New micellar morphologies from amphiphilic block copolymers: disks, toroids and bicontinuous micelles
Simon J.
Holder
*a and
Nico A. J. M.
Sommerdijk
*b
aFunctional Materials Group, School of Physical Sciences, University of Kent, Canterbury, Kent CT2 7NH, UK. E-mail: S.J.Holder@kent.ac.uk; Tel: +44 (0)-1227-823547
bLaboratory of Materials and Interface Chemistry and Soft Matter Cryo-TEM Research Unit, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: N.Sommerdijk@tue.nl; Tel: +31 40 247 5870
First published on 26th January 2011
Abstract
Amphiphilic AB and ABA block copolymers have been demonstrated to form a variety of self-assembled aggregate structures in dilute solutions where the solvent preferentially solvates one of the blocks. The most common structures formed by these amphiphilic macromolecules are spherical micelles, cylindrical micelles and vesicles (polymersomes). Interest into the characterisation and controlled formation of block copolymer aggregates has been spurred on by their potential as surfactants, nano- to micro-sized carriers for active compounds, for the controlled release of encapsulated compounds and for inorganic materials templating, amongst numerous other proposed applications. Research in the past decade has focussed not only on manipulating the properties of aggregates through control of both the chemistry of the constituent polymer blocks but also the external and internal morphology of the aggregates. This review article will present an overview of recent approaches to controlling the self-assembly of amphiphilic block copolymers with a view to obtaining novel micellar morphologies. Whilst the article touches upon multi-compartment micelles particular focus is placed upon control of the overall shape of micelles; i.e. those systems that expand the range of accessible morphologies beyond ‘simple’ spherical and cylindrical micelles namely disk-like, toroidal and bicontinuous micelles.
 Simon J. Holder | Dr Simon J. Holder is Senior Lecturer in Organic Chemistry in the Functional Materials Group at the School of Physical Sciences at the University of Kent. In 1994 he completed his PhD at the University of Hull in the field of Polymer and Organic Chemistry. After postdoctoral work on silicon-containing polymers, supramolecular chemistry and polymer self-assembly he moved to the University of Kent in 1999. His research focuses on the synthesis and applications of block copolymers and silicon-containing polymers and the self-assembly of amphiphilic block copolymers. |
 Nico A. J. M. Sommerdijk | Dr Nico A. J. M. Sommerdijk is associate professor in Materials and Interface Chemistry in the department of Chemical Engineering and Chemistry at the Eindhoven University of Technology. In 1995 he completed his PhD at the University of Nijmegen in the field of Organic and Supramolecular Chemistry. After postdoctoral work on inorganic materials and polymer self-assembly he moved to Eindhoven University of Technology in 1999 to work on Biomimetic Materials Chemistry. His research focuses on the use of cryoTEM in the study of (macro)molecular assemblies and their use as templates in biomimetic mineralization studies. |
1. Introduction
Amphiphilic AB and ABA block copolymers have been demonstrated to form a variety of self-assembled aggregate structures in dilute solutions where the solvent preferentially solvates one of the blocks.1 The most common structures formed by these amphiphilic macromolecules are spherical micelles, cylindrical micelles and vesicles (polymersomes).2–12 The morphology of the aggregate depends upon the relative volume ratio of the different blocks and upon the packing parameter (p), where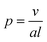 | (1) |
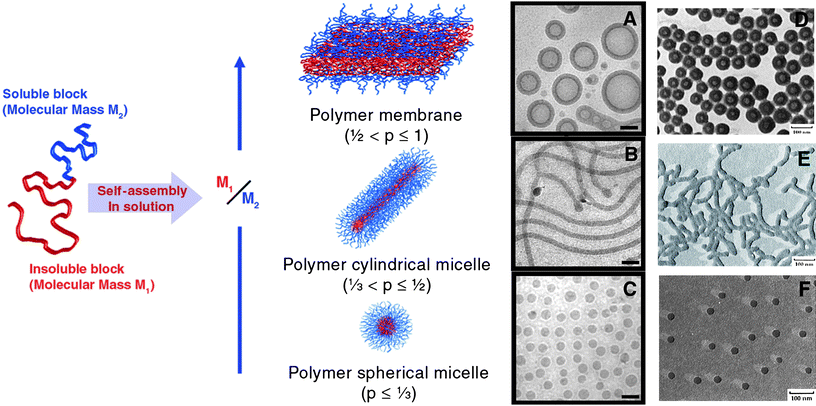 | ||
| Fig. 1 (Left) Schematic illustrating organisation of block copolymers in spherical and cylindrical micelles and vesicles. (Right) (A–C), cryoTEM micrographs of PB-b-PEO aggregates; (D–F), TEM micrographs of PS-b-PAA aggregates. (A and D) show vesicles, (B and E) show cylindrical micelles and (C and E) show spherical micelles. Scale bars (A to C) = 100 nm. Reproduced with permission from ref. 8,18 and 19 ©2008 Elsevier; 2003 American Association for the Advancement of Science; 1996 American Chemical Society. | ||
Interest into the characterisation and controlled formation of block copolymer aggregates has been spurred on by their potential as surfactants, nano- to micro-sized carriers for active compounds e.g.pharmaceuticals, for the controlled release of encapsulated compounds (again largely pharmaceuticals and biologically active compounds) and for inorganic materials templating, amongst numerous other proposed applications.20,21 The demands of all of these applications have meant that research in the past decade has focussed not only on manipulating the properties of aggregates through control of both the chemistry of the constituent polymer blocks but also the external and internal morphology of the aggregates. Control of chemistry of copolymers whereby a range of monomers can be utilised in the construction of new copolymers has expanded enormously in the past two decades through dramatic advances in controlled radical polymerisations and to a lesser extent through advances in living ionic polymerisations. Readers are directed to the cited reviews for further information on these topics.22–30
More recently increasing emphasis has been placed on controlling the external and internal morphologies of aggregates. This review article will present an overview of recent approaches to controlling the self-assembly of amphiphilic block copolymers with a view to obtaining novel micellar morphologies. Whilst this article will touch upon multi-compartment micelles particular focus will be placed upon control of the overall shape of micelles; i.e. those systems that expand the range of accessible morphologies beyond ‘simple’ spherical and cylindrical micelles namely disk-like, toroidal and bicontinuous micelles.
2. Multi-compartment micelles
Of particular recent interest has been the design and construction of multi-compartment micelles with water soluble shells and internally segregated micellar cores, where two (or more) separate types of hydrophobic regions exist. The most obvious advantage of these structures would be that these distinct core domains could be used to store two or more incompatible compounds in different nanocompartments within the core. This would be of particular interest in the delivery of more than one pharmaceutical or bioactive agent which were otherwise incompatible, to the same site simultaneously.31 The simplest structure for a multicompartment micelle is that of a core–shell–corona morphology (Fig. 2a) most commonly formed by linear ABC type block copolymers where A is the hydrophilic block and B and C are hydrophobic blocks with poor thermodynamic compatibility.32–34 It is this high thermodynamic incompatibility, represented by a high Flory–Huggins interaction parameter (χ), which drives the phase separation within the micellar hydrophobic core. The desire for the phase separated regions to solubilise different materials and the concomitant need for block incompatibility, triggered a widespread use of alkyl- and perfluoro-copolymer hydrophobic blocks as the immiscible components in multi-compartment micelles (e.g.Fig. 2c).35 A further approach to compartmentalised micelles was developed by Lutz et al. through the aqueous complexation of an amphiphilic block copolymer AB containing a hydrophobic segment A and a polyanionic segment B (poly(n-butyl acrylate)-b-poly(sodium 2-acrylamido-2-methylpropanesulfonate)), with a double hydrophilic block copolymer CD (poly[N,N,N-trimethylaminoethyl acrylate chloride]-b-poly[oligo(ethylene glycol) acrylate]) containing a polycationic block C and a non-ionic block D. This led to a core–shell–corona morphology where the shell region was formed from the polyionic complex between the cationic and anionic blocks. In a related vein, Liu et al. showed that by manipulating the ionic character of the ABC miktoarm terblock copolymerμ-[polystyrene][poly(ethylene oxide)] [poly(2-(dimethylamino)ethyl acrylate)] through pH changes, multicompartment micelles (PEO corona; PS + PDMAEA core) could also be generated.36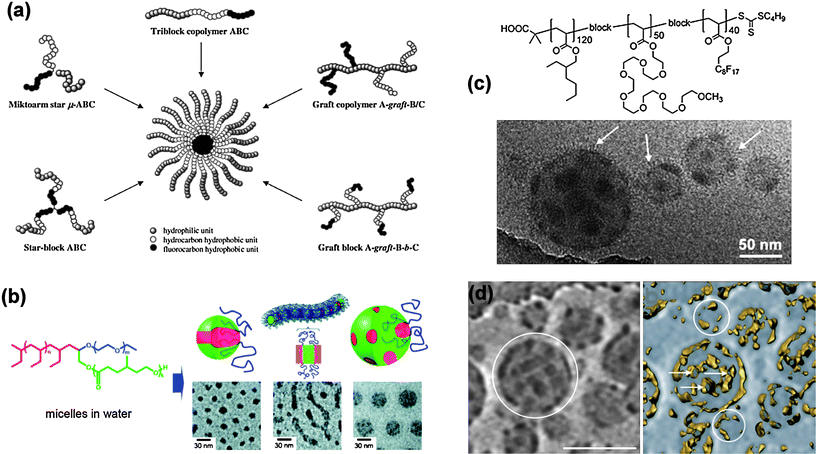 | ||
| Fig. 2 (a) Strategies for building multicompartment micelles via the aqueous self-assembly of various segmented amphiphilic copolymers. (b) Structure of a mikto-arm block copolymer and representations and cryo-TEMmicrographs of self-assembled micellar structures of (i) hamburger, (ii) segmented wormlike and (iii) raspberry micelles. (c) Structure of an ABCblock copolymer that forms raspberry micelles illustrated in accompanying TEM micrograph. (d) Cryo-TEM and ET images and of raspberry micelles in (c), scale bar = 112 nm. Reproduced with permission from ref. 33,35 and 38. ©2005 Wiley-VCH Verlag GmbH & Co. KGaA; 2009 Royal Society of Chemistry; 2008 American Chemical Society. | ||
More complex internal morphologies have been achieved through the use of miktoarm star block, star and graft block copolymers (Fig. 2b).37–39 Morphologies observed have included ‘hamburgers’ where a lamellar region is sandwiched between two other regions, segmented cylindrical micelles where the hydrophobic regions alternate along the length of the micelles and ‘raspberry’ micelles where one region adopts spherical shapes embedded in the matrix of the other.
CryoTEM has played a key role in observing the interior segregation in these micelles which would otherwise not be apparent from the simple contrast given by traditional TEM techniques (e.g. negative staining) and was first used by Hillmyer and Lodge to demonstrate segregation in aggregates formed from a mikto-arm star-block copolymer (Fig. 2b).40 Although cryoTEM has now been established as an important technique for the visualization of a large range of self-assembled structures in solution,41–43 it cannot unambiguously identify the morphology of 3D objects. This can be done, however, using cryo-electron tomography (cryoET). CryoET has been recognized as a strong and emerging technique in the biological sciences,44–46 but is still virtually unexplored for the analysis of samples from synthetic origin.47 It involves the acquisition of a series of cryoTEM images under different tilt angles and the subsequent computer-assisted reconstruction of the original 3D volume. Recently Laschewsky et al. have utilised cryoTEM (Fig. 2d) and cryo-electron tomography (cryo-ET) to investigate and conclusively demonstrate the phase separated internal structure of raspberry-like micelles (Fig. 2d) formed by an ABC linear block copolymer with comb-like hydrophilic block (Fig. 2c).35
3. Disk-like and toroidal micelles
After the finding that AB(C)-type polystyrene-poly(isocyanopeptide) block copolymers form helical aggregates,48 in the past decade further external morphologies have been observed for amphiphilic block copolymer aggregates in dilute solutions, including disk-like and toroidal micelles. Disk-like (or oblate spherical) micelles, whilst remaining relatively rare, have been observed to form from a number of ABC-type amphiphilic copolymers with non-ionic49,50 as well as ionic hydrophilic blocks.51,52In the former case a poly(ethylene oxide)-block-polystyrene-block-1,2-poly(butadiene) (PEO–PS–PB) copolymer was observed to form standard core–shell micelles with mixed PB–PS cores, however, fluorination of the PB component (PEO–PS–PB(F)) gave disk-like micelles due to the strong segregation between the perfluoro-block component and the PS. The formation of disk-like micelles from poly(acrylic acid)-block-poly(methyl acrylate)-block-polystyrene (PAA–PMA–PS) was dependent upon the presence of diamino counterions (ethylenediamine, EDA or ethylenedioxy-bis-ethylenediamine, EDDA), the amount of THF (acting as a plasticiser) remaining in the surrounding aqueous/THF medium and the length of the hydrophobic blocks (Fig. 3). Manipulation of these variables led to a high degree of control over micellar morphology enabling the switching between disk-like, cylindrical and spherical micelles. The interfacial curvature between the hydrophobic and hydrophilic components is dictated by volume and conformational differences between hydrophobic and charged hydrophilic blocks together with the interfacial energy between them. Correspondingly, the diamino counter-ions complexed with the PAA block altered both the volume and interfacial energy; the principle cause of disk formation being due to the condensed hydrophilic corona volume after complexation according to the authors. Nevertheless, the disk-like micelles were not thermodynamically stable in 100% water (in contrast to those observed for the PEO–PS–PB(F)copolymer) and required a plasticised core (supplied by the presence of THF in the dispersion medium).
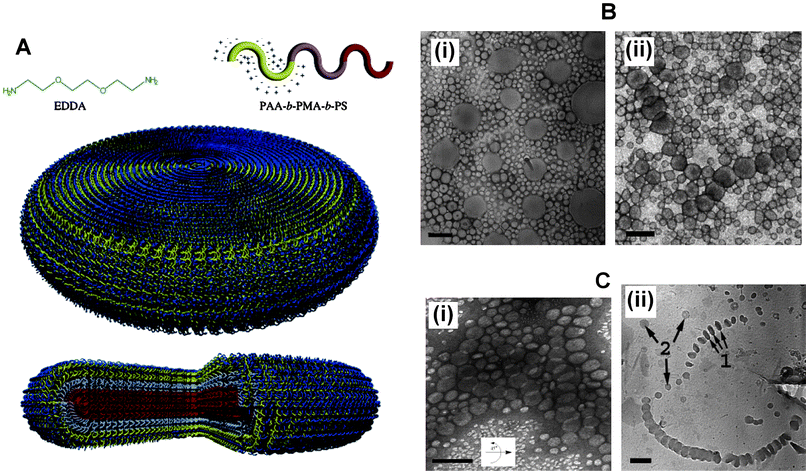 | ||
Fig. 3 (A) Schematic illustrating structure of ABCblock copolymer, counterion and internal organisation of copolymers within a disk-like micelle. (B) TEM micrographs showing disk-like micelles from dilute solutions of PAA-b-PMA-b-PS triblock copolymers with amine-to-acid functional group ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and solvent with 40% water and 60% THF: (i) EDA as the counterion; (ii) EDDA as the counterion. (C) (i) Tilted TEM micrograph of disk-like micelles formed with EDDA as the counterion, amine-to-acid functional group ratio of 0.3:1, and solvent with 40% water and 60% THF. (ii) Cryo micrograph for same sample solution: arrows 1, the disks are parallel to the electron beam axis; and arrows 2, disks are perpendicular to the electron beam axis. All scale bars are equal to 200 nm. Reproduced with permission from ref. 52. ©2005 American Chemical Society. :1, and solvent with 40% water and 60% THF: (i) EDA as the counterion; (ii) EDDA as the counterion. (C) (i) Tilted TEM micrograph of disk-like micelles formed with EDDA as the counterion, amine-to-acid functional group ratio of 0.3:1, and solvent with 40% water and 60% THF. (ii) Cryo micrograph for same sample solution: arrows 1, the disks are parallel to the electron beam axis; and arrows 2, disks are perpendicular to the electron beam axis. All scale bars are equal to 200 nm. Reproduced with permission from ref. 52. ©2005 American Chemical Society. | ||
It should be noted that disk-like micelles in the form of oblate (or prolate) spheroids are not stable morphologies for micelles according to the packing parameter model proposed by Israelachvili for simple hydrocarbonamphiphiles (Fig. 4); to paraphrase, “the unacceptability of an oblate spheroid comes about because the peripheral regions have too great a curvature while the central regions are too thick”. This is due to thermodynamic packing considerations whereby such shapes lead to high energy (and consequently unstable) packing, instead globular or flattened micelles with rounded rims are predicted. Thus the formation of oblate spheroidal and disk-like morphologies should be due to additional factors other than straightforward consideration of the packing parameter of the hydrophobic chains. Where disk-like micelles are formed from molecular surfactants these are usually mixed systems with two or more molecular components or exist in strongly ionic solutions.53–63 Lodge et al. note that for disk formation strong segregation between block components is needed, which indeed seems to be the case in other reported examples to date and is supported by theory and modelling.64–66 Further disk-like micelles (or discrete platelets) have been observed to form via internal crystallization processes: for PE containing diblock copolymers67 in decane and in water;68 for a polypeptidediblock copolymer;69 and via ionic complex formation between AB and BCdiblock copolymers.70 These examples cannot be considered using the packing parameter model due to strong interactions between blocks; i.e.crystallisation is driving the self-assembly process. In contrast to disk-like micelles the packing parameter model predicts toroidal micelles for p ≈ 0.44 a value which lies between that for spheres (p ≈ 0.33) and that of cylinders (p ≈ 0.5). The earliest examples of toroidal micelles formed were reported by Wooley et al. and resulted from the self-assembly of the same PAA–PMA–PScopolymers that formed the disk-like micelles but with different EDA and THF quantities.71 Since then a number of ABC, ABA and AB amphiphilic block copolymers have been observed to form toroidal micelles.72–81 In all cases the toroidal micelles are essentially looped cylindrical micelles although their formation has been proposed to arise by different pathways. Wooley et al. observed the formation of toroids from cylindrical and disk-like micelles through the elimination of high free energy end caps of cylindrical micelles or energetically unfavourable spherical micelles but also via the perforation of disc-like micelles82 (Fig. 5C).
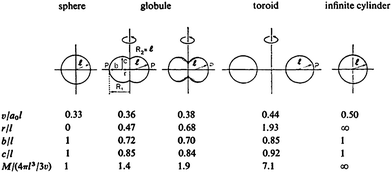 | ||
| Fig. 4 Approximate transition shapes from spheres to cylinders, based on purely geometric considerations for simple hydrocarbon based amphiphiles. A full explanation of the geometric parameters can be found in ref. 13. Reproduced with permission from ref. 13. ©1976 Royal Society of Chemistry. | ||
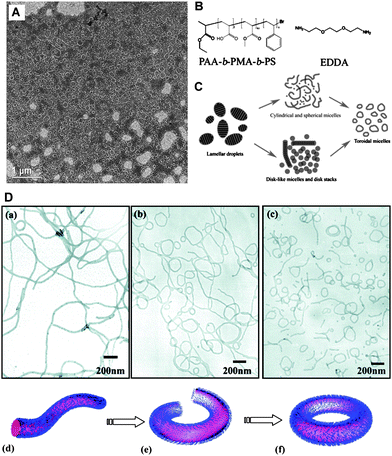 | ||
| Fig. 5 (A) TEM micrograph of toroidal micelles and cylindrical micelles formed from PAA-b-PMA-b-PS and EDDA (B). (C) Proposed routes to toroidal assembly for the PAA-b-PMA-b-PS and EDDA system. (D) (a)–(c) TEM micrographs illustrating formation of toroidal micelles from cylindrical micelles from PS-b-PVPblock copolymers, (a)–(c) represents increased annealing time; (d)–(f) schematic illustrating formation of toroidal micelles from cylindrical micelles. Reproduced with permission from ref. 82 and 83. ©2009 Royal Society of Chemistry; 2009 American Chemical Society. | ||
In contrast Ju and Wang reported that the stirring rate was the principle factor in the formation of toroids from a poly(4-vinypyridine)-block-polystyrene-block-poly(4-vinypyridine) (P4VP–PS–P4VP) in a dioxane/water mixture (from which the dioxane was removed by dialysis); slow stirring rates led to the formation of cylindrical micelles via end-to-end cylinder connection and faster rates led to increasing numbers of toroids formed through a cylinder–sphere–vesicle–ring transformation.83 The latter mechanism had previously been proposed by Liang et al. from experiment and real-space self-consistent field theory (Fig. 5a–f).84
The majority of samples of toroidal micelles reported to date have wide size distributions (and are often accompanied by significant quantities of cylindrical micelles and/or cylindrical–toroidal networks) recently the formation of toroidal micelles highly uniform in size (diameters = 70 ± 3 nm) and free of other morphologies has been reported from a polyisoprene-b-poly(2-vinylpyridine) (PI-b-P2VP) diblock copolymer in a THF/ethanolsolvent mixture.85 The production of samples of ‘pure’ highly uniform toroidal micelles opens up the possibility for their application considerably and it is to be hoped that such uniformity might be reached in aqueous solutions in the immediate future.
4. Bicontinuous micelles
A bicontinuous morphology in a discrete amphiphilic block copolymer aggregate in dilute solution was first observed by Eisenberg et al. formed from a 4.3% PS190-b-PAA20 solution in a 8.5% water–DMF mixture.86 The exact internal morphology of this aggregate appeared to consist of interconnected rods but further details were not presented and no subsequent data have been presented in the literature. Subsequently the possibility of forming bicontinuous aggregates (or micelles) was proposed by Fraaije and Sevink based on self-consistent-field simulations of dispersed droplets of amphiphilic block copolymers.87 The structures were generated by quenching a homogeneous droplet of a diblock copolymerAN–MBM with N = 20, in an aqueous bath and then relaxing the structure by a dynamic variant of self-consistent-field theory. The solvent was weakly selective and segregation was mild, χAS = 1.7, χABN = 40, and χAS − χBS = 0.3, so that A was slightly more solvophobic and B slightly more solvophilic. These parameters correlated with concentrated poly(propylene oxide)–poly(ethylene oxide) aqueous solutions in ambient conditions. These simulations resulted in a range of morphologies illustrated in Fig. 6. In all cases the droplets developed an outer fuzzy layer of the solvophilic B block. The internal structures depended on the size ratio f = M/N from f = 0.35 to f = 0.15 and changed from onion micelles with alternating A and B layer to a bicontinuous phase to a cylindrical phase and finally an inverted micellar phase. Too asymmetric polymersf = 0.1 did not form any internal structure. Interestingly the nature of bicontinuous phase formed at f = 0.25 was found not to vary with droplet size (Fig. 5b). | ||
| Fig. 6 (A) Morphologies of AN–MBMpolymersurfactant nanodroplets (isosurfaces partly removed for visualization) for A. Solvophobic block A concentration field for different block ratios f = M/N. 0.35 (a), 0.30 (b), 0.25 (c), 0.20 (d), 0.15 (e), 0.10 (f); (B) f = M/N = 0.25 for different initial radii R0. From left to right: R0 = 33, 30, 26, 23, 20 (in units of polymer bead size). Reproduced with permission from ref. 87. ©2003 American Chemical Society. | ||
Despite the relative simplicity of the linear AB block copolymer used in the Fraaije and Sevink simulations, to date the only bicontinuous aggregates demonstrated to form have been based on ABC structures, either linear or comb-like (Fig. 7). The first conclusive observed bicontinuous phase in a discrete amphiphilic block copolymer aggregate was presented by Wooley et al. from an ABCblock copolymer similar to the one that forms toroidal and disk-like micelles. PAA99-b-PMA73-b-PS203 when complexed with 2,2′-(ethylenedioxy)diethylamine formed bicontinuous aggregates in a mixture of THF:water in a volume ratio 1:0.2.88 As illustrated in Fig. 8 the bicontinuous nature of the aggregates was demonstrated by negative staining where the dark portions of the aggregates represent stained PAA blocks and the lighter portions represent hydrophobic blocks. The authors did not put forward an internal organisational model for these bicontinuous aggregates but did so for related porous aggregates (Fig. 8B). When the THF:water content was increased to 1:0.8 lamellar aggregates were observed. The authors suggested that as the water content increased the PAA chains become more swollen with water and created flatter interfaces within the particles resulting in an internal lamellar phase separation, as opposed to residing on a concave interface at lower water content.
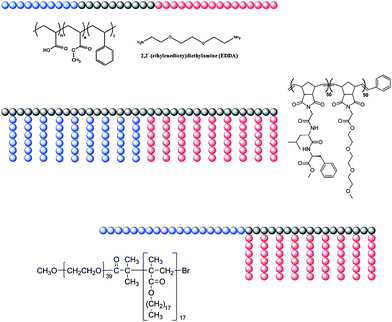 | ||
| Fig. 7 Schematic and chemical structures of copolymers found to form bicontinuous aggregates in solution. | ||
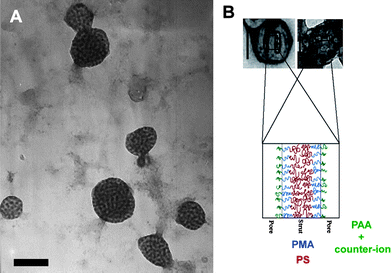 | ||
| Fig. 8 (A) TEM micrograph of bicontinuous micelles formed from PAA-b-PMA-b-PS and EDDA in THF/water; scale bar = 200 nm. (B) Proposed internal organisation of related structures. Reproduced with permission from ref. 88. ©2008 American Chemical Society. | ||
The first discrete bicontinuous aggregates where the internal morphology was conclusively demonstrated were observed for an amphiphilic polynorbornene-based block copolymer with comb-like segments of oligo(ethylene glycol)methyl ether (OEG) and a tri-peptideglycine-leucine-phenylalanine (GLF) (Fig. 9).89 These aggregates were prepared by the dropwise addition of water to a DMSO solution of the copolymer followed by the removal of the organic solvent by dialysis against pure water. Conventional TEM using negative staining of the dried aggregates indicated the formation of internally structured nanospheres with outer diameters varying between 50 and 450 nm. Their existence in solution was confirmed by cryoTEM, and the internal structure of the aggregates was further investigated by cryo-ET (Fig. 10a and b).
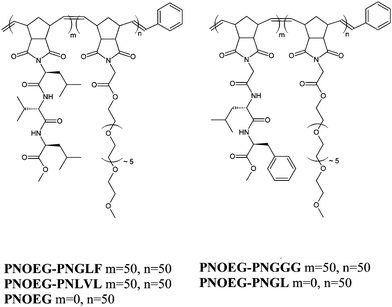 | ||
| Fig. 9 Polynorbornene (oligoethylene glycol) based double graft block copolymers containing glycine-leucine-phenylalanine (PNOEG–PNGLF), glycine-glycine-glycine (PNOEG–PNGGG), leucine-valine-leucine (PNOEG–PNLVL), glycine-leucine (PNOEG–PNGL) based segments and the single graft copolymer without peptide side chains (PNOEG). Reproduced with permission from ref. 89. ©2008 Wiley-VCH Verlag GmbH & Co. KGaA. | ||
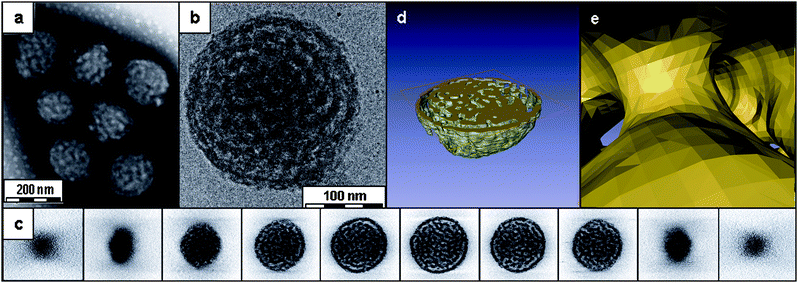 | ||
| Fig. 10 TEM analysis of aggregates of PNOEG–PNGLF. (a) Conventional TEM using negative staining, (b) cryoTEM image of a vitrified film, (c) gallery of z slices showing different cross-sections of a 3D SIRT reconstruction of a tomographic series recorded from the vitrified film in (b). Visualization of the segmented volume showing (d) a cross-section of the aggregate and (e) a view from within the hydrated channels. Reproduced with permission from ref. 89. ©2010 American Chemical Society. | ||
The 3D visualization of the reconstructed volume revealed that these nanospheres contained an interior consisting of a bicontinuous assembly in which the branched network of worm-like hydrophobic peptide-containing segments is segregated from channels containing the hydrated OEG moieties. Cross-sections (Fig. 10c and d) through the reconstructed volume revealed that the hydrophobic domains had diameters of ∼20 nm separated by water channels with diameters of ∼15 nm. In contrast to the predicted structures from Fraaije and Sevink, the cross-sections also showed that the shell which encloses the bicontinuous network has perforations connecting the internal and external aqueous phases. This is clearly demonstrated by segmentation presented in Fig.10d, which highlights both the bicontinuous structure and the perforations in the encapsulating shell. The 1H NMR spectra in D2O demonstrated that the hydrophobic regions of the aggregates were formed by both the peptide side chain and the PN backbone. These observations further suggest that in an aqueous medium the OEG-modified PN folds back onto the peptide-modified PN part, together forming the hydrophobic domains. This underlines the fact that this copolymer cannot be considered as a simple AB diblock amphiphilic copolymer and is better described as an (A)B(C)block copolymer.
Changing the tripeptide side chain from glycine-leucine-phenylalanine (PNOEG–PNGLF) to leucine-valine-leucine (PNOEG–PNLVL), i.e. changing the polymer's composition but not its hydrophilic/hydrophobic balance, led to the formation of single, tightly folded and branched worm-like micelles. With both polymers having the same weight fraction of OEG-grafts (WOEG = 0.33) and comparable molecular weights this difference in aggregation was attributed to the specific amino acid sequence of the peptide graft. Similarly PNOEG–PNGGG and PNOEG–PNGL which both have the same molecular weight (45 kg mol−1) and similar WOEG (0.38 and 0.39, respectively) formed small clustered micelles, and similar bicontinuous micelles to PNOEG–PNLVL respectively. The latter observation suggests that the presence of the glycine-leucine sequence, rather than the precise value of WOEG or the value of the packing parameter p, is critical in the formation of the bicontinuous internal structure in this case. This might imply that the formation of these aggregates is specifically related to their chemical structure; which may result from differences in the χ parameters between segments and/or may be a consequence of peptide associations. The dependence of bicontinuous micelle formation on an ABCblock copolymer structure of some form was further reinforced for a completely different amphiphilic block copolymer.
Aggregate dispersions of the semi-crystalline AB(C) comb-like block copolymer poly(ethylene oxide)-block-poly(octadecyl methacrylate) (PEO39-b-PODMA17, PEO–PODMA) (Fig. 7)90 were formed by slow addition of water to THF solutions at 35 °C and subsequent dialysis against water at the same temperature. DLS at 35 °C indicated that the size of the aggregates was concentration dependent, giving diameters of ∼350 nm for 5 wt% and ∼275 nm for 1 wt% solutions. Furthermore, DSC analysis and fluorescence experiments of the 5 wt% solution revealed a thermal transition at Ttrans = 22 °C, assigned to the melting and crystallisation of portions of the octadecyl chains in the aggregates. CryoTEM allowed the variable temperature analysis of aggregate morphology by plunge freezing of a sample equilibrated at different temperatures. The 2D cryoTEM projection images of the 5 wt% solution vitrified at 4 °C (below Ttrans) showed round aggregates that possessed an ordered internal microphase-separated structure (Fig. 11a). Samples vitrified at the transition point (22 °C) showed spherical aggregates with a variety of internal structures with lower apparent order compared to those present at 4 °C (Fig. 11b). Also the projection images recorded at 45 °C (above Ttrans) showed round objects; however, these showed poor contrast with the surrounding vitrified ice matrix, and an ordered internal structure could no longer be observed (Fig. 11c). Cryo-ET was performed both at 4 °C and at 45 °C to further analyze the internal structural transitions within the aggregates.
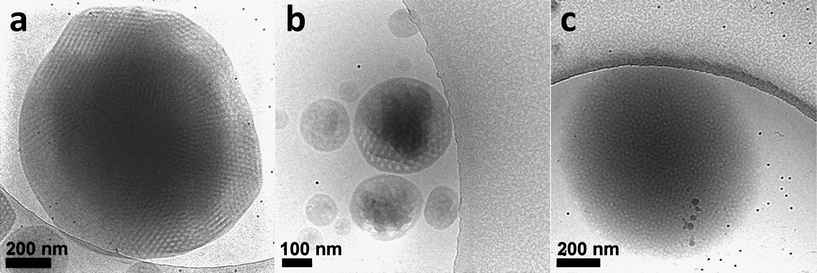 | ||
| Fig. 11 cryoTEM 2D projection images of 5 wt% solution of PEO39-b-PODMA17 (PEO–PODMA) aggregates vitrified at (a) 4 °C, (b) 22 °C and (c) 45 °C. Reproduced with permission from ref. 90. ©2010 American Chemical Society. | ||
The 3D visualization of the reconstructed volumes revealed that spherical aggregates below Ttrans possessed a sponge-like mainly bicontinuous network structure of intertwined water-filled and carbon-rich channels (both ∼13 nm in thickness/diameter, Fig. 11a and b). Similarly to the PNOEG–PNGLF aggregates the aqueous channels were in contact with the surrounding medium. While the majority of structural component of the aggregates at 4 °C was observed to be bicontinuous, some internal lamellar organization was observed in place (Fig. 12a and b). The tilt series and 3D reconstructions (Fig. 12c and d) recorded from the samples vitrified at 45 °C still showed some residual but highly disordered microphase-separated internal structure, again with ∼13 nm dimensions.
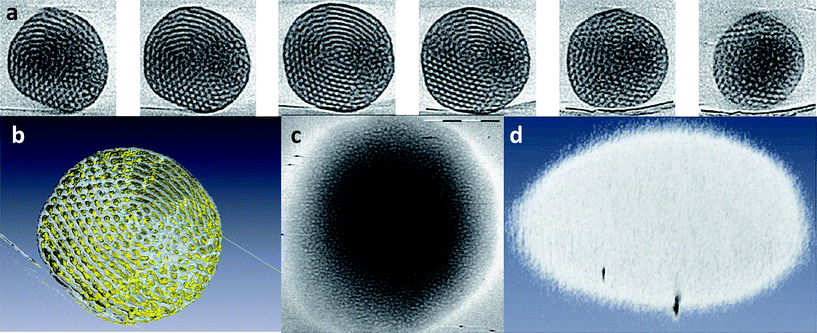 | ||
| Fig. 12 cryoET of a 5 wt% solution of PEO39-b-PODMA17 (PEO–PODMA) aggregates. (a) Gallery of z slices (left to right) and (b) computer visualization of a 3D reconstruction of a particle showing its internal structure at 4 °C. (c) z-Slice of a particle showing its internal structure and (d) computer visualization of a 3D reconstruction of a particle showing its external shape at 45 °C. Reproduced with permission from ref. 90. ©2010 American Chemical Society. | ||
The tomograms further showed that the order–disorder thermal transition is accompanied by a flattening of the aggregates to a more planar oblate spheroid morphology which, along with the amorphous nature of the block copolymer, also explains the reduced electron density observed in the 2D images. The resolution and contrast of the reconstructions, however, were not sufficient to determine whether the observed residual compartments were interconnected throughout the interior of the aggregates, as was the case below Ttrans.
As previously noted the self-assembly of amphiphilic block copolymers without strongly attractive segments can be considered as being described by the packing parameter defined by Israelachvili et al. (eqn (1)).13,14 The principal necessity for formation of bicontinuous cubic phases is that the packing parameter, p, has a value greater than one and consequently the volume of the hydrocarbon/hydrophobic ‘wedge’ is substantially larger than the product of a and l. According to Hyde91 the packing parameter can be related to the Gaussian curvature through eqn (2). Where 〈K〉 < 0 as a consequence of one of the two principal radii of curvatures (R1 and R2) being negative (〈K〉 = 1/(R1R2) and is the surface-averaged Gaussian curvature) leading to the wedge-like shape of the overall molecule forming the aggregate.
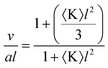 | (2) |
Further modifications to the packing parameter concept have allowed for the role of the hydrocarbon tail group to be included in predicting aggregate shapes and properties.92 This enables a preliminary interpretation of the occurrence of bicontinuous micelles for PAA-b-PMA-b-PS and PEO–PODMA. In both cases since it is expected that l scales with v, the formation of the bicontinuous phase results from a relatively reduced interfacial head group cross-sectional area (a) relative to v, giving a negative packing parameter value and negative (concave) curvature at the water–hydrophobe interface. It is notable that in the former case a transition from a bicontinuous to a lamellar organization results from an increase in water concentration relative to THF. This may result in an increase in the hydration of the PAA–EDDA corona and hence an increase in hydrophilic volume and the interfacial area (a), but it may also lead to a decrease in the relative hydrocarbon volume v, through deswelling of the hydrophobic component. Given that various weight fractions of the three blocks in the PAA-b-PMA-b-PS have been studied as reported in a number of publications51,52,71,82,88,93 and that only one example of a bicontinuous phase is given, this suggests that the appropriate balance between a and v is difficult to attain for these linear copolymers. The PEO–PODMAcopolymer in contrast possesses an intrinsic high volume hydrocarbon region relative to the PEO component which has a relatively smaller cross-sectional area when hydrated. That an order–disorder thermal transition leads to the disappearance of the bicontinuous phase suggests that crystallization of the nano-phase separated side-chains is necessary for the bicontinuous phase. Essentially crystallization increases v and or decreases a, and thus decreases the packing parameter; this is easy to envisage since crystallization of the side chains necessitates an extended all trans-form in contrast to the coiling liquid chains above the melting point. Concomitantly, melting of the side-chains leads to a reduction in volume and increase in head group area.
These internally structured self-assembled nanospheres can be considered the polymeric analogues of cubosomes, aggregates that have liquid crystalline interiors with cubic or hexagonal order. Typically cubosomes are formed from low-molecular-weight compounds that are often present as mixtures and often require stabilizers.94–101 The high degree of order found in low molecular weight cubosomes was only met in the aggregates formed by PEO–PODMA, the origin of which whilst not yet fully understood is thought to lie with the bulky comb-like block acting as the surfactant tail.
5. Conclusions
As may be surmised from the discussion above, it is a particularly exciting time to be studying block copolymerself-assembly. The shear range of macromolecular structures available through contemporary synthetic techniques means that the design of defined aggregate structures in solution with quite specific physical and chemical properties is entirely achievable and will continue apace. Whilst the Israelachvili packing parameter model is still of considerable use and can be utilised in the design of aggregates the available chemistries available to the synthesist means that tailored chemical and physical interactions in the self-assembled structures can over-ride this model and induce aggregate morphologies that might otherwise remain inaccessible. All of the aggregate morphologies described above deserve further study not only to elucidate the principles of self-assembly but also to ascertain differences in physical properties compared to their ‘classical’ counterpart morphologies (such as spherical and cylindrical micelles). Applications immediately suggest themselves in all cases and of particular interest are controlled delivery and organic/inorganic templating.Molecular cubosomes are currently being explored as delivery agents of bioactive components and the polymer cubosomes presented above may provide future delivery agents with prolonged stability, suited for long term slow release applications. Particularly interesting is the possibility to have block copolymer aggregates with temperature-responsive structure and morphology. Low molecular weight cubosomes have also been suggested as mineralization templates. The present polymeric counterparts may be interesting alternatives in which the reduced dynamics of polymer aggregates may prevent structural rearrangement upon exposure to mineral ions. The high degree of order found in low molecular weight cubosomes was only met in the aggregates formed by PEO–PODMA, the origin of which whilst not yet fully understood is thought to lie with the bulky comb-like block acting as the surfactant tail. Dendritic, branched and comb-like hydrophobic polymer chains would appear to the ideal targets for the specific design of future polymer cubosomes therefore. Notably a recent report on dendritic amphiphiles also described cubosome formation.102
Notes and references
- Z. Tuzar and P. Kratochvil, in Surface and Colloid Science, ed. E. Matijevic, Plenum Press, New York, 1993, vol. 15, ch. 1, pp. 1–83 Search PubMed.
- B. M. Discher, D. A. Hammer, F. S. Bates and D. E. Discher, Curr. Opin. Colloid Interface Sci., 2000, 5, 125–131 CrossRef CAS.
- P. Alexandridis and T. A. Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- P. Bahadur and K. Pandya, Langmuir, 1992, 8, 2666–2670 CrossRef CAS.
- K. Nakashima and P. Bahadur, Adv. Colloid Interface Sci., 2006, 123, 75–96 CrossRef.
- N. J. Jain, V. K. Aswal, P. S. Goyal and P. Bahadur, J. Phys. Chem. B, 1998, 102, 8452–8458 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Polym. Adv. Technol., 1998, 9, 677–699 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, J. Am. Chem. Soc., 1996, 118, 3168–3181 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- J. F. Lutz, Polym. Int., 2006, 55, 979–993 CrossRef CAS.
- J. F. Gohy, in Block Copolymers II, Springer-Verlag Berlin, Berlin, 2005, vol. 190, pp. 65–136 Search PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- M. Antonietti and S. Forster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- I. R. Schmolka, J. Am. Oil Chem. Soc., 1977, 54, 110–116 CrossRef CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, J. Controlled Release, 2002, 82, 189–212 CrossRef CAS.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544–3561 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- C. L. McCormick, S. E. Kirkland and A. W. York, Polym. Rev., 2006, 46, 421–443 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- T. P. Lodge, A. Rasdal, Z. B. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS.
- S. Kubowicz, J. F. Baussard, J. F. Lutz, A. F. Thunemann, H. von Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262–5265 CrossRef CAS.
- J. F. Lutz and A. Laschewsky, Macromol. Chem. Phys., 2005, 206, 813–817 CrossRef CAS.
- K. Stahler, J. Selb and F. Candau, Langmuir, 1999, 15, 7565–7576 CrossRef.
- H. Von Berlepsch, C. Bottcher, K. Skrabania and A. Laschewsky, Chem. Commun., 2009, 2290–2292 RSC.
- C. Liu, M. A. Hillmyer and T. P. Lodge, Langmuir, 2009, 25, 13718–13725 CrossRef CAS.
- Z. B. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 765–771 CrossRef CAS.
- Z. B. Li, M. A. Hillmyer and T. P. Lodge, Langmuir, 2006, 22, 9409–9417 CrossRef CAS.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS.
- N. Saito, C. Liu, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2008, 41, 8815–8822 CrossRef CAS.
- H. Cui, T. K. Hodgdon, E. W. Kaler, L. Abezgauz, D. Danino, M. Lubovsky, Y. Talmon and D. J. Pochan, Soft Matter, 2007, 3, 945–955 RSC.
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2010, 49, 7850–7858 CrossRef CAS.
- F. Nudelman, G. de With and N. A. J. M. Sommerdijk, Soft Matter, 2011, 7, 17–24 RSC.
- W. Baumeister, Biol. Chem., 2004, 385, 865–872 CrossRef CAS.
- R. McIntosh, D. Nicastro and D. Mastronarde, Trends Cell Biol., 2005, 15, 43–51 CrossRef CAS.
- S. Nickell, C. Kofler, A. P. Leis and W. Baumeister, Nat. Rev. Mol. Cell Biol., 2006, 7, 225–230 CrossRef CAS.
- A. J. B. Koster, in Electron Tomography: Methods for the Three-Dimensional Visualization of Structures in the Cell, ed. J. Frank, Springer Science + Business Media, LLC, New York, 2nd edn, 2006, ch. 4, pp. 113–162 Search PubMed.
- J. Cornelissen, M. Fischer, N. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427–1430 CrossRef CAS.
- W. F. Edmonds, Z. B. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 4526–4530 CrossRef CAS.
- T. P. Lodge, M. A. Hillmyer, Z. L. Zhou and Y. Talmon, Macromolecules, 2004, 37, 6680–6682 CrossRef CAS.
- H. G. Cui, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Macromolecules, 2006, 39, 6599–6607 CrossRef CAS.
- Z. Li, Z. Chen, H. Cui, K. Hales, K. Qi, K. L. Wooley and D. J. Pochan, Langmuir, 2005, 21, 7533–7539 CrossRef CAS.
- K. Edwards and M. Almgren, Langmuir, 1992, 8, 824–832 CrossRef CAS.
- M. Silvander, G. Karlsson and K. Edwards, J. Colloid Interface Sci., 1996, 179, 104–113 CrossRef CAS.
- M. Swanson-Vethamuthu, E. Feitosa and W. Brown, Langmuir, 1998, 14, 1590–1596 CrossRef CAS.
- M. Bergstrom and J. S. Pedersen, Phys. Chem. Chem. Phys., 1999, 1, 4437–4446 RSC.
- M. Almgren, Biochim. Biophys. Acta, Biomembr., 2000, 1508, 146–163 CrossRef CAS.
- T. H. Bayburt, Y. V. Grinkova and S. G. Sligar, Nano Lett., 2002, 2, 853–856 CrossRef CAS.
- A. Shioi and T. A. Hatton, Langmuir, 2002, 18, 7341–7348 CrossRef CAS.
- D. Danino, D. Weihs, R. Zana, G. Oradd, G. Lindblom, M. Abe and Y. Talmon, J. Colloid Interface Sci., 2003, 259, 382–390 CrossRef CAS.
- M. Johnsson and K. Edwards, Biophys. J., 2003, 85, 3839–3847 CrossRef CAS.
- M. Singh, C. Ford, V. Agarwal, G. Fritz, A. Bose, V. T. John and G. L. McPherson, Langmuir, 2004, 20, 9931–9937 CrossRef CAS.
- A. Sayyed-Ahmad, L. M. Lichtenberger and A. A. Gorfe, Langmuir, 2010, 26, 13407–13414 CrossRef CAS.
- S. S. Soni, N. V. Sastry, J. George and H. B. Bohidar, J. Phys. Chem. B, 2003, 107, 5382–5390 CrossRef CAS.
- S. S. Soni, N. V. Sastry, J. V. Joshi, E. Seth and P. S. Goyal, Langmuir, 2003, 19, 6668–6677 CrossRef CAS.
- A. N. Semenov, I. A. Nyrkova and A. R. Khokhlov, Macromolecules, 1995, 28, 7491–7500 CrossRef CAS.
- A. Ramzi, M. Prager, D. Richter, V. Efstratiadis, N. Hadjichristidis, R. N. Young and J. B. Allgaier, Macromolecules, 1997, 30, 7171–7182 CrossRef CAS.
- W. Wang, R. Liu, Z. Li, C. Meng, Q. Wu and F. Zhu, Macromol. Chem. Phys., 2010, 211, 1452–1459 CrossRef CAS.
- A. Constancis, R. Meyrueix, N. Bryson, S. Huille, J. M. Grosselin, T. Gulik-Krzywicki and G. Soula, J. Colloid Interface Sci., 1999, 217, 357–368 CrossRef CAS.
- I. K. Voets, A. de Keizer, P. de Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2006, 45, 6673–6676 CrossRef CAS.
- D. J. Pochan, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS.
- I. LaRue, M. Adam, M. Pitsikalis, N. Hadjichristidis, M. Rubinstein and S. S. Sheiko, Macromolecules, 2006, 39, 309–314 CrossRef CAS.
- J. T. Zhu, Y. G. Liao and W. Jiang, Langmuir, 2004, 20, 3809–3812 CrossRef CAS.
- I. C. Reynhout, J. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2332 CrossRef CAS.
- Y. L. Liu, Y. H. Chang and W. H. Chen, Macromolecules, 2008, 41, 7857–7862 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, D. Wouters, S. Hoeppener and U. S. Schubert, Macromolecules, 2008, 41, 1581–1583 CrossRef CAS.
- L. Liu, J. K. Kim, R. Gunawidjaja, V. V. Tsukruk and M. Lee, Langmuir, 2008, 24, 12340–12346 CrossRef CAS.
- J. Fu, D. H. Kim and W. Knoll, ChemPhysChem, 2009, 10, 1190–1194 CrossRef CAS.
- C. Tsitsilianis, Y. Roiter, I. Katsampas and S. Minko, Macromolecules, 2008, 41, 925–934 CrossRef CAS.
- E. Lee, Y. H. Jeong, J. K. Kim and M. Lee, Macromolecules, 2007, 40, 8355–8360 CrossRef CAS.
- W. Wei, H. F. Xu, X. Z. Qu, X. L. Ji, W. Jiang and Z. Z. Yang, Macromol. Rapid Commun., 2007, 28, 1122–1127 CrossRef CAS.
- H. G. Cui, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2009, 5, 1269–1278 RSC.
- H. Z. Yu and W. Jiang, Macromolecules, 2009, 42, 3399–3404 CrossRef CAS.
- Y. Jiang, J. T. Zhu, W. Jiang and H. J. Liang, J. Phys. Chem. B, 2005, 109, 21549–21555 CrossRef CAS.
- H. Huang, B. Chung, J. Jung, H. W. Park and T. Chang, Angew. Chem., Int. Ed., 2009, 48, 4594–4597 CrossRef CAS.
- K. Yu, L. F. Zhang and A. Eisenberg, Langmuir, 1996, 12, 5980–5984 CrossRef CAS.
- J. Fraaije and G. J. A. Sevink, Macromolecules, 2003, 36, 7891–7893 CrossRef CAS.
- K. Hales, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Nano Lett., 2008, 8, 2023–2026 CrossRef CAS.
- A. L. Parry, P. H. H. Bomans, S. J. Holder, N. Sommerdijk and S. C. G. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS.
- B. E. McKenzie, F. Nudelman, P. H. H. Bomans, S. J. Holder and N. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS.
- S. T. Hyde, J. Phys. Chem., 1989, 93, 1458–1464 CrossRef CAS.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- H. G. Cui, Z. Y. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS.
- J. Barauskas, M. Johnsson, F. Joabsson and F. Tiberg, Langmuir, 2005, 21, 2569–2577 CrossRef CAS.
- J. Barauskas, M. Johnsson and F. Tiberg, Nano Lett., 2005, 5, 1615–1619 CrossRef CAS.
- W. Buchheim and K. Larsson, J. Colloid Interface Sci., 1987, 117, 582–583 CrossRef CAS.
- T. Landh, J. Phys. Chem., 1994, 98, 8453–8467 CrossRef CAS.
- K. Larsson, J. Phys. Chem., 1989, 93, 7304–7314 CrossRef CAS.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5, 64–69 CrossRef CAS.
- P. T. Spicer, Curr. Opin. Colloid Interface Sci., 2005, 10, 274–279 CrossRef CAS.
- R. H. Templer, Curr. Opin. Colloid Interface Sci., 1998, 3, 255–263 CAS.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim, F. S. Bates, K. P. Davis, T. P. Lodge, M. L. Klein, R. H. DeVane, E. Aqad, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, Science, 2010, 328, 1009–1014 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |