Polymerization of substituted acetylenes and features of the formed polymers
Masashi
Shiotsuki
*a,
Fumio
Sanda
a and
Toshio
Masuda
b
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University Katsura Campus, Kyoto, 615-8510, Japan. E-mail: shiotsuki@adv.polym.kyoto-u.ac.jp
bDepartment of Environmental and Biological Chemistry, Faculty of Engineering, Fukui University of Technology, 3-6-1 Gakuen, Fukui, 910-8505, Japan
First published on 22nd December 2010
Abstract
Progress in the polymerization of substituted acetylenes and the properties and functions of the formed polymers that have been synthesized in the past several years are surveyed. Polymerizationcatalysts for substituted acetylenes, new monomers and polymers, controlled polymerizations, and photoelectronic functions and separation membranes of substituted polyacetylenes are discussed. A focus is placed on the development of novel rhodiumcatalysts for the polymerization of phenylacetylenes, the helical structures of the polymers obtained from chiral monosubstituted acetylenes, and highly gas-permeable polymers prepared from disubstituted acetylenes, in which great advances have been made recently.
 Masashi Shiotsuki | Masashi Shiotsuki has been an Assistant Professor at the Graduate School of Engineering at Kyoto University since 2003. He received his DEng degree in 2003 from the Graduate School of Engineering at Kyoto University under the supervision of Professor Take-aki Mitsudo. His research interests include transition metal-catalyzed polymerization and conjugated polymers, including substituted polyacetylenes. |
 Fumio Sanda | Fumio Sanda is currently an Associate Professor at Kyoto University. His research interests include amino acid based polymers, optically active conjugated polymers, and precisely controlled polymerization. He is an Editor of Polymer Reviews and Journal of the Adhesion Society of Japan. |
 Toshio Masuda | Toshio Masuda is currently a Professor at the Fukui University of Technology. His research interests include substituted polyacetylenes, transition metal catalyzed polymerization, gas separation membranes, and polymeric functional materials. He is an Associate Editor of Polymer. |
1 Introduction
Substituted acetylenes, which are typically mono- or disubstituted, can be polymerized by chain growth in the presence of suitable transition metal catalysts to yield high molecular weight polymers (eqn (1) and (2)). The mono- and disubstituted acetylenes are classified as aliphatic or aromatic and then further categorized as hydrocarbon-based or heteroatom-containing monomers. The polymers possess carbon–carbon alternating double bonds along the main chain, in which the extent of the conjugation depends on the number, type, and bulkiness of the side groups.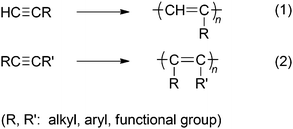
Transition metals of various groups in the periodic table, including Nb, Ta, Mo, W, Fe, Ru, Rh, and Pd, are effective in the polymerization of substituted acetylenes. The types of monomers that are polymerizable with a particular catalyst are rather restricted; hence, it is important to recognize the characteristics of each catalyst. Two types of reaction mechanisms may be involved, depending on the polymerizationcatalysts used. One is the metathesis mechanism, whereby the active species are metal carbenes, namely, species having a metal–carbon double bond and the other is the insertion mechanism in which the active species are alkyl metals, namely, species having a metal–carbon single bond. These mechanisms can be distinguished from each other according to the catalysts used but are rather difficult to distinguish based on the polymer structure. Recently, the controlled polymerizations of phenylacetylene (PA), including living polymerization, have been intensively researched.
The carbon–carbon alternating double bonds in the main chain of these polymers can exhibit unique properties, including electrical conductivity, nonlinear optical properties, magnetic properties, gas permeability, and photo- and electroluminescent properties, which are not accessible from the corresponding vinyl polymers. The structure and properties of poly(phenylacetylene) [poly(PA)] have been studied in detail, particularly with respect to its geometric structure, helical structure, and photoluminescence. In contrast, disubstituted acetylenepolymers are less conjugated due to steric hindrance, and so their photoelectronic functions have not been studied as much as those of monosubstituted acetylenepolymers. Instead, disubstituted acetylenepolymers are known as highly gas-permeable membrane materials due to their stiff main chain structure and the presence of spherical side groups.
This brief review surveys the polymerization of substituted acetylenes, focusing on the research performed over the past five years. Monomers and polymers, polymerizationcatalysts, controlled polymerizations, and functional polyacetylenes are discussed. Readers are encouraged to access other reviews and studies for a more comprehensive review of the polymerization of substituted acetylenes and the features of the substituted polyacetylenes that are formed.1–7
1.1 Recent advances in polymerizationcatalysts
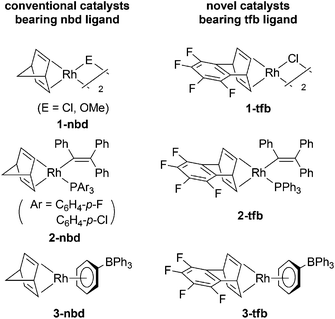
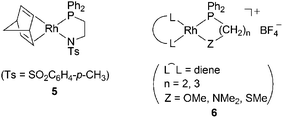
In the early study of substituted polyacetylene chemistry, various transition metal catalysts were examined, but for the most part, early transition metal catalysts were found to be effective. In particular, metal halides such as MoCl5, WCl6, TaCl5, and NbCl5 that are activated by alkylating agents successfully polymerize mono- and disubstituted acetylenes, resulting in high molecular weight polymers. In contrast, great efforts have been made in developing late transition metal catalysts in the past decade. Late transition metal catalysts complement early transition metal catalysts with respect to high tolerance towards air and moisture and the accessibility to well-defined catalysts suitable not only for living polymerization but also for the mechanistic studies of polymerization of substituted acetylenes. In this section, the late transition metal catalysts newly developed, mainly since 2006, are discussed, along with some improvements of early transition metal catalysts.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh2}(PPh3)] (2-tfb) and [(tfb)Rh(η6-Ph)BPh3] (3-tfb). Catalyst2-tfb polymerizes PA in a living fashion to accomplish the most narrow molecular weight distribution (i.e., polydispersity index 1.03) reported thus far.9Catalyst3-tfb shows higher activity than conventional 3-nbd.10 Optically pure dimethyl-introduced zwitterionic complexes 3-(S,S)-tfbMe2 and 3-(R,R)-tfbMe2 are effective for the helix-sense-selective polymerization of monosubstituted acetylene monomer 4 (Scheme 1).11 Complex 3-tfb is converted into a cationic derivative [(tfb)Rh(PPh3)2][BPh4] by the reaction of 3-tfb with PPh3, which also induces the living polymerization of PA in the presence of amines.12
CPh2}(PPh3)] (2-tfb) and [(tfb)Rh(η6-Ph)BPh3] (3-tfb). Catalyst2-tfb polymerizes PA in a living fashion to accomplish the most narrow molecular weight distribution (i.e., polydispersity index 1.03) reported thus far.9Catalyst3-tfb shows higher activity than conventional 3-nbd.10 Optically pure dimethyl-introduced zwitterionic complexes 3-(S,S)-tfbMe2 and 3-(R,R)-tfbMe2 are effective for the helix-sense-selective polymerization of monosubstituted acetylene monomer 4 (Scheme 1).11 Complex 3-tfb is converted into a cationic derivative [(tfb)Rh(PPh3)2][BPh4] by the reaction of 3-tfb with PPh3, which also induces the living polymerization of PA in the presence of amines.12
 | ||
| Scheme 1 Helix-sense-selective polymerization of PA derivative 4 by the chiral Rh zwitterionic catalyst, 3-(S,S)-tfbMe2 and 3-(R,R)-tfbMe2. | ||
A few reports have addressed rhodiumcatalysts that have a variety of bidentate ligands are composed of two different types of coordination sites (Chart 2). Jia and coworkers have demonstrated the activity of a neutral Rhcatalyst bearing a phosphinosulfonamido ligand, [(nbd)Rh(Ph2PCH2CH2NTs)] (5, Ts = SO2C6H4-p-Me), in the polymerization of PA.13 Jiménez et al. investigated a series of cationic Rhcatalysts coordinated by hemilabile Ph2P(CH2)nZ-type bidentate ligands (6, n = 2 or 3; Z = OMe, NMe2, SMe) that polymerize PA and its derivatives efficiently.14 Using NMR spectroscopy, they were able to directly observe the initiating species in the polymerization of PA using the catalyst containing the phosphinoamino ligand. The phenylethynyl Rh species 6b is formed in the reaction of 6a (diene = 1,5-cyclooctadiene (cod), n = 3, Z = NMe2) with PA, along with the formation of an ammonium moiety derived from the bidentate ligand and an acetylenic terminal proton of PA. Prior to this, it had been revealed that Rh acetylide-type complexes form in the reaction of certain complexes with acetylenic monomers.15,16 This is the first case demonstrating that a reaction forming the Rh-acetylide is driven by the formation of ammonium salt (Scheme 2). Complex 6 with diene = tfb, n = 3, and Z = NMe2 can achieve quasi-living polymerization of PA in the presence of 4-(N,N-dimethylamino)pyridine. This work also contributes to the following study regarding branched poly(PA) formed with the same catalyst as reported by Jiménez and Collins et al.17
 | ||
| Chart 2 The Rhcatalysts bearing P–L type bidentate ligands. | ||
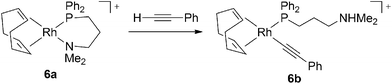 | ||
| Scheme 2 The formation of phenylethynyl species 6b by the reaction of 6a and excess PA. | ||
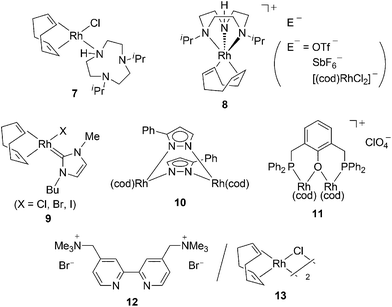
Other polymerizationcatalysts based on Rh complexes are summarized in Chart 3. Rh complexes bearing 1,4,7-triazacyclononane (7 and 8) have been well-characterized and are utilized as polymerizationcatalysts for PA.18 The Rhcarbene complex 9 is synthesized by the reaction of [(cod)Rh(µ-OMe)]2 with an ionic liquid, namely, 1-butyl-3-methylimidazolium halide, and is active in the catalyticpolymerization of PA.19 New bimetallic complexes 10 and 11 show catalytic activity in the polymerization of PA.20,21Water-soluble cationic bipyridine12 enables the recovery and reuse of a conventional catalyst, [(cod)RhCl]2 (13), which polymerizes PA in aqueous conditions under air.22 It has also been reported that the conventional complex 13 is incorporated in a spherical protein called ferritin. The apo-Fr-containing Rh–nbd complex obtained induces the polymerization of PA in the case of the protein to give the corresponding polymer with a narrower molecular weight distribution than that with catalyst13 alone.23
 | ||
| Chart 3 Rh-based catalysts for the polymerization of monosubstituted acetylene monomers. | ||
Recently, a few heterogeneous Rhcatalysts have been reported. Trzeciak's group demonstrated rhodiumnanoparticles stabilized by polyvinylpyrrolidone exhibit catalytic activity in the polymerization of PA.24 The stereochemistry of the polymer produced with this catalyst is purely cis-transoidal. The progress in polymerization can be monitored by atomic force microscopy (AFM) and transmission electron microscopy (TEM). This report includes the first detection of a spectacular helical PPA using AFM imaging. Son and Sweigart's group reported that the nanoparticles composed of the (benzoquinone)Rh(cod) complex and aluminium compounds catalyze the polymerization of PA. The catalystnanoparticles can be recovered by centrifugation, and the recovered nanoparticles show almost the same activity.25

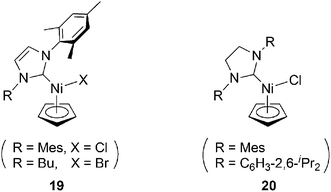
Ni-based catalysts (19 and 20, shown in Chart 5) are activated by excess MAO (methylaluminoxane) to oligomerize PA.28 This yields a corresponding oligomer, with up to 60% yield. Catalyst20 with R = C6H3-2,6-iPr2 results in the highest molecular weight polymer (Mn = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600) but also forms lower molecular weight oligomers, while the other catalysts yield monodisperse polymers with a smaller size range, with average molecular weights of less than 1600.
600) but also forms lower molecular weight oligomers, while the other catalysts yield monodisperse polymers with a smaller size range, with average molecular weights of less than 1600.
 | ||
| Chart 5 Novel Nicatalysts bearing imidazol-2-ylidene moieties. | ||
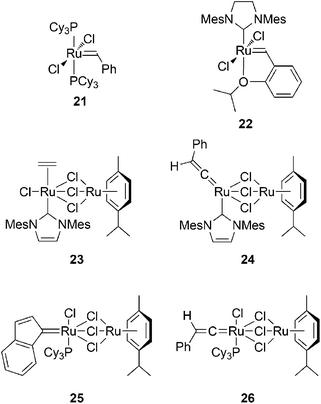 | ||
| Chart 6 Ruthenium carbene catalysts for the polymerization of monosubstituted acetylenic monomers. | ||
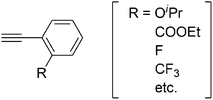
Another Rucarbene complex 27 (Chart 8) was reported to catalyze the oligomerization of PA and its derivatives into linear oligomers containing both positively charged and uncharged imidazolium end-groups.30 The Mw values of the formed oligomers are less than 670.
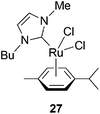 | ||
| Chart 8 Ru imidazolylidenecatalyst. | ||
It was shown that a molybdenum amide complex having the β-agostic NSi–H⋯Mo complex28 (Chart 9) polymerizes PA with moderate efficiency (TON = 34 and TOF = 2).32
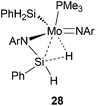 | ||
| Chart 9 Structure of a Mo amido complex having agostic NSi–H⋯Mo moiety. | ||
Heterogeneous Mocatalysts supported by siliceous mesoporous molecular sieves can successfully polymerize alkynes.33 Two types of Mocatalysts have been studied, which are based on the MoO3 and Schrock carbenes, Mo(CHCMe2Ph)(N–C6H3–2,6-iPr2)[OCMe(CF3)2]2. The latter exhibits high activity in the polymerization of 1-hexyne.
The quadruply bonded ditungsten complexes Na4[W2Cl8(THF)x] and [WCl4(thf)2] have been demonstrated to show catalytic activity in the polymerization of mono- and disubstituted acetylene monomers.34 In particular, Na4[W2Cl8(THF)x] shows high activity and achieves high yields of corresponding polymers with high molecular weights, with a Mn of 105000 at most in the case of poly(tert-butylacetyelene). Related triply bonded ditungsten complexes such as A3[W2(µ-Cl)3Cl6] (A+ = Bu4N+) and Na[W2(µ-Cl)3Cl4(THF)2] also show catalytic activity on the polymerization of several monosubstituted acetylenes.35
2 Monosubstituted acetylenes: secondary structures and functions of monosubstituted acetylenepolymers
Naturally occurring biomacromolecules, including proteins and DNA, commonly contain helical conformations, which are essential for carrying out their sophisticated and fundamental functions. Synthetic helical polymers have attracted much attention due to their ability to exhibit sophisticated features based on regulated structures that are similar to biomacromolecules. In particular, conjugated helical polymers such as polyisocyanides,36 polysilanes,37–39 and polyacetylenes40–43 have been intensively studied because of their photo-electronic functions, which are useful in industrial applications.Helical polymers of monosubstituted acetylenes were first synthesized with an Fecatalyst.44 Since the development of Rh(I) complexes45–47 and [Rh(nbd)Cl]2–triethylamine48 as catalysts for PA polymerization, these Rh complexes have been most commonly used as catalysts for the stereospecific polymerization of monosubstituted acetylenes due to their high tolerance towards various polar functional groups. The substituted polyacetylenes that are formed have cis-stereoregular double bonds in the main chains. The head-to-tail contents of Rh-based monosubstituted acetylenepolymers are estimated to be around 90% according to pyrolysis gas chromatography.49 This section overviews the recent studies on the secondary structures and functions of Rh-based monosubstituted acetylenepolymers.
2.1. Poly(PA) derivatives
A wide variety of poly(PA) derivatives adopt helical conformations (Chart 10). Achiral poly(PA)s having carboxylic groups and crown ether moieties (29a and 29b) predominantly induce one-handed helical structures by the addition of optically active compounds such as esters and the ammonium salts of amino acids.43 Helix-sense-selective polymerization is achieved using optically active amines as cocatalysts to yield poly(PA)s substituted by bis(hydroxymethyl)groups with biased helix sense,50–52 wherein intramolecular hydrogen bonding between the hydroxy groups stabilizes the helical structure. Anion recognition systems are constructed based on the interaction between 29c53 and 29d.54Alanine-derived 29e furnishes the twisting cables, spiral ribbons, spherical vesicles, and helical nanotubes.55N-Methylvaline-derived 29f catalyzes the asymmetric reduction of aromatic ketimines.56 The color and helical structures of 29g film are tuned by exposure to organic solvent vapor and heat.57,58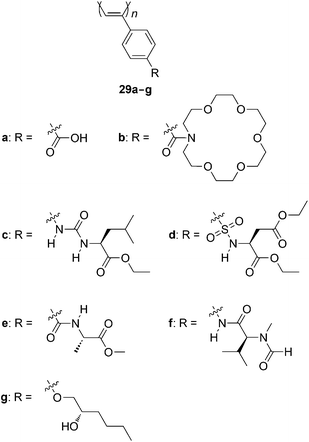 | ||
| Chart 10 Poly(PA) derivatives that adopt helical conformations. | ||
Chart 11 summarizes some recent examples of poly(PA) derivatives that exhibit electronic and photo-functions.59–61 The relationship between the spin–orbit coupling constant and g-values of poly(para-haloPA)s is examined.62Carbon nanotubes interact with 29h and 29i, which have carbazole/fluorene moieties by electron transfer,63 and form hybrids with 29j and 29k, which have pyrene64 and ferrocene65 moieties. A film of 29h undergoes electrochemical crosslinking.66 Polyradicals 29l67 and 29m68 exhibit redox properties and excellent reversible charge/discharge properties and thus are expected to become organic polymer batteries.
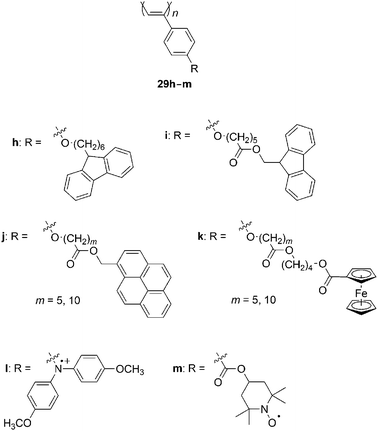 | ||
| Chart 11 Electronic and photo-functional poly(PA) derivatives. | ||
In addition to poly(PA) derivatives, polyacetylenes substituted with naphthalene,69pyrene,70 and carbazole59,71–73 moieties are synthesized, and their photoelectronic properties are examined. These polymers feature UV-vis absorption at long wavelength regions and exhibit large luminescence due to the long conjugation between the polyacetylene backbones and the polyaromatic side chains compared to poly(PA) derivatives.
2.2. Poly(N-propargylamide) derivatives
N-Propargylamides (30) efficiently undergo polymerization with Rhcatalysts to give cis-stereoregular poly(N-propargylamide)s (31, Scheme 3). When the substituent R is optically active, 31 predominantly forms helices with a one-handed screw sense due to the steric repulsion between the side chains as well as intramolecular N–H⋯OC hydrogen bonding (Fig. 1).74 Since the first report of helix formation of 31 in 2001, ca. 50 papers related to poly(N-propargylamide)s have been published.
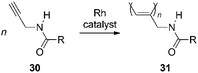 | ||
| Scheme 3 Polymerization of N-propargylamide. | ||
![Possible conformations of tightly (top) and loosely (bottom) twisted helical cis-stereoregular poly(N-propargylamide) [–CHC(CH2NHCOH)–]n, which accompany helically arranged intramolecular hydrogen-bonding strands (dotted lines) formed between the amidegroups at the ith and (i + 3)th units (top) and the ith and (i + 2)th units (bottom). Methine and methylenehydrogen atoms are omitted for clarity.](/image/article/2011/PY/c0py00333f/c0py00333f-f1.gif) | ||
| Fig. 1 Possible conformations of tightly (top) and loosely (bottom) twisted helical cis-stereoregular poly(N-propargylamide) [–CHC(CH2NHCOH)–]n, which accompany helically arranged intramolecular hydrogen-bonding strands (dotted lines) formed between the amidegroups at the ith and (i + 3)th units (top) and the ith and (i + 2)th units (bottom). Methine and methylenehydrogen atoms are omitted for clarity. | ||
Recent examples of helical poly(N-propargylamide)s include 32a, which is derived from optically active hydroxycarboxylic acids (Chart 12). Polymer32a tends to adopt a helical conformation in polar solvents such as DMF, while this does not happen in nonpolar solvents such as CHCl3.75 The opposite trend is true for poly(N-propargylamide)s that do not contain hydroxy groups. It is likely that the hydroxy groups of 32a prevent polar solvents from disturbing the intramolecular hydrogen bonding between the amidegroups. Gels that consist of mainly helically twisted 32a (m = 0) derived from lactic acid recognize chirality more prominently than analogous polymer gels that do not contain helices. The hydroxy groups of 32a can be protected by esterification. The bulkiness of the R′ substituent of 32b affects the helical conformation. Comparing 32b with R′ = –CH2Ph, –CHPh2, and –CPh3, it can be seen that the polymer bearing more phenyl groups forms a looser helix.76 Bulky side chains make the polymer chain flatter. Polymer32c, which carries azobenzene moieties in the side chains, is synthesized from lactic acid.77 The CD spectra simulated by the molecular orbital method agree with the experimental spectra and indicate the arrangement of azobenzene moieties in a mutual chiral geometry of a one-handed screw sense. The trans-azobenzene moieties in the side chain isomerize into cis-forms upon UV irradiation, while the helical structure of the main chain is not affected as much. The cis-azobenzene moieties reisomerize into trans-forms upon visible-light irradiation. Simultaneously, the azobenzene moieties recover the initial chiral arrangement at the side chains. The polymer forms a cholesteric liquid crystal.
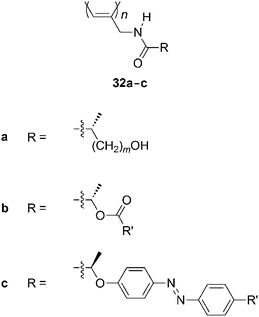 | ||
| Chart 12 Poly(N-propargylamide)s derived from optically active hydroxycarboxylic acids. | ||
Amino acids are a versatile source of chirality for organic synthesis. Synthetic polymers containing amino acid moieties show useful functions such as chiral separation, bioactivity, and stimuli-responsiveness in a manner similar to peptides and proteins.78 A series of helical polyacetylenes substituted with amino acid moieties are synthesized, some of which invert the helical sense by external stimuli such as temperature,79solvent,80,81 and pH.82 Fluorescence emission can be controlled according to the secondary structure of the polymers.83,84Ornithine- and lysine-based helical poly(N-propargylamide)s 32d and 32e can change the helix degree in response to an acid (Chart 13).85 Gels based on these polymers recognize chirality.86
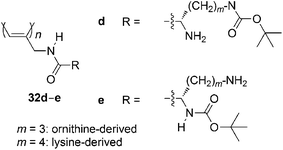
Synthetic polymers that have sugar residues have attracted much attention due to their potential applications in biologically active materials. Fluorescent-dye-labelled amphiphilic helical copolymers (33) are synthesized by the copolymerization of N-propargylamides that contain the galactose residue,87lauryloylgroup, and rhodamine B dye moieties (Chart 14).88 Human aortic endothelial cells (HAECs) are cultured in a medium containing 33. Cell uptake of the copolymer is confirmed by red fluorescence emission from each of the HAECs. An amylose-grafted polyacetylene is also synthesized chemoenzymatically, utilizing the polymerization of N-propargylamide.89
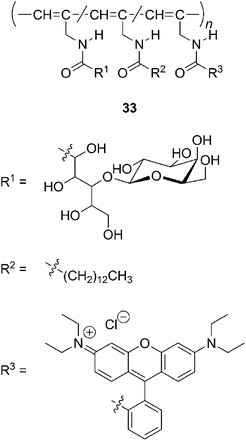 | ||
| Chart 14 Amphiphilic copolymers of N-propargylamides labelled with a fluorescent dye. | ||
The microemulsion polymerization of N-propargylamides provides nanoscale particles consisting of helical polymers in aqueous medium.90 These particles show a higher preference for a one-handed screw sense compared with the polymers that are synthesized in organic solvents. The subsequent radical polymerization of vinyl monomers yields nanoparticles consisting of a core that is composed of a substituted helical polyacetylene and a shell that is composed of a vinyl polymer.91 Achiral N-propargylamides undergo aqueous emulsion polymerizations in chiral micelles consisting of sodium dodecyl sulfate and an amino acid to yield optically active helical polymer emulsions.92
Optically active N-propargylphosphonamidates (34) having a P-chiral center are synthesized and polymerized to obtain the polymers shown in Chart 15.93–95 The formed polymers adopt a helical conformation that is stabilized by intramolecular N–H⋯OP hydrogen bonding. The predominance of the screw sense is determined by the P-chirality rather than the C-chirality.
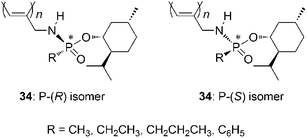 | ||
| Chart 15 P-Chiral poly(N-propargylphosphonamidate)s. | ||
2.3. Poly(1-methylpropargyl ester) derivatives
Propargyl alcohol is the most simple acetylene monomer that has a hydroxy group; it undergoes polymerization with Pd and Nicatalysts.96–1001-Methylpropargyl alcohol (35) is a chiral derivative of propargyl alcohol and has various applications in the field of organic chemistry; e.g., it has been utilized for the regioselective carbometalation with Grignard reagents affording 2-substituted allylic alcohols,101 in the synthesis of 2-substituted indolesviaSonogashira couplingcyclization,102 as a precursor of chiral allenylzinc and indium reagents,103 and in the synthesis of phosphinoyl 1,3-diene.104 The polymerization of 35 and the ester derivatives (i.e., 37) was first reported in 2007 (Scheme 4).105 The obtained polymers36 and 38 form helices, and the helical conformation of 38 having estergroups is thermally more stable than that of 36, which contains hydroxy groups.106 The remarkable ability of such a small chiral moiety to induce helicity is most likely due to the location of the chiral group adjacent to the main chain. In other words, the presence of a chiral group in close proximity to the main chain has enough of an effect to induce a helix that is stabilized by steric repulsion between the side chains.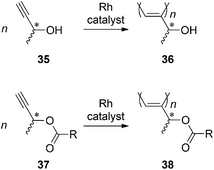 | ||
| Scheme 4 Polymerization of 1-methylpropargylalcohols and esters. | ||
Helical poly(1-methylpropargyl ester)s having various functional groups are synthesized. They include liquid crystalline polymers having cholesterylgroups,107,108 fluorescent polymers having carbazole109 and pyrenyl110 groups, photo-isomerizable polymers having azobenzene moieties,111 and stimuli-responsive polymers having amino acid moieties.112Graft copolymers consisting of a helical polyacetylene backbone and poly(methyl methacrylate)/poly(styrene) side chains are synthesized by the polymerization of 1-methylpropargyl ester-based macromonomers.113,114
Poly(1-methylpropargyl-N-alkylcarbamate)s (39, Chart 16) utilize intramolecular N–H⋯OC hydrogen bonding in addition to the steric repulsion between the methyl groups adjacent to the main chain.115 The polymers form a pseudohexagonal columnar structure by self-assembly or self-organization in the solid state. Thus, chiral 1-methylpropargyl alcohol is a simple but extremely powerful and useful helical source for substituted polyacetylenes.
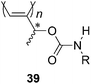 | ||
| Chart 16 Poly(1-methylpropargyl-N-alkylcarbamate)s. | ||
3 Disubstituted acetylenes
3.1 Polymerization
The polymerization behavior of disubstituted acetylene monomers has been sufficiently clarified, and the related studies and reviews are cited for ref. 2–4 and 6. The general relationship between the type of catalyst and the structure of the disubstituted acetylene monomers is as follows. In general, disubstituted acetylenes are sterically more crowded than their monosubstituted counterparts, and consequently, their effective polymerizationcatalysts are restricted virtually to group 5 and 6 transition metal catalysts; Rhcatalysts are generally not effective. Among disubstituted acetylenes, those with less steric hindrance, specifically internal alkynes, polymerize with Mo and W catalysts, while they tend to yield cyclotrimers with Nb and Tacatalysts. 1-Chloro-alkynes and 1-chloro-2-phenylacetylene polymerize with only Mocatalysts. In contrast, sterically crowded disubstituted acetylenes such as 1-trimethylsilyl-1-propyne do not polymerize with Mo or W catalysts, but they do polymerize with Nb and Tacatalysts. Diphenylacetylene and its ring-substituted derivatives are even more sterically hindered and polymerize only with the TaCl5-cocatalyst system. 1-Phenyl-1-alkynes have intermediate steric hindrance and hence polymerize with Nb, Ta, and W catalysts.The polymers from disubstituted acetylenes that have two identical groups or two groups of similar sizes are generally insoluble in any solvent. Most polymers from disubstituted acetylenes are colorless, although some aromatic polymers are yellow in color. While the controlled polymerization of monosubstituted acetylenes is applied to precision polymer syntheses such as living polymers, star polymers, and polymer brushes, this is not the case with disubstituted monomers because living polymerization is still not possible with disubstituted acetylene monomers. Cylindrical polymer brushes composed of a poly(diphenylacetylene) main chain and poly(oxyethylene) side chains have been prepared using the macromonomer and so-called ‘graft-from’ methods.116
3.2 Gas-permeable polymers
Polymers from disubstituted acetylenes have been intensively examined for their practical application as gas-permeable materials.117–120 These studies are motivated by the extremely high gas permeability of poly(1-trimethylsilyl-1-propyne) (PTMSP, Chart 17),120 which is the most permeable material available among all polymers. The oxygen permeability coefficient (PO2) of PTMSP ranges from 4000 to 9000 barrers, which is about ten times larger than that of poly(dimethylsiloxane). In addition to its high permeability, the ability of PTMSP to yield a freestanding film and its gas permeation mechanism, which is different from that of poly(dimethylsiloxane), have attracted much attention among membrane scientists. Poly[1-phenyl-2-p-(trimethylsilyl)phenylacetylene] (PTMSDPA, Chart 17) is a typical, highly gas-permeable poly(diphenylacetylene) and features high thermal stability compared to PTMSP.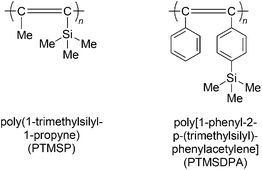 | ||
| Chart 17 Structures of PTMSP and PTMSDPA. | ||
Examples of highly gas-permeable substituted polyacetylenes are shown in Table 1. The PO2 values and oxygen/nitrogen selectivities (PO2/PN2) (25 °C) of about 100 substituted polyacetylenes have been measured so far.117,118,120 Among these substituted polyacetylenes, many of the polymers with large PO2 values contain spherical substituents, such as t-Bu, Me3Si and Me3Gegroups. In contrast, a majority of the less permeable polyacetylenes possess long n-alkyl groups. When the phenyl group is a main substituent, the gas permeability of the resulting polyacetylenes is usually considerably lower. Among commercially available oxygen-permeable polymermembranes, poly(dimethylsiloxane) is known to be most permeable to oxygen, with a PO2 value of 600 barrers (25 °C). As seen in Table 1, substituted polyacetylenes are very permeable to oxygen. The high gas permeability of polyacetylenes is attributable to their high free volume, which is presumably derived from their low cohesive energy structure, stiff main chain and spherical substituents.
| No |
|
PO2/barrera | PO2/PN2 | Reference | |
|---|---|---|---|---|---|
| R1 | R2 | ||||
| a 1 barrer = 1 × 10−10 cm3(STP) cm/(cm2 s cmHg). | |||||
| 1 | Me | SiMe3 | 4 × 103 to 9 × 103 | 1.8 | 120 |
| 2 | Me | SiEt3 | 860 | 2.0 | 121 |
| 3 | Me | SiMe2Et | 500 | 2.2 | 121 |
| 4 | Me | SiMe2-i-C3H7 | 460 | 2.7 | 121 |
| 5 | Me | GeMe3 | 7800 | 122 | |
| 6 | Ph | C6H4-p-SiMe3 | 1100–1550 | 2.1 | 117 and 118 |
| 7 | Ph | Ph | 910 | 2.2 | 123 |
| 8 | Ph | C6H4-m-SiMe3 | 1200 | 2.0 | 124 |
| 9 | Ph | C6H4-m-GeMe3 | 1100 | 2.0 | 117 and 118 |
| 10 | Ph | C6H4-p-t-C4H9 | 1100 | 2.2 | 117 and 118 |
| 11 | β-Naphthyl | C6H4-p-SiMe3 | 3500 | 1.8 | 117 and 118 |
| 12 | β-Naphthyl | Ph | 4300 | 1.6 | 117 and 118 |
| 13 | C6H4-p-F | C6H4-p-SiMe3 | 2900 | 1.5 | 124 |
| 14 | C6H4-p-F | Ph | 3000 | 1.4 | 124 |
| 15 | C6H3-m,p-F2 | C6H4-p-SiMe3 | 3600 | 1.5 | 124 |
| 16 | C6H3-m,p-F2 | Ph | 3800 | 1.3 | 124 |
| 17 | Ph | C6H4-p-OSiMe2-t-Bu | 160 | 3.2 | 125 |
| 18 | Ph | C6H4-p-OH | 8.0 | 3.3 | 125 |
| 19 | Ph |
|
1100 | 2.1 | 126 |
| 20 | Ph |

|
1400 | 1.9 | 126 |
| 21 | Ph |
|
4800 | 1.5 | 127 |
| 22 | C6H3-m,p-F2 |
|
6600 | 1.3 | 127 |
| 23 | Ph |
|
14400 |
1.2 | 128 |
| 24 | C6H3-m,p-F2 |
|
18700 |
1.1 | 128 |
Poly(diphenylacetylene)s are thermally very stable (T0 > 400 °C) and possess a film-forming ability. The ease in modifying ring substituents provides an opportunity to tune the permeability as well as the solubility and second-order conformation of the polymer. The permeability of poly(diphenylacetylene)s depends significantly on the shape of the ring substituents.117,118 Specifically, those with bulky ring substituents such as t-Bu, Me3Si and Me3Gegroups (nos. 6 and 8–10 in Table 1) exhibit very large PO2 values of up to 1000–1500 barrers, which is about one fourth of that of PTMSP and approximately twice as large as that of poly(dimethylsiloxane).
While poly(diphenylacetylene) is insoluble in any solvent, its derivatives with bulky ring-substituents are usually soluble in common solvents such as toluene and chloroform and derived membranes by solution casting. A poly(diphenylacetylene) membrane has been prepared by the desilylation of a PTMSDPA membrane that was catalyzed by trifluoroacetic acid.123 The prepared polymermembrane shows high thermal stability, insolubility in any solvent, and high gas permeability (e.g., an oxygen permeability of 910 barrers at 25 °C; no. 7 in Table 1). The high gas permeability of poly(diphenylacetylene) seems to be due to the generation of molecular-scale voids. In a similar way, poly(diphenylacetylene)s that contain various silyl groups such as Me2-i-PrSi, Et3Si and Me2-n-C8H17Si are soluble in common solvents, and poly(diphenylacetylene) membranes can be obtained by desilylation from those membranes. These oxygen permeability coefficients (120–3300 barrers) are quite different from one another, despite having the same polymer structure. When the bulkier silyl groups are removed, the oxygen permeability tends to increase to a larger extent.
Poly[1-aryl-2-p-(trimethylsilyl)phenylacetylene]s [aryl = naphthyl, fluorenyl, phenanthryl] are soluble in common solvents and afford freestanding membranes.117,118 These Si-containing polymermembranes are desilylated to yield the membranes of poly[1-aryl-2-phenylacetylene]s. Both the starting and the desilylated polymers show very high thermal stability and high gas permeability. For instance, the T0 and PO2 values of poly(1-β-naphthyl-2-phenylacetylene) are 470 °C and 4300 barrers, respectively (no. 12 in Table 1).117,118 Poly(diphenylacetylene)s with silyl groups and fluorine atoms are highly gas-permeable.124 The fractional free volume (FFV) of poly[1-(4-fluoro)phenyl-2-p-(trimethylsilyl)phenylacetylene] is 0.28 and appreciably large (e.g., 0.26 of PTMSDPA). The oxygen permeability coefficient of poly[1-(4-fluoro)phenyl-2-p-(trimethylsilyl)phenylacetylene] is as high as 2900 barrers, which is about twice that of PTMSDPA (no. 13 in Table 1). The incorporation of fluorine atoms into PTMSDPA generally enhances gas permeability.
Disubstituted acetylenes with hydroxy groups do not polymerize because Ta and Nbcatalysts are deactivated by polar groups such as hydroxy groups. In contrast, a protected monomer, that is, 1-phenyl-2-p-(tert-butyldimethylsiloxy)phenylacetylene, polymerizes to give a high molecular weight polymer.125 This polymer is soluble in common organic solvents and provides a freestanding membrane. Desilylation of a poly[1-phenyl-2-p-(tert-butyldimethylsiloxy)phenylacetylene] membrane yields a poly(diphenylacetylene) that has free hydroxy groups. This is the first example of a highly polar group-carrying poly(diphenylacetylene). Unlike the starting polymer, poly(1-phenyl-2-p-hydroxyphenylacetylene) is insoluble in nonpolar solvents such as toluene and chloroform. The PCO2/PCH4 and PCO2/PN2 permselectivity ratios of the poly(1-phenyl-2-p-hydroxyphenylacetylene) membrane can be as large as ca. 46, while the PCO2 is kept relatively high at 110 barrers.
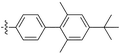
Diarylacetylene monomers containing substituted biphenyl and anthrylgroups have been synthesized and then polymerized with TaCl5–n-Bu4Sncatalyst to produce the corresponding poly(diarylacetylene)s.126 The formed polymers are soluble in common organic solvents such as cyclohexane, toluene, and chloroform and have high thermal stability over 400 °C according to thermogravimetric analysis. These polymermembranes, especially those with twisted biphenylgroups, exhibit high gas permeability, e.g., their PO2 values range from 130 to 1400 barrers. The membranes that have two methyl or chlorine atoms in the biphenylgroup show fairly high gas permeability (PO2 1100 and 1400 barrers, respectively), mostly likely because the twisted biphenyl structure is useful in generating molecular scale voids (no. 19 and 20 in Table 1).
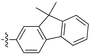
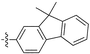
Diarylacetylenes having fluorenylgroups and other substituents (i.e., trimethylsilyl, tert-butyl, bromine, and fluorine) also polymerize with TaCl5–n-Bu4Sn, forming high molecular weight polymers (Mw 105 to 106) in approximately 10–60% yields.127 These polymers are soluble in common organic solvents and yield tough freestanding membranes by solution casting. These polymermembranes show high gas permeability, e.g., the PO2 value of the polymer that contains 9,9-dimethylfluorenyl and phenyl groups is as large as 4800 barrers (no. 21 in Table 1). The polymermembrane that possesses two fluorine atoms at the meta and para positions of the phenyl ring displays the highest oxygen permeability (PO2 6600 barrers) among these types of polymers.
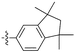
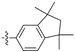
Acetylenic monomers containing indan and other groups also polymerize with the TaCl5–n-Bu4Sncatalyst.128 Most of the formed polymers are soluble in common organic solvents and afford freestanding membranes by solution casting. Despite the absence of bulky spherical groups, polymethylated indan-containing polymermembranes show extremely high gas permeability. For instance, the PO2 value of the polymer bearing 1,1,3,3-tetramethylindan and phenyl groups can reach 14400 barrers (no. 23 in Table 1). In particular, the PO2 values of the polymers having 1,1,3,3-tetramethylindan and either p-fluorophenyl or p,m-difluorophenylgroups reach 17900 and 18700 barrers, respectively, which are clearly larger than that of PTMSP.
PTMSP, which has long been known as the most gas-permeable polymer, is still being investigated with respect to various aspects of its permeation of gases and liquids. Research subjects in this area include membranes based on PTMSP for liquid–liquid separation;129 the effect of direct-current discharge treatment on the surface properties of a PTMSPmembrane;130 crosslinking and stabilization of nanoparticle-filled PTMSP nanocomposite membranes for gas separations;131 crosslinking PTMSP and its effect on physical stability;132gas transport properties of MgO-filled PTMSP nanocomposites;133bromination of PTMSP with different microstructures and properties of bromine-containing polymers;134 pure and mixed gas CH4 and n-C4H10 permeability and diffusivity in PTMSP;135gas transport properties of PTMSP and ethylcellulose filled with different molecular weight trimethylsilylsaccharides and the impact on fractional free volume and chain mobility;136Fourier transform infrared spectroscopy of PTMSPaging;137 and free volume and interstitial mesopores in silica filled PTMSP nanocomposites.138
PTMSDPA, a disubstituted glassy acetylene-based polymer, exhibits higher permeabilities to organic vapors than to permanent gases due to its rigid polyacetylene backbone and bulky side groups, which provide a relatively high FFV value of 0.26.139 The gas permeability and desilylation effect of poly(diphenylacetylene)s that have trimethylsilyl and alkyl groups have been studied.140Sulfonic acidgroups have been introduced into poly(diphenylacetylene)s to yield ionic and hydrophilic polyacetylenes.141,142 The degree of sulfonation usually ranges from 0.5 to 1.5 per repeating unit, and freestanding membranes can be obtained from the sulfonated polymers. Application of the membranes as proton-conducting fuelcell membranes has been examined.141 These membranes can also be used as CO2 separation membrane materials.142 The sulfonated polymers exhibit high CO2 permselectivity; e.g., their CO2/N2 separation factors are over 31. The sulfonated poly(diphenylacetylene) with the highest degree of sulfonation displays the highest CO2/N2 ratio of 75.
3.3 Photoelectronic functions
In regard to photoelectronic functions, monosubstituted acetylenepolymers have been studied more than disubstituted acetylenepolymers because they are generally more conjugated and coloured due to their less sterically demanding structure. However, considering the higher stability of the disubstituted acetylenepolymers, they may be more suited for practical applications.New poly(diphenylacetylene)s with alkoxy, silyl and fluorinegroups (e.g., 40 in Chart 18) have been synthesized using W and Tacatalysts.143 The polymer solutions emit a strong bluish-green light when photoexcited. The polymers containing electron-donating alkoxygroups show slightly longer fluorescence maxima compared to the polymers with the electron-withdrawing fluorine atoms. The effect of the alkyl chain length on the fluorescence of the poly(diphenylacetylene)s containing alkylsilane moieties (41 in Chart 18) in their side chains has been studied.144 Longer alkyl groups in the side chains of the polymer lead to longer fluorescence lifetimes. A longer alkyl group is also shown to be more effective than a shorter one in aligning the polymer chain parallel to the shearing direction.
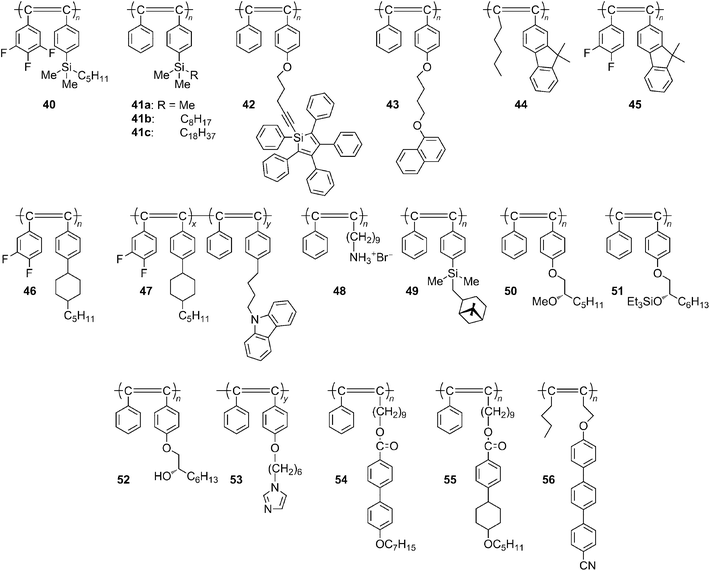
A poly(diphenylacetylene) containing 1,2,3,4,5-pentaphenylsilole (SiC4Ph5) pendants (42 in Chart 18) has been synthesized, and its photoluminescence has been studied.145 The ethynylgroup of the diphenylacetylene moiety polymerizes exclusively, resulting in a soluble polymer. The chloroform solution of the polymer shows a backbone emission centering at 522 nm, whereas the silole pendant is nonradiative at room temperature. Intramolecular rotations of the Ph groups on the silole moieties are responsible for the nonradiative decay of the silolechromophore. The intramolecular rotations, however, can be largely restricted through a cooling process of the polymer solution, which shows cooling-enhanced emission. Thus, the silole emission becomes dominant at lower temperatures.
α-Naphthalene-containing poly(diphenylacetylene)s with methylene spacers of different lengths (m = 4, 6, and 8) (e.g., 43 in Chart 18) have been synthesized. Although the TaCl5–n-Bu4Sncatalyst results in insoluble products in low yields, the WCl6–Ph4Sn catalyst furnishes soluble polymers with high molecular weights (Mw up to 5.0 × 104) in satisfactory yields of up to 62%.146 When the polymers are photoexcited in THF solution, they emit strong green lights with high efficiencies up to 98%. No significant shifts in the photoluminescence spectra are observed, even though the polymers are cast into thin solid films, suggesting little involvement of aggregative or excimeric emission. A multilayer EL device has been constructed that emits a green light of 520 nm with a maximum external quantum efficiency of 0.16%. The spectral stability is outstanding; no recognizable change is observed in the EL spectrum, even when the device current is raised.
Novel fluorene-containing polymers, poly[(1-pentyl-2-(9,9-dimethylfluoren-2-yl)acetylene)] (44 in Chart 18), and poly[1-(3,4-difluorophenyl)-2-(9,9-dimethylfluoren-2-yl)acetylene] (45 in Chart 18), have been synthesized using TaCl5–n-Bu4Sn as the catalyst.147 These polymers show emission peaks from 402 to 590 nm. In addition, their electroluminescent properties have been studied in heterostructure light-emitting diodes (LEDs), using these polymers as an emitting layer. A device based on 45 exhibits an orange-red emission at 602 nm with a maximum luminescence of 923 cd m−2 at 8 V. A device with the ITO/PEDOT/a mixture of 44 and 45 (98:2 wt ratio)/Ca/Al shows near-white emission. Its maximum luminance and current efficiency are 450 cd m−2 at 15 V and 1.3 cd A−1, respectively.
For organic light-emitting diode (OLED) applications, novel poly(diphenylacetylene)s (e.g., 46 in Chart 18), which exhibit air stability, better solubility in common organic solvents and higher luminescence than polyacetylene, have been examined as an emitter.148 The devices have a maximum brightness of 827 candela cd m−2 at 12 V and a maximum current efficiency of 0.78 cd A−1 at 9 V with a maximum luminescence at 536 nm.
Fluorine-containing poly(diphenylacetylene) (46 in Chart 18) shows a large red shift in UV-vis absorption and PL emission and a very high luminescent efficiency compared to its counterpart, which lacks the two fluorine atoms.149 The device performance can be improved using a light-emitting copolymer composed of 46 and a carbazole-bearing unit (47 in Chart 18). A light-emitting diode of ITO/PEDOT/47/Ca/Al displays a maximum luminescence of 4230 cd m−2 at 14 V and a maximum current efficiency of 3.37 cd A−1 at 7 V.
Nanohybridization of inorganic semiconductors with organic conjugated polymers is expected to lead to the creation of new hybrids with combined advantages of the two components, namely, the high charge mobility of the inorganics and the ready processability of the organics. Poly(diphenylacetylene)s containing ammonium bromide moieties (48) and PbBr2 provide a functional perovskite nanohybrid that shows a higher photoconductivity than its parent polymer48 alone.150
Optically active poly(diphenylacetylene) derivatives, poly(4-((S)-2-methoxyoctyloxy)diphenylacetylene) (50 in Chart 18), poly(4-((S)-2-triethylsiloxyoctyloxy)diphenylacetylene) (51), and poly(4-((S)-2-hydoxyoctyloxy)diphenylacetylene) (52) have been synthesized, and their chiroptical and liquid crystalline properties have been examined.152 The mirror image of the circular dichroic (CD) spectra of 50 and 51 in dilute solution indicates that their polymer backbones adopt a helical conformation with the opposite handedness. Polymer52 prepared from 51 by the deprotection of the triethylsilylgroup shows the same helical handedness as in 51. All of these polymers have a lyotropic liquid crystalline property, while thermotropic liquid crystalline behavior is observed in 50 and 52. The spin-cast films of 50–52 show strong bisignate CD signals centered at the absorption band of the polymer backbone, suggesting the formation of a chiral organization.
An imidazole-functionalized disubstituted acetylenepolymer (53 in Chart 18) has been synthesized via a postfunctional strategy153 and evaluated as a sensor for copper ions and α-amino acids by fluorescence quenching. Fluorescence quenching is observed at a low Cu2+ (7.0 × 10−7 mol L−1) concentration. The fluorescence intensity sharply decreases with an increase in Cu2+ concentration. The addition of α-amino acids to the solution of the 53/Cu2+ complex enhances the fluorescence of 53, assuming that the α-amino acid removes copper ions from the complex. Upon addition of glycine, the quenched fluorescence turns on immediately. The detection limit of glycine is as low as 6.0 × 10−5 mol L−1.
The liquid crystalline properties and optical anisotropy of poly[1-phenyl-2-p-(dimethyl-n-octadecylsilylphenyl)acetylene] (41c) have been investigated in detail.154Polymer41c exhibits unexpected smectic phase liquid crystallinity in highly concentrated aromatic organic solvents such as toluene. The two major absorption bands located at 430 and 370 nm are attributable to the π–π* transition parallel to the main chain and the localized π–π* transition with a charge-transfer characteristic among mesogenic repeating units perpendicular to the main chain axis, respectively. Polymer41c exhibits highly polarized absorption and fluorescence bands in a sheared film. The main chain axis of the polymer is aligned parallel to the shearing direction, whereas the long axis of the stilbene-like side group is perpendicular to the shearing direction.
A novel acetylene monomer containing a cyanoterphenylgroup, namely, 1-[(4′-cyano-4-terphenyl)oxy]-3-octyne, has been polymerized with WCl6–PhSn4catalyst to yield a liquid crystalline aliphatic polyacetylene (56 in Chart 18).155Polymer56 exhibits a nematic phase according to a polarizing optical microscope and shows a strong emission at 411 nm.
References
- Review: J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 100, 1645–1681 Search PubMed.
- T. Masuda, F. Sanda and M. Shiotsuki, in Comprehensive Organometallic Chemistry III, ed. R. Crabtree and M. Mingos, Elsevier, Oxford, 2007, vol. 11, ch. 16, pp. 557–593 Search PubMed.
- Review: T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165–180 Search PubMed.
- Review: M. G. Mayershofer and O. Nuyken, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5723–5747 Search PubMed.
- Review: J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 Search PubMed.
- T. Masuda and F. Sanda, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Wheinheim, 2003, vol. 3, pp. 375–406 Search PubMed.
- Review: J. Sedlacek and J. Vohlidal, Collect. Czech. Chem. Commun., 2003, 41, 1745–1790 Search PubMed.
- I. Saeed, M. Shiotsuki and T. Masuda, Macromolecules, 2006, 39, 8977–8981 CrossRef CAS.
- I. Saeed, M. Shiotsuki and T. Masuda, Macromolecules, 2006, 39, 8567–8573 CrossRef CAS.
- N. Onishi, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2009, 42, 4071–4076 CrossRef CAS.
- T. Nishimura, Y. Ichikawa, T. Hayashi, N. Onishi, M. Shiotsuki and T. Masuda, Organometallics, 2009, 28, 4890–4893 CrossRef CAS.
- M. Shiotsuki, N. Onishi, F. Sanda and T. Masuda, Chem. Lett., 2010, 244–245 CrossRef CAS.
- P. Xue, H. S. Y. Sung, I. D. Williams and G. Jia, J. Organomet. Chem., 2006, 691, 1945–1953 CrossRef CAS.
- M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, E. Vispe, F. J. Lahoz and L. A. Oro, Macromolecules, 2009, 42, 8146–8156 CrossRef CAS.
- H. Komatsu, Y. Suzuki and H. Yamazaki, Chem. Lett., 2001, 998–999 CrossRef CAS.
- Y. Kishimoto, T. Miyatake, T. Ikariya and R. Noyori, Macromolecules, 1996, 29, 5054–5055 CrossRef CAS.
- M. Angoy, M. I. Bartolomé, E. Vispe, P. Lebeda, M. V. Jiménez, J. J. Pérez-Torrente, S. Collins and S. Podzimek, Macromolecules, 2010, 43, 6278–6283 CrossRef CAS.
- A. L. Gott, P. C. McGowan and C. N. Temple, Organometallics, 2008, 28, 2852–2860 CrossRef.
- W. Gil, T. Lis, A. Trzeciak and J. J. Ziółkowski, Inorg. Chim. Acta, 2006, 359, 2835–2841 CrossRef CAS.
- T. Morawitz, S. Bao, M. Bolte, H.-W. Lerner and M. Wagner, J. Organomet. Chem., 2008, 693, 3878–3884 CrossRef CAS.
- C. Zhu, N. Yukimura and M. Yamane, Organometallics, 2010, 29, 2098–2013 CrossRef CAS.
- Y.-H. Wang and F.-Y. Tsai, Chem. Lett., 2007, 1492–1493 CrossRef CAS.
- S. Abe, K. Hirata, T. Ueno, K. Morino, N. Shimizu, M. Yamamoto, M. Takata, E. Yashima and Y. Watanabe, J. Am. Chem. Soc., 2009, 131, 6958–6960 CrossRef CAS.
- M. Kopaczyńska, J. H. Fuhrhop, A. M. Trzeciak, J. J. Ziółkowski and R. Choukroun, New J. Chem., 2008, 32, 1509–1512 RSC.
- K. H. Park, K. Jang, S. U. Son and D. A. Sweigart, J. Am. Chem. Soc., 2006, 128, 8740–8741 CrossRef CAS.
- S. O. Ojwach, I. A. Guzei, J. Darkwa and S. F. Mapolie, Polyhedron, 2007, 26, 851–861 CrossRef CAS.
- K. Li, M. S. Mohlala, T. V. Segapelo, P. M. Shumbula, I. A. Guzei and J. Darkwa, Polyhedron, 2008, 27, 1017–1023 CrossRef CAS.
- W. Buchowicz, W. Wojtczak, A. Pietrzykowski, A. Lupa, L. B. Jerzykiewicz, A. Makal and K. Woźniak, Eur. J. Inorg. Chem., 2010, 648–656 CrossRef CAS.
- T. Katsumata, M. Shiotsuki, F. Sanda, X. Sauvage, L. Delaude and T. Masuda, Macromol. Chem. Phys., 2009, 210, 1891–1902 CrossRef CAS.
- P. Csabai, F. Joó, A. M. Trzeciak and J. J. Ziółkowski, J. Organomet. Chem., 2006, 691, 3371–3376 CrossRef CAS.
- N. S. Santhosh and G. Sundararajan, Eur. Polym. J., 2007, 43, 4306–4315 CrossRef CAS.
- A. Y. Khalimon, R. Simionescu, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Angew. Chem., Int. Ed., 2008, 47, 7701–7704 CrossRef CAS.
- H. Balcar, P. Topka, J. Sedláček, J. Zedník and J. Čejka, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2593–2599 CrossRef CAS.
- G. Floros, N. Saragas, P. Pararaskevopoulou, I. Choinopoulos, S. Koinis, N. Psaroudakis, M. Pitsikalis and K. Metris, J. Mol. Catal. A: Chem., 2008, 289, 76–81 CrossRef CAS.
- N. Saragas, G. Floros, P. Paraskevopoulou, N. Psaroudakis, S. Koinis, M. Pitsikalis and K. Mertis, J. Mol. Catal. A: Chem., 2009, 303, 124–131 CrossRef CAS.
- Review: M. Suginome and Y. Ito, Adv. Polym. Sci., 2004, 171, 77–136 Search PubMed.
- Review: M. Fujiki, J. Organomet. Chem., 2003, 685, 15–34 Search PubMed.
- Review: M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J. (Tokyo, Jpn.), 2003, 35, 297–344 Search PubMed.
- Review: T. Sato, K. Terao, A. Teramoto and M. Fujiki, Polymer, 2003, 44, 5477–5495 Search PubMed.
- Review: T. Aoki, T. Kaneko and M. Teraguchi, Polymer, 2006, 47, 4867–4892 Search PubMed.
- Review: K. Akagi, Chem. Rev., 2009, 109, 5354–5401 Search PubMed.
- Review: J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 Search PubMed.
- Review: E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 Search PubMed.
- F. Ciardelli, S. Lanzillo and O. Poeroni, Macromolecules, 1974, 7, 174–179 CrossRef CAS.
- Y. Kishimoto, M. Itou, T. Miyake, T. Ikariya and R. Noyori, Macromolecules, 1995, 28, 6662–6666 CrossRef CAS.
- A. Furlani, C. Napoletano, M. V. Russo, A. Camus and N. Marsich, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 75–86 CAS.
- A. Furlani, C. Napoletano, M. V. Russo and W. J. Feast, Polym. Bull. (Heidelberg, Ger.), 1986, 16, 311–317 Search PubMed.
- M. Tabata, W. Yang and K. Yokota, Polym. J. (Tokyo, Jpn.), 1990, 22, 1105–1107 CrossRef CAS.
- Z. Doležal, R. Kubinec, J. Sedláček, V. Pacáková and J. Vohlídal, J. Sep. Sci., 2007, 30, 731–739 CrossRef CAS.
- T. Aoki, T. Kaneko, N. Maruyama, A. Sumi, M. Takahashi, T. Sato and M. Teraguchi, J. Am. Chem. Soc., 2003, 125, 6346–6347 CrossRef CAS.
- T. Kaneko, Y. Umeda, T. Yamamoto, M. Teraguchi and T. Aoki, Macromolecules, 2005, 38, 9420–9426 CrossRef CAS.
- T. Kaneko, Y. Umeda, H. Jia, S. Hadano, M. Teraguchi and T. Aoki, Macromolecules, 2007, 40, 7098–7102 CrossRef CAS.
- R. Kakuchi, S. Nagata, R. Sakai, I. Otsuka, H. Nakade, T. Satoh and T. Kakuchi, Chem.–Eur. J., 2008, 14, 10259–10266 CrossRef CAS.
- R. Kakuchi, T. Kodama, R. Shimada, Y. Tago, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 3892–3897 CrossRef CAS.
- K. K. L. Cheuk, B. S. Li, J. W. Y. Lam, Y. Xie and B. Z. Tang, Macromolecules, 2008, 41, 5997–6005 CrossRef CAS.
- K. Terada, T. Masuda and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4971–4981 CrossRef CAS.
- T. Fukushima, K. Takachi and K. Tsuchihara, Macromolecules, 2006, 39, 3103–3105 CrossRef CAS.
- T. Fukushima, H. Kimura and K. Tsuchihara, Macromolecules, 2009, 42, 8619–8626 CrossRef CAS.
- J. Qu, R. Kawasaki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2006, 47, 6551–6559 CrossRef.
- K. Tamura, T. Fujii, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2008, 49, 4494–4501 CrossRef CAS.
- H.-P. Xu, B.-Y. Xie, W.-Z. Yuan, J.-Z. Sun, F. Yang, Y.-Q. Dong, A. Qin, S. Zhang, M. Wang and B. Z. Tang, Chem. Commun., 2007, 1322–1324 RSC.
- A. Miyasaka, T. Sone, Y. Mawatari, S. Setayesh, K. Müllen and M. Tabata, Macromol. Chem. Phys., 2006, 207, 1938–1944 CrossRef CAS.
- H. Zhao, W. Z. Yuan, J. Mei, L. Tang, X. Q. Liu, J. M. Yan, X. Y. Shen, J. Z. Sun, A. Qin and B. Z. Tang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4995–5005 CrossRef CAS.
- W. Z. Yuan, J. Z. Sun, Y. Dong, M. Ha1ussler, F. Yang, H. P. Xu, A. Qin, J. W. Y. Lam, Q. Zheng and B. Z. Tang, Macromolecules, 2006, 39, 8011–8020 CrossRef CAS.
- W. Z. Yuan, J. Z. Sun, J. Z. Liu, Y. Dong, Z. Li, H. P. Xu, A. Qin, M. Hussler, J. K. Jin, Q. Zheng and B. Z. Tang, J. Phys. Chem. B, 2008, 112, 8896–8905 CrossRef CAS.
- T. Fulghum, S. M. Abdul Karim, A. Baba, P. Taranekar, T. Nakai, T. Masuda and R. C. Advincula, Macromolecules, 2006, 39, 1467–1473 CrossRef CAS.
- H. Murata, D. Miyajima and H. Nishide, Macromolecules, 2006, 39, 6331–6335 CrossRef CAS.
- J. Qu, T. Katsumata, M. Satoh, J. Wada, J. Igarashi, K. Mizoguchi and T. Masuda, Chem.–Eur. J., 2007, 13, 7965–7973 CrossRef CAS.
- J. G. Rodríguez and J. L. Tejedor, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2038–2047 CrossRef CAS.
- J. M. Reyna-González, M. Aguilar-Martínez, A. García-Concha, C. Palomar and E. Rivera, Synth. Met., 2009, 159, 659–665 CrossRef CAS.
- F. Sanda, R. Kawasaki, M. Shiotsuki and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4450–4458 CrossRef CAS.
- J. Qu, R. Kawasaki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 467–476 CrossRef CAS.
- F. Sanda, R. Kawasaki, M. Shiotsuki, T. Takashima, A. Fujii, M. Ozaki and T. Masuda, Macromol. Chem. Phys., 2007, 208, 765–771 CrossRef CAS.
- R. Nomura, J. Tabei and T. Masuda, J. Am. Chem. Soc., 2001, 123, 8430–8431 CrossRef CAS.
- F. Sanda, T. Fujii, J. Tabei, M. Shiotsuki and T. Masuda, Macromol. Chem. Phys., 2008, 209, 112–118 CrossRef CAS.
- F. Sanda, T. Fujii, M. Shiotsuki and T. Masuda, Polym. J. (Tokyo, Jpn.), 2008, 40, 768–774 CrossRef CAS.
- T. Fujii, M. Shiotsuki, Y. Inai, F. Sanda and T. Masuda, Macromolecules, 2007, 40, 7079–7088 CrossRef CAS.
- F. Sanda and T. Endo, Macromol. Chem. Phys., 1999, 200, 2651–2661 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 8888–8892 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Polymer, 2006, 47, 2596–2602 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 253–261 CrossRef CAS.
- F. Sanda, K. Terada and T. Masuda, Macromolecules, 2005, 38, 8149–8154 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 8893–8896 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Polymer, 2006, 47, 1584–1589 CrossRef CAS.
- R. Liu, F. Sanda and T. Masuda, Polymer, 2007, 48, 6510–6518 CrossRef CAS.
- R. Liu, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4175–4182 CrossRef CAS.
- J. Kadokawa, K. Tawa, M. Suenaga, Y. Kaneko and M. Tabata, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43, 1179–1187 Search PubMed.
- M. Suenaga, Y. Kaneko, J. Kadokawa, T. Nishikawa, H. Mori and M. Tabata, Macromol. Biosci., 2006, 6, 1009–1018 CrossRef CAS.
- Y. Sasaki, Y. Kaneko and J. Kadokawa, Polym. Bull. (Heidelberg, Ger.), 2009, 62, 291–303 Search PubMed.
- J. Deng, B. Chen, X. Luo and W. Yang, Macromolecules, 2009, 42, 933–938 CrossRef CAS.
- B. Chen, J. Deng, X. Liu and W. Yang, Macromolecules, 2010, 43, 3177–3182 CrossRef CAS.
- X. Luo, L. Li, J. Deng, T. Guo and W. Yang, Chem. Commun., 2010, 46, 2745–2747 RSC.
- D. Yue, T. Fujii, K. Terada, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Rapid Commun., 2006, 27, 1460–1464 CrossRef CAS.
- D. Yue, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 68–73 CrossRef CAS.
- D. Yue, T. Fujii, K. Terada, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1515–1524 CrossRef CAS.
- M. Yang, M. Zheng, A. Furlani and M. V. Russo, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2709–2713 CrossRef CAS.
- A. Furlani, M. V. Russo and A. Longo, Polymer, 1997, 38, 183–189 CrossRef CAS.
- M. V. Russo, A. Furlani, P. Altamura and I. Fratoddi, Polymer, 1997, 38, 3677–3690 CrossRef CAS.
- X. Zhan, M. Yang and H. Sun, Catal. Lett., 2000, 70, 79–82 CrossRef CAS.
- X. Zhan, M. Yang and H. Sun, Macromol. Rapid Commun., 2001, 22, 530–534 CrossRef CAS.
- Z. Lu and S. Ma, J. Org. Chem., 2006, 71, 2655–2660 CrossRef CAS.
- S. S. Palimkar, P. H. Kumer, R. J. Lahoti and K. V. Srinivasan, Tetrahedron, 2006, 62, 5109–5115 CrossRef CAS.
- J. M. Marshall, P. Eidam and H. S. Eidam, J. Org. Chem., 2006, 71, 4840–4844 CrossRef CAS.
- L. B. Han, Y. Ono and H. Yazawa, Org. Lett., 2005, 7, 2909–2911 CrossRef CAS.
- Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2007, 40, 1864–1867 CrossRef CAS.
- Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Chem.–Asian J., 2008, 3, 2075–2081 CrossRef CAS.
- J. Qu, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2007, 208, 823–832 CrossRef CAS.
- J. Qu, Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2007, 208, 1992–1999 CrossRef CAS.
- J. Qu, Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 4628–4636 CrossRef.
- J. Qu, Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 6491–6500 CrossRef CAS.
- J. Qu, F. Jiang, H. Chen, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4749–4761 CrossRef CAS.
- J. Qu, F. Sanda and T. Masuda, Eur. Polym. J., 2009, 45, 448–454 CrossRef CAS.
- M. Shiotsuki, W. Zhang and T. Masuda, Polym. J. (Tokyo, Jpn.), 2007, 39, 690–695 CrossRef CAS.
- W. Zhang, M. Shiotsuki and T. Masuda, Macromol. Rapid. Commun., 2007, 28, 1115–1121 CrossRef CAS.
- Y. Shirakawa, Y. Suzuki, K. Terada, M. Shiotsuki, T. Masuda and F. Sanda, Macromolecules, 2010, 43, 5575–5581 CrossRef CAS.
- Z. Qin, Y. Chen, W. Zhou, X. He, F. Bai and M. Wan, Eur. Polym. J., 2008, 44, 3732–3740 CrossRef CAS.
- K. Nagai, Y. M. Lee and T. Masuda, in Macromolecular Engineering, ed. K. Matyjaszewsky, Y. Gnanou and L. Leibler, Wiley-VCH, Weinheim, Germany, 2007, part 4, ch. 19 Search PubMed.
- T. Masuda and K. Nagai, in Materials Science of Membranes, ed. Y. Yampolskii, I. Pinnau and B. D. Freeman, Wiley, Chichester, UK, 2006, ch. 8 Search PubMed.
- Review: M. Ulbricht, Polymer, 2006, 47, 2217–2262 Search PubMed.
- Review: K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721–798 Search PubMed.
- L. M. Robeson, W. F. Burgoyne, M. Langsam, A. C. Savoca and C. F. Tien, Polymer, 1994, 35, 4970–4978 CAS.
- G. Kwak and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2964–2969 CrossRef CAS.
- T. Sakaguchi, K. Yumoto, M. Shiotsuki, F. Sanda, M. Yoshikawa and T. Masuda, Macromolecules, 2005, 38, 2704–2709 CrossRef CAS.
- T. Sakaguchi, M. Shiotsuki, F. Sanda, B. D. Freeman and T. Masuda, Macromolecules, 2005, 38, 8327–8332 CrossRef CAS.
- Y. Shida, T. Sakaguchi, M. Shiotsuki, F. Sanda, B. D. Freeman and T. Masuda, Macromolecules, 2005, 38, 4096–4102 CrossRef CAS.
- Y. Hu, T. Shimizu, K. Hattori, M. Shiotsuki, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 861–868 CrossRef CAS.
- A. Fukui, K. Hattori, Y. Hu, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2009, 50, 4159–4165 CrossRef CAS.
- Y. Hu, M. Shiotsuki, F. Sanda, B. D. Freeman and T. Masuda, Macromolecules, 2008, 41, 8525–8532 CrossRef CAS.
- A. V. Volkov, V. V. Volkov and V. S. Khotimskii, Vysokomol. Soedin., Ser. A Ser. B, 2009, 51, 2113–2128 Search PubMed.
- Yu. V. Kostina, A. B. Gil'man, A. V. Volkov, M. S. Piskarev, S. A. Legkov, E. G. Litvinova, V. S. Khotimskii and G. N. Bondarenko, High Energy Chem., 2009, 43, 509–511 CrossRef CAS.
- L. Shao, J. Samseth and M.-B. Haegg, J. Appl. Polym. Sci., 2009, 113, 3078–3088 CrossRef CAS.
- S. D. Kelman, B. W. Rowe, C. W. Bielawski, S. J. Pas, A. J. Hill, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2008, 320, 123–134 CrossRef CAS.
- S. Matteucci, V. A. Kusuma, S. D. Kelman and B. D. Freeman, Polymer, 2008, 49, 1659–1675 CrossRef CAS.
- A. A. Masalev, V. S. Khotimskii, G. N. Bondarenko and M. V. Chirkova, Vysokomol. Soedin., Ser. A Ser. B, 2008, 50, 47–53 Search PubMed.
- R. D. Raharjo, B. D. Freeman, D. R. Paul and E. S. Sanders, Polymer, 2007, 48, 7329–7344 CrossRef CAS.
- J. Qiu, J. M. Zheng and K. V. Peinemann, Macromolecules, 2007, 40, 3213–3222 CrossRef CAS.
- V. L. Khodzhaeva and V. G. Zaikin, J. Appl. Polym. Sci., 2007, 103, 2523–2527 CrossRef CAS.
- P. Winberg, K. DeSitter, C. Dotremont, S. Mullens, I. F. J. Vankelecom and F. H. J. Maurer, Macromolecules, 2005, 38, 3776–3782 CrossRef CAS.
- R. D. Raharjo, H. J. Lee, B. D. Freeman, T. Sakaguchi and T. Masuda, Polymer, 2005, 46, 6316–6324 CrossRef CAS.
- A. Takeda, T. Sakaguchi and T. Hashimoto, Polymer, 2009, 50, 5031–5036 CrossRef CAS.
- H. Ito, E. Akiyama, H. Yokota and K. Takedai, US Pat., Appl. Publ., 2007, 20070231654.
- T. Sakaguchi, K. Kameoka and T. Hashimoto, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6463–6471 CrossRef CAS.
- G. Kwak, H.-Q. Wang, K.-H. Choi, K.-H. Song, S.-H. Kim, H.-S. Kim, S.-J. Lee, H.-Y. Cho, E.-J. Yu, H.-J. Lee, E.-J. Park and L.-S. Park, Macromol. Rapid Commun., 2007, 28, 1317–1324 CrossRef CAS.
- G. Kwak, M. Minakuchi, T. Sakaguchi, T. Masuda and M. Fujiki, Macromolecules, 2008, 41, 2743–2746 CrossRef CAS.
- J. Chen, H. S. Kwok and B. Z. Tang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2487–2498 CrossRef CAS.
- C. C. W. Law, J. W. Y. Lam, A. Qin, Y. Dong, H. S. Kwok and B. Z. Tang, Polymer, 2006, 47, 6642–6651 CrossRef CAS.
- C. Huang, S. Yang, K. Chen and C. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 519–531 CrossRef CAS.
- C.-H. Huang, C.-W. Lee, C.-S. Hsu, C. Renaud and T.-P. Nguyen, Thin Solid Films, 2007, 515, 7671–7674 CrossRef CAS.
- S.-H. Yang, C.-H. Huang, C.-H. Chen and C.-S. Hsu, Macromol. Chem. Phys., 2009, 210, 37–47 CrossRef CAS.
- H. Xu, J. Sun, A. Qin, J. Hua, Z. Li, Y. Dong, H. Xu, W. Yuan, Y. Ma, M. Wang and B. Z. Tang, J. Phys. Chem. B, 2006, 110, 21701–21709 CrossRef CAS.
- M. Teraguchi, J. Suzuki, T. Kaneko, T. Aoki and T. Masuda, Macromolecules, 2003, 36, 9694–9697 CrossRef CAS.
- T. Fukushima and K. Tsuchihara, Macromolecules, 2009, 42, 5453–5460 CrossRef CAS.
- Q. Zeng, C. K. W. Jim, J. W. Y. Lam, Y. Dong, Z. Li, J. Qin and B. Z. Tang, Macromol. Rapid Commun., 2009, 30, 170–175 CrossRef CAS.
- G. Kwak, M. Minakuchi, T. Sakaguchi, T. Masuda and M. Fujiki, Chem. Mater., 2007, 19, 3654–3661 CrossRef CAS.
- L. Chen, Y. Chen, W. Zhou and X. He, Synth. Met., 2009, 159, 576–582 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |