (2.18 eV) analogues and 50 (1.85 eV).111,127,128 Next to 50, 55 exhibited the highest power conversion efficiencies among the other CBz derivatives. The low efficiencies of 51 and 53 were probably due to low molecular weight and mobilities.129 The slightly lower Voc value for 54 is due to the higher HOMO energy level of the polymer since it has been suggested that Voc can be correlated with the difference between LUMO level of the acceptor and HOMO energy level of the donor units. Better PCE values were obtained when PC61BM was replaced by PC71BM which has stronger absorption in the visible region (Table 3).130
| Number | Device architecture | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| 42 | ITO/PEDOT:PSS/Polymer:PC61BM/LiF/Al | 1.04 | 4.66 | 46 | 2.2 |
| 43a | ITO/PEDOT:PSS/Polymer:PC61BM (1:3)/Ba/Al |
0.85 | 2.53 | 32.7 | 0.91 |
| 43b | ITO/PEDOT:PSS/Polymer:PC71BM (1:3)/LiF/Al |
0.94 | 6.2 | 0.46 | 2.7 |
| 44 | ITO/PEDOT:PSS/Polymer:PC71BM (1:3)/Ca/Al |
0.95 | 8.4 | 44 | 3.50 |
| 45 | ITO/PEDOT:PSS/Polymer:PC71BM (1:3)/Ca/Al |
0.97 | 9.1 | 51 | 4.50 |
| 46 | ITO/PEDOT:PSS/Polymer:PC71BM (1:3)/Ca/Al |
0.52 | 5.0 | 34.3 | 0.89 |
| 47 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/PFN/Al |
1.0 | 2.64 | 51.5 | 1.30 |
| 48 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.95 | 3.0 | 56 | 1.80 |
| 49 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.90 | 2.6 | 44 | 1.10 |
| 50 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.86 | 6.8 | 56 | 3.60 |
| 50 | ITO/PEDOT:PSS/Polymer:PC71BM/TiOx/Al | 0.88 | 10.6 | 66 | 6.10 |
| 51 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.71 | 2.9 | 32 | 0.70 |
| 52 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.96 | 3.7 | 60 | 2.40 |
| 53 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.85 | 1.4 | 60 | 0.80 |
| 54 | ITO/PEDOT:PSS/Polymer:PC61BM (1:4)/Al |
0.80 | 4.15 | 44 | 1.46 |
| 54 | ITO/PEDOT:PSS/Polymer:PC71BM (1:4)/Al |
0.75 | 7.23 | 45 | 2.58 |
| 55 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.90 | 5.57 | 42 | 2.1 |
| 55 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.80 | 3.91 | 48.4 | 1.51 |
| 55 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/PFN/Al |
0.90 | 4.68 | 65.3 | 2.75 |
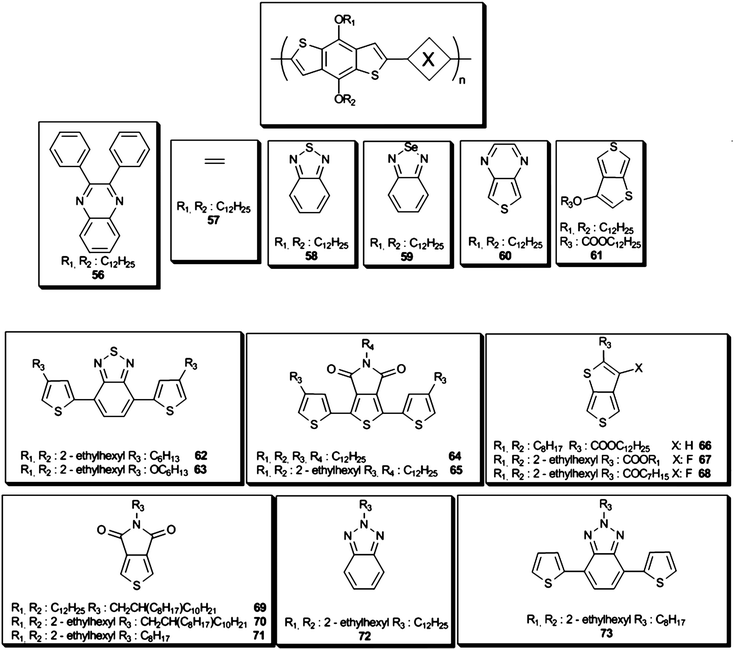
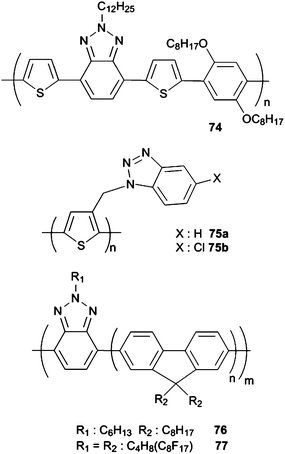
In order to modulate the band gap and energy levels, different copolymers of BTd and BSe were designed and synthesized. For instance, copolymers with fluorene, CBz, dithienylsilole, dithienylpyrrole and benzodithiophene (BDT) were used with strong electron acceptor benzazoles such as BTd and BSe.96,126,131,132 Among others, BDT comprising copolymers have become of great interest since 2008. Hou et al. synthesized and characterized a series of 4,8-bis-dodecyloxy-BDTcopolymers56–60 (Scheme 8) to investigate the effects of copolymerized functional groups on band gap.133 Changing the acceptor unit, band gap lowering abilities of the electron deficient units were determined as thieno[3,4-b]pyrazine (TPZ) ≫ BSe > 2,3-diphenylquinoxaline (DPQ) > BTd. The power conversion efficiencies of the devices were only up to 0.9% with BTdas the acceptor. In 2009, Liang and co-workers synthesized a COOR3 (R3: C12H25) substituted thienothiophene and BDT derivative 61 (Scheme 8) (PCE %: 5.30),134 and the copolymer exhibited 5.6% efficiency with a corrected Jsc of 15.6 mA cm−2 by NREL. Hou and his group, in 2009, synthesized two other BDT derivatives 62 and 63 (Scheme 8) with alkyl and alkoxy chains on the thiophene units to examine the side chain effect.135 After annealing both 62 (1.95% PCE) and 63 (1.28% PCE) revealed higher photovoltaic performances with PC71BM. Szarko and co-workers also focused on BDT with various side chains and obtained 7.4% PCE with 2-ethylhexyl and 2-ethylhexyloxy branched alkyl chains (67) (Scheme 8) using both PC61BM and PC71BM.136 Liang et al. also reported a BDT derivative 67 showing 7.4% power conversion efficiency with 14.50 mA cm−2Jsc using chlorobenzene and 1,8-diiodobenzene (DIB).137 By changing the fluorinated electron acceptor unit to an alkyl chain a poly[4,8-bis-substituted-benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl-alt-4-substituted-thieno[3,4-b]thiophene-2,6-diyl] derivative 68 (Scheme 8) was synthesized. It revealed a very high PCE % of 7.73 which certified by NREL as 6.77%.138 Alternating BDT and N-alkylated dioxopyrrolothiophenecopolymers (69 and 70 (Scheme 8)) were also used for OPVs and 3.42% and 4.79% PCE were achieved with 2% diiodooctane (DIO) and PC71BM as the acceptor.139 Recently, Leclerc’s group raised the efficiency of the BDT and thieno[3,4-c]pyrrole-4,6-dione (71) copolymer (Scheme 8) to 5.5% using PC71BM.140 Frechet and co-workers improved the efficiency to 6.8% with 1:1.5 polymer:PC61BM ratio using a chlorobenzene/DIO (99/1, v/v) solution.141 Alternating bisthiophene-phthalimide (PhBT) and BDTcopolymers64 and 65 (Scheme 8) were also synthesized viaStille coupling by Zhang et al. recently; however, only 1.08% and 1.54% maximum efficiencies were recorded which were not as high for the previous BDT derivatives.142 Lately, combination of BDT with BTz was also utilized and their photovoltaic properties were investigated in detail.143 The polymer72 (was obtained viaStille coupling and additional Th units added to 72 yielded the low band gap polymer73(Scheme 8) . Since the polymers were only p-type dopable, LUMO energies were estimated from the optical band gap and HOMO values. Photovoltaic devices were fabricated using PC61BM and PC71BM with a device architecture of ITO/(40 nm) PEDOT:PSS/active layer/(20 nm) Ca/(60 nm) Al. The optimal results for BDT and BTz based copolymer/PC61BM devices were 0.61 V Voc, 3.1 mA cm−2Jsc, 37% FF and 0.7% PCE. For 73/PC61BM devices best results were obtained with a 1:3 ratio (Voc: 0.61 V, Jsc: 4.4 mA cm−2, FF: 55%, PCE: 1.5%). When the acceptor unit was replaced by PC71BM the power conversion efficiencies were raised up to 1.4% and 1.7% for 72 and 73, respectively due to the higher absorption coefficient of PC71BM.130 Later the BDT unit in 73 was replaced with alkoxybenzene (74 (Scheme 9)) and the optical band gap was lowered as well as the HOMO–LUMO energy levels.111 It was surprising that although the HOMO level was lowered ca. 0.14 eV, Voc for 74:PC61BM devices decreased to 0.55 V compared to 73 (0.61 V) and could not even be improved with PFN/Al cathode modification bilayer. Maximum 1.39% power conversion efficiencies were reported for 74:PC61BM devices with 4.5 mA cm−2Jsc (Table 4).
 | ||
| Scheme 8 | ||
 | ||
| Scheme 9 | ||
| Number | Device architecture | V oc (V) | J sc (mAcm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| 56 | ITO/PEDOT:PSS/Polymer:PC61BM (1:1)/Ca/Al |
0.60 | 1.54 | 26 | 0.23 |
| 57 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.56 | 1.16 | 38 | 0.25 |
| 58 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.68 | 2.97 | 44 | 0.90 |
| 59 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.55 | 1.05 | 32 | 0.18 |
| 60 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/Al |
0.22 | 1.41 | 35 | 0.11 |
| 61 | ITO/PEDOT:PSS/Polymer:PC61BM (1:1)/Ca/Al |
0.58 | 12.5 | 65.4 | 4.76 |
| 61 | ITO/PEDOT:PSS/Polymer:PC71BM (1:1)/Ca/Al |
0.56 | 15.0 | 63.3 | 5.30 |
| 62 | ITO/PEDOT:PSS/Polymer:PC61BM (1:3)/Ca/Al (Annealed) |
0.84 | 6.25 | 36.9 | 1.95 |
| 63 | ITO/PEDOT:PSS/Polymer:PC61BM (1:3)/Ca/Al (Annealed) |
0.40 | 5.27 | 60.6 | 1.28 |
| 64 | ITO/PEDOT:PSS/Polymer:PC61BM (1:1)/LiF/Al |
0.90 | 2.40 | 50 | 1.08 |
| 65 | ITO/PEDOT:PSS/Polymer:PC71BM (1:1)/LiF/Al |
0.93 | 2.96 | 56 | 1.54 |
| 66 | ITO/PEDOT:PSS/Polymer:PC71BM (1:1.2)/Ca/Al |
0.56 | 15.0 | 63.3 | 5.60 |
| 67 | ITO/PEDOT:PSS/Polymer:PC71BM (1:1.5)/Ca/Al |
0.74 | 14.5 | 68.9 | 7.40 |
| 68 | ITO/PEDOT:PSS/Polymer:PC71BM (1:1.5)/Ca/Al + 3% DIO |
0.76 | 15.2 | 66.9 | 7.73 |
| 69 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/LiF/Al + 2% DIO |
0.93 | 6.58 | 56 | 3.42 |
| 70 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/LiF/Al + 2% DIO |
0.91 | 10.34 | 51 | 4.79 |
| 71 | ITO/PEDOT:PSS/Polymer:PC71BM (1:2)/LiF/Al |
0.85 | 9.81 | 66 | 5.50 |
| 71 | ITO/PEDOT:PSS/Polymer:PC61BM (1:1.5)/Ca/Al + 2% DIO |
0.85 | 11.5 | 68 | 6.80 |
| 72 | ITO/PEDOT:PSS/Polymer:PC61BM (1:3)/Ca/Al |
0.61 | 3.10 | 37 | 0.70 |
| 72 | ITO/PEDOT:PSS/Polymer:PC71BM (1:3)/Ca/Al |
0.61 | 4.80 | 47 | 1.40 |
| 73 | ITO/PEDOT:PSS/Polymer:PC61BM (1:3)/Ca/Al |
0.61 | 4.40 | 55 | 1.50 |
| 73 | ITO/PEDOT:PSS/Polymer:PC71BM (1:4)/Ca/Al |
0.61 | 4.50 | 62 | 1.70 |
| 74 | ITO/PEDOT:PSS/Polymer:PC61BM (1:2)/PFN/Al |
0.55 | 4.50 | 56.1 | 1.39 |
In order to increase the power conversion efficiencies electron donorgroups were widely used with BTd, benzoquinoxaline144 and thieno(3,4-b)pyrazine cores.145 Nevertheless, photovoltaic and photophysical investigations of BTzhomopolymers were not carried out yet. Recently, BTz and a 3HT derivative 21 was polymerized by our group using FeCl3 as the oxidizing agent and characterized in terms of its photovoltaic properties.146 The polymer was unique since BTz was used as the acceptor moiety in OSCs for the first time. The polymer21 was fluorescent and both p and n-type dopable. HOMO–LUMO levels and electrochemical band gap values were calculated from the EVS and CV measurements as −5.45 eV and −2.95 eV and 2.55 eV, respectively. Using different PC61BM loadings, several solar cells were produced with 21 and preliminary results showed that 2.35 mA cm−2 short circuit density and Voc values up to 0.85 V could be achieved. Once it has been realized that BTz incorporated donor–acceptor type polymers can be used for photovoltaic applications, a chemically synthesized BTz derivative homopolymer19 was also used as the donor material in BHJ solar cells with PC61BM. 19 showed that 19:PC61BM blends can yield a maximum Voc of 250 mV and Jsc of 1.5 mA cm−2 under AM 1.5 conditions. The low open circuit potential of the blends may be attributed to the high HOMO energy level of 19 compared to other donor materials which lie between −5.2 and −5.8 eV.52 Ultimately, a BTz and Th bearing chemically polymerized homopolymer20 was also utilized in organic photovoltaics as the donor material with PC61BM. The preliminary results showed that the devices with different feed ratios can have 0.65 V Voc, 4.02 mA cm−2Jsc and 0.8% PCE using dichlorobenzene.147 Due to its dual properties, the polymer was also used as an n-type material but no significant results were obtained.
Organic light emitting diodes (OLEDs)
After the breakthrough discovery of electroluminescence (EL) in conjugated polymers reported in 1990,148 a considerable effort has been devoted to develop conjugated materials as the active layers in OLEDs for use in display fabrication.149Polymer LEDs which can be deposited on large areas show high efficiency and fine color variation whereas these are not attainable with inorganic materials.150 Nevertheless, suitable processing methods can provide a wide range of pattering options for OLEDs.151The earliest example for BTz containing fluorescent conjugated polymer is 75a which contains BTz as the electron-accepting pendant group.152 Its showed orange-red electroluminescence (EL) (λmax = 580 nm) with an efficiency much higher than that of octyl substituted Ths.153 Cl substitution on BTz unit as in 75b resulted in a lower EL efficiency than that of 42a.
Although Yamamoto et al. reported the synthesis and photoluminescence properties of PFO and BTzcopolymers, electroluminescence properties of 75a and 75b (Scheme 9) were investigated by Cao et al.154 Three different polymers were prepared with different fractions of BTz units in the polymer backbone by random copolymerization. Increasing BTz content resulted in a slight decrease in the optical band gap and HOMO/LUMO levels of copolymers. It was shown that 1.62% external quantum efficiency and 2.69 of cd A−1 luminance efficiency with high color stability could be obtained when 75b was used.
![(a) Polymer structures of RF-B, RF-G, and RF-Rpolymers. (b) The energy diagram of ITO, PEDOT:PSS, poly[(9,9-dioctylfluorene)- co -(4-butylphenyldiphenylamine)] (TFB), Poly(9,9-dioctylfluorene) (F8), RF-B, RF-G, RF-R, and Ca. Adapted with permission from ref. 156, Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA.](/image/article/2011/PY/c1py00007a/c1py00007a-f3.gif) | ||
| Fig. 3 (a) Polymer structures of RF-B, RF-G, and RF-Rpolymers. (b) The energy diagram of ITO, PEDOT:PSS, poly[(9,9-dioctylfluorene)- co -(4-butylphenyldiphenylamine)] (TFB), Poly(9,9-dioctylfluorene) (F8), RF-B, RF-G, RF-R, and Ca. Adapted with permission from ref. 156, Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Ober et al. synthesized PFO and BTzcopolymer76 and its semiperfluoroalkyl substituted derivatives 77 (Scheme 9) and RF-B (Fig. 3).155,156 These fluorinated polymers in film form remained unaffected when exposed to organic solvents and spin coated from hydrofluoroether solution on to a prepatterned ITO substrate to show polymers are patternable under conventional photolithographic conditions. This patterned device allowed pixel array operation and gave light emission in 100 μm scale. (Fig. 4) After this report, the same group also achieved the photolithographic patterning of multilayer solution processed devices.156 The authors claimed that using fluorinated alkyl chains they overcome the organic solvent damage on light emitting polymers which are solution processed and photolithographically patterned.
 | ||
| Fig. 4 Operating pixels of the patterned EL device described in a) Adapted with permission from ref. 155, Copyright 2010 American Chemical Society; b) Adapted with permission from ref. 156, Copyright 2010 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Outlook
A number of BTz containing CPs are described in the literature and briefly highlighted in this review. Polymers with BTz in main chain are great candidates for use in organic electronic devices. Especially in terms of electrochromic applications, these polymers were proven to have the most important desired properties such as multicolored electrochromism, transmissive state and photoluminescence with a single polymer. However, for the display technology switching times of these polymers should be shortened which might allow fast response of polymers to external stimuli change. For OPVs and OLEDs BTz bearing polymers are still hiding their mystery because of the lack of scientific invention. Device performance is directly affected by the construction conditions, thus optimization studies can provide higher efficiencies in such devices which have not been completely studied yet. A number of different derivatives to vary the electron accepting ability of the BTz unit can be synthesized and their potentials in organic electronics can be investigated due to the promising optoelectronic properties of BTz. From the application point of view, new materials should be designed and tested for these devices in order to connect high efficiency to state of the art devices. Although only optoelectronic properties of BTz bearing CPs are being reported, biochemical applications of these polymers can be a topic of investigation. With all these results and curiosity about BTz bearing polymers, they will attract more attention in the near future.References
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- D. A. M. Egbe, B. Carbonnier, E. Birckner and U. W. Grummt, Prog. Polym. Sci., 2009, 34, 1023–1067 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS.
- C. A. Thomas, K. Zong, K. A. Abboud, P. J. Steel and J. R. Reynolds, J. Am. Chem. Soc., 2004, 126, 16440–16450 CrossRef CAS.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55–57, 299–306 CrossRef CAS.
- U. Salzner and M. E. Köse, J. Phys. Chem. B, 2002, 106(10), 9221–9226 CrossRef CAS.
- A. R. Katritzky, X. Lan, J. Z. Yang and O. V. Denisko, Chem. Rev., 1998, 98, 409–548 CrossRef CAS.
- A. R. Katritzky and B. V. Rogovoy, Chem.–Eur. J., 2003, 9, 4586–4593 CrossRef CAS.
- A. R. Katritzky and S. Rachwal, Chem. Rev., 2010, 110, 1564–1610 CrossRef CAS.
- T. Yamamoto, K. Sugiyama, T. Kanbara, H. Hayashi and H. Etori, Macromol. Chem. Phys., 1998, 199, 1807–1813 CrossRef CAS.
- T. Kanbara and T. Yamamoto, Chem. Lett., 1993, 22, 419–422 CrossRef.
- A. Tanimoto and T. Yamamoto, Macromolecules, 2006, 39, 3546–3552 CrossRef CAS.
- A. Tanimoto and T. Yamamoto, Adv. Synth. Catal., 2004, 346, 1818–1823 CrossRef CAS.
- T. Yasuda, T. Imase and T. Yamamoto, Macromolecules, 2005, 38, 7378–7585 CrossRef CAS.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 2010, 83, 431–455 CrossRef CAS.
- Y. Tsubata, T. Suzuki, T. Miyashi and Y. Yamashita, J. Org. Chem., 1992, 57, 6749–6755 CrossRef CAS.
- H. Wettach, F. Pasker and S. Höger, Macromolecules, 2008, 41, 9513–9515 CrossRef CAS.
- A. Balan, G. Gunbas, A. Durmus and L. Toppare, Chem. Mater., 2008, 20, 7510–7513 CrossRef CAS.
- J. Roncali, P. Blanchard and P. Frere, J. Mater. Chem., 2005, 15, 1589–1610 RSC.
- S. Celebi, D. Baran, A. Balan and L. Toppare, Electrochim. Acta, 2010, 55, 2373–2376 CrossRef CAS.
- A. T. Taskin, A. Balan, Y. A. Udum and L. Toppare, Smart Mater. Struct., 2010, 19, 065005 CrossRef.
- A. Durmus, G. E. Gunbas, P. Camurlu and L. Toppare, Chem. Commun., 2007, 3246–3248 RSC.
- A. Cihaner and F. Algı, Adv. Funct. Mater., 2008, 18(22), 3583–3589 CrossRef CAS.
- G. Sonmez, C. K. F. Shen, Y. Rubin and F. Wudl, Angew. Chem., Int. Ed., 2004, 43, 1498–1502 CrossRef CAS.
- A. Durmus, G. E. Gunbas and L. Toppare, Chem. Mater., 2007, 19, 6247–6251 CrossRef CAS.
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Mater., 2008, 20, 691–695 CrossRef CAS.
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Funct. Mater., 2008, 18, 2026–2030 CrossRef CAS.
- A. Berlin, G. Zotti, S. Zecchin, G. Schiavon, B. Vercelli and A. Zanelli, Chem. Mater., 2004, 16, 3667–3676 CrossRef CAS.
- H. Akpınar, A. Balan, D. Baran, E. K. Ünver and L. Toppare, Polymer, 2010, 51, 6123–6131 CrossRef.
- A. L. Dyer, M. R. Craig, J. E. Babiarz, K. Kiyak and J. R. Reynolds, Macromolecules, 2010, 43, 4460–4467 CrossRef CAS.
- R. M. Walczak and J. R. Reynolds, Adv. Mater., 2006, 18, 1121–1131 CrossRef CAS.
- G. Sonmez, H. B. Sonmez, C. K. F. Shen and F. Wudl, Adv. Mater., 2004, 16, 1905–1908 CrossRef.
- G. Sonmez, Chem. Commun., 2005, 5251–5259 RSC.
- A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt and L. Toppare, Chem. Commun., 2009, 6768 RSC.
- J. Xu, J. Hou, S. Zhang, G. Nie, S. Pu, L. Shen and Q. Xiao, J. Electroanal. Chem., 2005, 578, 345 CrossRef CAS.
- G. Cetin, A. Balan, G. Gunbas, A. Durmus and L. Toppare, Org. Electron., 2009, 10, 34 CrossRef.
- A. F. Diaz and K. K. Kanazawa, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- A. F. Diaz and J. I. Castillo, J. Chem. Soc., Chem. Commun., 1980, 397–398 RSC.
- A. F. Diaz, J. I. Castillo, J. A. Logan and W. Y. J. Lee, J. Electroanal. Chem., 1981, 129, 115 CrossRef CAS.
- E. M. Genies, G. Bidan and A. F. Diaz, J. Electroanal. Chem., 1983, 149, 101 CrossRef CAS.
- D. Baran, A. Balan, B. M. Esteban, H. Neugebauer, N. S. Sariciftci and L. Toppare, Macromol. Chem. Phys., 2010, 211, 2602–2610 CrossRef CAS.
- S. Celebi, A. Balan, B. Epik, D. Baran and L. Toppare, Org. Electron., 2009, 10, 631 CrossRef CAS.
- A. T. Taskin, A. Balan, B. Epik, E. Yildiz, Y. A. Udum and L. Toppare, Electrochim. Acta, 2009, 54, 5449 CrossRef CAS.
- Y. Mazaki and K. Kobayashi, Tetrahedron Lett., 1989, 30, 3315–3318 CrossRef CAS.
- N. Akbasoglu, A. Balan, D. Baran, A. Cirpan and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5603–5610 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. Mac-Donald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- O. Gidron, Y. D. Posner and M. Bendikov, J. Am. Chem. Soc., 2010, 132, 2148–2150 CrossRef CAS.
- A. Gandini, Polym. Chem., 2010, 1, 245–251 RSC.
- A. Gandini, Macromolecules, 2008, 41(24), 9491–9504 CrossRef CAS.
- J. K. Politis, J. C. Nemes and M. D. Curtis, J. Am. Chem. Soc., 2001, 123, 2537–2547 CrossRef CAS.
- A. Balan, D. Baran, N. S. Sariciftci and L. Toppare, Sol. Energy Mater. Sol. Cells, 2010, 94, 1797–1802 CrossRef CAS.
- M. Içli, M. Pamuk, F. Algi, A. M. Önal and A. Cihaner, Chem. Mater., 2010, 22, 4034–4044 CrossRef CAS.
- C. M. Amb, P. M. Beaujuge and J. R. Reynolds, Adv. Mater., 2010, 22, 724–728 CrossRef CAS.
- B. Yigitsoy, S. M. A. Karim, A. Balan, D. Baran and L. Toppare, Synth. Met., 2010, 160, 2534–2539 CrossRef CAS.
- M. Li, Y. Sheynin, A. Patra and M. Bendikov, Chem. Mater., 2009, 21, 2482–2488 CrossRef CAS.
- A. Kumar, D. M. Welsh, M. C. Morvant, F. Piroux, K. A. Abboud and J. R. Reynolds, Chem. Mater., 1998, 10, 896–902 CrossRef CAS.
- B. Yigitsoy, S. M. A. Karim, A. Balan, D. Baran and L. Toppare, Electrochim. Acta, 2011, 56, 2263–2268 CrossRef CAS.
- A. Balan, D. Baran and L. Toppare, J. Mater. Chem., 2010, 20, 9861–9866 RSC.
- G. Hızalan, A. Balan, D. Baran and L. Toppare, J. Mater. Chem., 2011, 21, 1804–1809 RSC.
- B. Friedel, C. R. McNeill and N. C. Greenham, Chem. Mater., 2010, 22, 3389–3398 CrossRef CAS.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. Boer, Polym. Rev., 2008, 48, 531–582 Search PubMed.
- P. Herguth, X. Jiang, M. S. Liu and A. K. Y. Jen, Proc. SPIE–Int. Soc. Opt. Eng., 2003, 4800, 138–147 CAS.
- A. Cirpan, L. Ding and F. E. Karasz, Polymer, 2005, 46, 811–817 CrossRef CAS.
- S. Beaupré, P. L. T. Boudreault and M. Leclerc, Adv. Mater., 2010, 22, E6–27 CrossRef CAS.
- M. H. Chen, J. Hou, Z. Hong, G. Yang, S. Sista, L. M. Chen and Y. Yang, Adv. Mater., 2009, 21, 4238–4242 CrossRef CAS.
- F. Huang, L. Hou, H. Shen, J. Jiang, F. Wang, Hongyu. Zhen and Y. Cao, J. Mater. Chem., 2005, 15, 2499–2507 RSC.
- S. Ko, R. Mondal, C. Risko, J. K. Lee, S. Hong, M. D. McGehee, J. L. Bredas and Z. Bao, Macromolecules, 2010, 43, 6685–6698 CrossRef CAS.
- E. Kaya, A. Balan, D. Baran, A. Cirpan and L. Toppare, Org. Electron., 2011, 12, 202–209 CrossRef CAS.
- A. A. Argun, P. H. Aubert, B. C. Thompson, I. Schwendeman, C. L. Gaupp, J. Hwang, N. J. Pinto, D. B. Tanner, A. G. MacDiarmid and J. R. Reynolds, Chem. Mater., 2004, 16, 4401–4412 CrossRef CAS.
- F. C. Krebs, Nat. Mater., 2008, 7, 766–767 CrossRef CAS.
- P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2003, 77, 117–133 CrossRef CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–38 CrossRef.
- P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7, 795–799 CrossRef CAS.
- P. Shi, C. M. Amb, E. P. Knott, E. J. Thompson, D. Y. Liu, J. Mei, A. L. Dyer and J. R. Reynolds, Adv. Mater., 2010, 22, 4949–4953 CrossRef CAS.
- M. İçli, M. Pamuk, F. Algı, A. M. Önal and A. Cihaner, Org. Electron., 2010, 11, 1255–1260 CrossRef CAS.
- Y. Udum, E. Yıldız, G. Gunbas and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3723 CrossRef CAS.
- G. Öktem, A. Balan, D. Baran and L. Toppare, Chem. Commun., 2011 10.1039/c0cc04934d.
- C. J. Brabec, M. Heeney, I. McCulloch and J. Nelson, Chem. Soc. Rev., 2011 10.1039/c0cs00045k.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS.
- P. K. Ho, D. S. Thomas, Richard H. Friend and N. Tessler, Science, 1999, 285, 233–236 CrossRef CAS.
- J. D. Yuen, R. Menon, N. E. Coates, E. B. Namdas, S. Cho, S. T. Hannahs, D. Moses and A. J. Heeger, Nat. Mater., 2009, 8, 572–575 CrossRef CAS.
- S. Shoaee, T. M. Clarke, C. Huang, S. Barlow, S. R. Marder, M. Heeney, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2010, 132, 12919–12926 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- H. Hoppe, T. Glatzel, N. Niggemann, W. Schwinger, F. Schaeffler, A. Hinsch, M. Ch, Lux-Steiner and N. S. Sariciftci, Thin Solid Films, 2006, 511–512, 587–592 CrossRef CAS.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–685 CrossRef CAS.
- B. C. Thompson and J. M. J. Frechet, Angew. Chem., 2008, 120, 62–68 CrossRef.
- C. J. Brabec, S. Gowrisanker, J. J. M. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839–3856 CrossRef CAS.
- A. W. Hains, Z. Liang, Michael A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- A. D. Bouillud, I. Levesque, Y. Tao, M. D'Iorio, S. Beaupre, P. Blondin, M. Ranger, J. Bouchard and M. Leclerc, Chem. Mater., 2000, 12, 1931–1936 CrossRef CAS.
- M. Inbasekaran, E. P. Woo, W. Wu and M. T. Bernius, WO Patent 0 046 321, 2000.
- D. M. Russell, A. C. Arias, R. H. Friend, C. Silva, C. Ego, A. C. Grimsdale and K. Müllen, Appl. Phys. Lett., 2002, 80, 2204–2206 CrossRef CAS.
- U. Asawapirom and U. Scherf, Macromol. Rapid Commun., 2001, 22, 746–749 CrossRef CAS.
- M. Svensson, F. Zhang, S. C. Venstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CrossRef CAS.
- J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS.
- C. R. McNeill, A. Abrusci, J. Zaumseil, R. Wilson, M. J. McKiernan, J. H. Burroughes, J. J. M. Halls, N. C. Greenham and R. H. Friend, Appl. Phys. Lett., 2007, 90, 193506–3 CrossRef.
- H. M. P. Wong, P. Wang, A. Abrusci, M. Svensson, M. R. Andersson and N. C. Greenham, J. Phys. Chem. C, 2007, 111, 5244–5249 CrossRef CAS.
- Z. G. Zhang, K. L. Zhang, G. Liu, C. X. Zhu, K. G. Neoh and E. T. Kang, Macromolecules, 2009, 42, 3104–3111 CrossRef CAS.
- F. Zhang, J. Bijleveld, E. Perzon, K. Tvingstedt, S. Barrau, O. Inganäs and M. R. Andersson, J. Mater. Chem., 2008, 18, 5468–5474 RSC.
- R. Yang, R. Tian, J. Yan, Y. Zhang, J. Yang, Q. Hou, W. Yang, C. Zhang and Y. Cao, Macromolecules, 2005, 38, 244–253 CrossRef CAS.
- J. Iuo, Q. Hou, J. Chen and Y. Cao, Synth. Met., 2006, 156, 470–475 CrossRef.
- O. Inganäs, F. Zhang and M. R. Andersson, Acc. Chem. Res., 2009, 42, 1731–1739 CrossRef CAS.
- S. Admassie, O. Inganäs, W. Mammo, E. Perzon and M. R. Andersson, Synth. Met., 2006, 156, 614–623 CrossRef CAS.
- X. Wang, E. Perzon, W. Mammo, F. Oswald, S. Admassie, N.-K. Persson, F. Langa, M. R. Andersson and O. Inganäs, Thin Solid Films, 2006, 511–512, 576–580 CrossRef CAS.
- C. M. B. Svanström, J. Rysz, A. Bernasik, A. Budkowski, F. Zhang, O. Inganäs, M. R. Andersson, K. O. Magnusson, J. J. Benson-Sminth, J. Nelson and E. Moons, Adv. Mater., 2009, 21, 4398–4403 CrossRef.
- E. Zhou, J. Cong, S. Yamakawa, Q. Wei, M. Nakamura, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2010, 43, 2873–2879 CrossRef CAS.
- S. Hellström, L. J. Lindgren, Y. Zhou, F. Zhang, O. Inganäs and M. R. Andersson, Polym. Chem., 2010, 1, 1272–1280 RSC.
- J. Hou, T. L. Chen, S. Zhang, H. Y. Chen and Y. Yang, J. Phys. Chem., 2009, 113, 1601–1605 Search PubMed.
- L. Zhang, C. He, J. Chen, P. Yuan, L. Huang, C. Zhang, W. Cai, Z. Liu and Y. Cao, Macromolecules, 2010, 43, 9771–9778 CrossRef CAS.
- W. Li, R. Qin, M. Andersson, F. Li, C. Zhang, B. Li, Z. Liu, Z. Bo and F. Zhang, Polymer, 2010, 51, 3031–3038 CrossRef CAS.
- F. Zhang, K. G. Jespersen, C. Björström, Svensson, M. R. Andersson, V. Sundström, K. Magnusson, E. Moons, A. Yartsev and O. Inganäs, Adv. Funct. Mater., 2006, 16, 667–674 CrossRef CAS.
- F. Zhang, W. Mammo, L. M. Andersson, S. Admassie, M. R. Andersson and O. Inganäs, Adv. Mater., 2006, 18, 2169–2173 CrossRef CAS.
- F. Wang, J. Luo, K. Yang, J. Chen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 2253–2260 CrossRef CAS.
- J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS.
- J. Li, F. Dierschke, J. Wu, A. C. Grimsdale and K. Müllen, J. Mater. Chem., 2006, 16, 96–100 RSC.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi and P. W. M. Blom, Appl. Phys. Lett., 2006, 88, 093511–093513 CrossRef.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–303 CrossRef CAS.
- B. Peng, A. Najari, B. Liu, P. Berrouard, D. Gendron, Y. He, K. Zhou, M. Leclerc and Y. Zou, Macromol. Chem. Phys., 2010, 211, 2026–2033 CrossRef CAS.
- H. A. M. Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. N-Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS.
- H. Wu, F. huang, Y. Mo, W. Yang, D. Wang, J. Peng and Y. Cao, Adv. Mater., 2004, 16, 1826–1830 CrossRef CAS.
- C. He, C. Zhong, H. Wu, R. Yang, W. Yang, F. Huang, G. C. Bazan and Y. Cao, J. Mater. Chem., 2010, 20, 2617–2622 RSC.
- W. Zhao, W. Cai, R. Xu, W. Yang, X. Gong, H. Wu and Y. Cao, Polymer, 2010, 51, 3196–3202 CrossRef CAS.
- R. B. Aich, N. Blouin, A. Bouchard and M. Leclerc, Chem. Mater., 2009, 21, 751–757 CrossRef CAS.
- P. L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- P. Schilinsky, U. Asawapirom, U. Scherf, M. Biele and C. J. Brabec, Chem. Mater., 2005, 17, 2175–2180 CrossRef CAS.
- M. M. Wien, J. M. Kroon, W. J. H. Verhees, J. Knlo, J. C. Hummelen, P. A. Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2009, 41, 1110–1119.
- L. Huo, H. Y. Chen, J. Hou, T. L. Chen and Y. Yang, Chem. Commun., 2009, 5570–5572 RSC.
- J. Hou, M. H. Park, S. Zhang, Y. Yao, L. M. Chen, J. H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Y. Liang, Y. Wu, D. Feng, S. T. Tsai, H. J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS.
- J. Hou, H. Y. Chen, S. Zhang and Y. Yang, J. Phys. Chem. C, 2009, 113, 21202–21207 CrossRef CAS.
- J. M. Szarko, J. Guo, Y. Liang, B. Lee, B. S. Rolczynski, J. Strzalka, T. Xu, S. Loser, T. J. Marks, L. Yu and L. X. Chen, Adv. Mater., 2010, 22, 5468–5472 CrossRef CAS.
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS.
- H. Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- G. Zhang, Y. Fu, Q. Zhang and Z. Xie, Chem. Commun., 2010, 46, 4997–4999 RSC.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupre, B. R. Aich, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS.
- C. Pilliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2010, 132, 7595–7597 CrossRef CAS.
- G. Zhang, Y. Fu, Q. Zhang and Z. Xie, Macromol. Chem. Phys., 2010, 211, 2596–2601 CrossRef CAS.
- Z. Zhang, B. Peng, B. Li, C. Pan, Y. Li, Y. He, K. Zhou and Y. Zou, Polym. Chem., 2010, 1, 1441–1447 RSC.
- S. Gunes, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt, N. S. Sariciftci and L. Toppare, Sol. Energy Mater. Sol. Cells, 2008, 92, 1162–1169 CrossRef.
- L. M. Campos, A. Tontcheva, S. Gunes, G. Sonmez, H. Neugebauer, N. S. Sariciftci and F. Wudl, Chem. Mater., 2005, 17, 4031–4033 CrossRef CAS.
- D. Baran, A. Balan, S. Celebi, B. M. Esteban, H. Neugebauer, N. S. Sariciftci and L. Toppare, Chem. Mater., 2010, 22, 2978–2987 CrossRef CAS.
- B. M. Esteban, A. Balan, D. Baran, J. Gasioroswski, H. Neugebauer, L. Toppare and N. S. Sariciftci, submitted.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- D. Braun and A. J. Heeger, Appl. Phys. Lett., 1991, 58, 1982–1984 CrossRef CAS.
- E. Menard, M. A. Meitl, Y. G. Sun, J. U. Park, D. J. L. Shir, Y. S. Nam, S. Jeon and J. A. Rogers, Chem. Rev., 2007, 107, 1117–1160 CrossRef CAS.
- S. H. Ahn, M. Z. Czae, E. R. Kim, H. Lee, S. H. Han, J. Noh and M. Hara, Macromolecules, 2001, 34, 2522–2527 CrossRef CAS.
- D. Braun, G. Gustafsson, D. McBranch and A. J. Heeger, J. Appl. Phys., 1992, 72, 564–568 CrossRef CAS.
- M. Sun, Q. Niu, R. Yang, B. Du, R. Liu, W. Yang, J. Peng and Y. Cao, Eur. Polym. J., 2007, 43, 1916–1922 CrossRef CAS.
- J. K. Lee, H. H. Fong, A. A. Zakhidov, G. E. McCluskey, P. G. Taylor, M. Santiago-Berrios, H. D. Abruna, A. B. Holmes, G. G. Malliaras and C. K. Ober, Macromolecules, 2010, 43, 1195–1198 CrossRef CAS.
- H. H. Fong, J. K. Lee, Y. F. Lim, A. A. Zakhidov, W. W. H. Wong, A. B. Holmes, C. K. Ober and G. G. Malliaras, Adv. Mater., 2011, 23, 735–739 CrossRef CAS.
Footnotes |
| † Current Address: Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513 5600 MB Eindhoven, The Netherlands |
| ‡ Current Address: Institute of Materials for Electronics and Energy Technology, Friedrich Alexander University Erlangen-Nurnberg, Martensstrasse 7, 91058 Erlangen, Germany |
| This journal is © The Royal Society of Chemistry 2011 |