Tuning the lower critical solution temperature of thermoresponsive polymers by biospecific recognition†
Jens
Buller
a,
André
Laschewsky
*a,
Jean-François
Lutz‡
b and
Erik
Wischerhoff
*b
aDepartment of Chemistry, Universität Potsdam, Karl-Liebknecht-Str. 24-25, D-14476, Potsdam-Golm, Germany
bFraunhofer Institute for Applied Polymer Research, Geiselbergstrasse 69, D-14476, Potsdam-Golm, Germany
First published on 15th February 2011
Abstract
A thermosensitive statistical copolymer based on oligo(ethylene glycol) methacrylates incorporating biotin was synthesized by free radical copolymerisation. The influence of added avidin on its thermoresponsive behaviour was investigated. The specific binding of avidin to the biotinylated copolymers provoked a marked increase of the lower critical solution temperature.
Thermoresponsive polymer systems are intensively investigated for bioapplications such as tissue engineering, drug delivery, protein chromatography or controlled bioadhesion.1–9 Most of these systems exhibit a lower critical solution temperature (LCST) in aqueous media: while they are compatible with water at low temperatures, they phase-separate above a certain temperature value. So-called switchable biosensors exploit this effect, modulating the accessibility of a polymer-tethered recognition group for an analyte by changing the temperature.10,11
In other situations, it is desirable to reverse the concept. In this case, the binding of an analyte is used to induce a change in the polymer properties. Though such analyte responsive polymers do not require temperature change as a trigger, they are often based on thermosensitive polymers.
While in the simplest case, such responsive systems are realised in form of soluble polymers, they are frequently implemented in form of hydrogels. In many cases of such analyte responsive polymeric hydrogels,2,12,13 a binding event on a polymer modifies the swelling behaviour. One typical strategy exploits crosslinking effects, as e.g. in thermoresponsive networks.14 Alternatively, the concept can be put into practice with permanently hydrophilic networks. When for instance physical antibody–antigen crosslinks are incorporated into a covalently crosslinked polyacrylamide hydrogel, the crosslinks can be broken by competitive binding of additional free antigens, thus provoking a reversible swelling response.15
Another approach utilises the inherent phase transition of thermosensitive hydrogels. In this strategy, the analyte binding results in a shift of the transition temperature. Under appropriate isothermal conditions near the transition temperature this shift will induce the phase transition. The polymer scaffold acts as actuator that transduces (and potentially amplifies) the signal generated by the binding event.
Several such systems have already been investigated so far. For instance, thermosensitive hydrogels incorporating phenyl boronic acid moieties were reported for the detection of glucose.16,17 A covalently crosslinked poly(N-isopropyl acrylamide (PNIPAM) hydrogels containing anti-fluorescein antibodies showed a swelling response when exposed to fluorescein.18 Similarly, a PNIPAM hydrogel containing a lectin swelled when the sodium salt of a dextran sulfate was added.19 However, a shift of a lower critical solution temperature due to binding of native proteins has not been observed, yet. Such a case, however, is of particular importance, since proteins are the most relevant and common analytes with regard to biomedical sensor systems. In fact, the use of water-soluble polymers for sensing proteins is a thriving topic at present.20
The avidin–biotin system is a widespread and well studied model affinity system for protein–ligand interactions.21 In the past, thermosensitive polymers have been conjugated to biotin, for modifying the specific recognition of avidin by modulating the temperature.22–27 Typically, biotin was introduced as end-group in such systems. Here, we demonstrate that it is possible to reverse the trigger effect, namely that binding of avidin to a thermosensitive polymer incorporating biotin moieties (cf.Scheme 1) can markedly shift the lower critical solution temperature. For this purpose, we incorporated the biotin moieties into the thermo-sensitive polymer throughout the polymer chain, and used only a short linker group for attaching the biotin moiety, in the attempt to avoid a potential spacer effect. Thus, we hoped to maximise the effect of the protein–ligand binding event on the phase transition temperature.
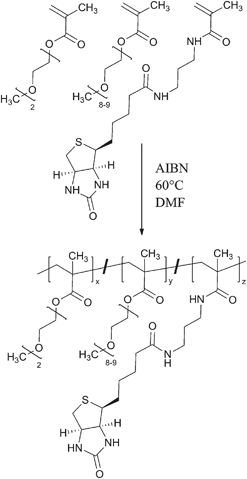 | ||
| Scheme 1 Free radical copolymerization of MEO2MA, OEGMA and biotin-3-aminopropyl methacrylamide yielding a biotin-containing and thermosensitive statistical copolymer. For the investigated copolymer: x = 0.68, y = 0.14 and z = 0.18. | ||
To selectively bind avidin to the polymer, biotinyl-3-aminopropyl methacrylamide was synthesised. This new monomer was prepared in analogy to a known homologue (see ESI†).28 Briefly, the N-hydroxy succinimide ester of biotin was reacted with 3-aminopropyl methacrylamide. The resulting functional monomer was copolymerized with 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and oligo(ethylene glycol) methacrylate (OEGMA, Mn = 475 g mol−1) by free radical polymerization with AIBN as thermal initiator (Scheme 1). Copolymers made from these monomers are well known to be thermosensitive and allow a simple adjustment of the LCST by varying the comonomer composition.29,30
When the polymers incorporate the biotinylated monomer, they are slightly less hydrophilic than pure poly(MEO2MA-co-OEGMA)s copolymers. Hence, the cloud point of a copolymer containing 18 mol% of biotinylated monomer is about 5 °C lower than the cloud point of a copolymer with the same ratio MEO2MA/OEGMA, but devoid of the biotinylated monomer. Still, by incorporating appropriate molar amounts of the OEGMA monomers in the ternary copolymer, the LCST can be adjusted at will, e.g. close to human body temperature of 37 °C (cf.Fig. 2).
While the reactivity of the methacrylate monomers MEO2MA and OEGMA in copolymerisation is nearly the same, the functional methacrylamide is expected to be slightly less reactive. In fact, binary model studies provided copolymerisation parameters r1 and r2 of 1.1 and 0.90 for MEO2MA and the hydrophilic model monomer 2-hydroxypropyl methacrylamide HPMA, respectively, while r1 and r2 values of 1.0 and 0.77 were found for OEGMA and HPMA. Accordingly, the copolymer produced is characterised by a moderate chemical heterogeneity, which might blur the phase transition somewhat.
The successful integration of biotinylated groups into the polymer was confirmed by 1H NMR spectroscopy. An assay based on 4-hydroxyazobenzene-2-carboxylic acid (HABA) was performed to assess to which extent the biotinylated groups are accessible to avidin.31 When binding to avidin, this azo dye forms a complex, which has a maximum VIS absorbance at 500 nm. UV-vis spectra of pure HABA, avidin complexed by HABA and a mixture of avidin and HABA with a sample of the biotinylated polymer are shown in Fig. 1.
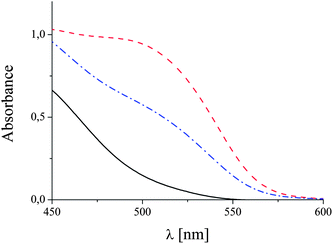 | ||
| Fig. 1 UV-vis spectra of the HABA/avidin reagent before (red dashed line) and after the addition of 0.05 g L−1 biotinylated polymer (blue dash dotted line). The continuous black curve shows the absorption of pure HABA. | ||
Due to its much higher affinity, biotin displaces HABA easily, when it is added to an avidin–HABA complex. Consequently, the absorption band at 500 nm decreases with increasing biotin concentration. This effect can be used to calculate the amount of accessible biotin in the system (see ESI†). The concentration of biotin in a 0.05 g L−1 solution of polymer was determined to be 8 μmol L−1. According to the compositional data from 1H NMR spectroscopy, this means that about 20% of the polymer bound biotin moieties are accessible to avidin. Possibly, once bound, the comparatively large avidin blocks the access of more protein to adjacent biotin groups of the polymer.
To elucidate the influence of bound avidin on the LCST of the polymer, equal volumes of biotinylated polymer and avidin solutions in phosphate buffered saline (PBS) were mixed. Turbidity measurements of mixtures with varying concentrations of avidin for a given concentration of biotinylated polymer were performed (Fig. 2). With increasing amounts of avidin, the cloud points continuously shift to higher temperatures. A maximum shift of the cloud point of 9 °C is reached for an avidin concentration of 60 μmol L−1. The observed increase in the transition temperature indicates that the avidin–polymer complex is more hydrophilic than the polymer by itself.
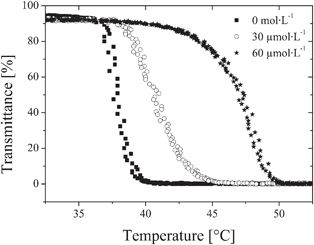 | ||
| Fig. 2 Temperature dependent transmittance of biotinylated polymer in phosphate buffered saline PBS (concentration 2.0 g L−1), with different concentrations of avidin. | ||
Noteworthy, our finding seems to be in contrast to the observations by Colonne et al., who reported the shrinking of biotinylated hydrogel nanospheres composed of N,N-diethyl acrylamide and 2-hydroxyethyl methacrylate upon addition of avidin.32 However, in the context of our findings, the reported contraction is likely to result from a crosslinking effect with the tetrafunctional avidin acting as a crosslinker, rather than from reduced hydrophilicity. In line with this explanation, we observed macroscopic flocculation of the polymer at ambient temperature, when the avidin concentration was increased above 60 μmol L−1. Interestingly, this concentration corresponds to a protein/ligand ratio of about one avidin molecule per polymer chain. The bulky white precipitate formed could not be redissolved, neither by dilution nor by reducing the temperature. In contrast, for lower avidin concentrations (from 5 to 60 μmol L−1), clear solutions were obtained, when avidin was added to the copolymer solutions. Even when heated beyond the phase transition temperature, the collapsed polymers stayed colloidally dispersed for quite some time, but sedimentation occurred after storing such samples overnight. Then again, the formed precipitate could not be redissolved by cooling. Presumably, the crosslinking in the collapsed state is more efficient, as both avidin and polymer chains get into a much closer contact with each other.
Since the carboxyl group of free biotin adds to the binding interactions with avidin, free biotin exhibits a higher binding constant to avidin, and thus is expected to displace avidin from the covalently attached biotin moieties in the polymer.33Fig. 3a shows clouding curves of a 2 g L−1 biotinylated polymer solution with 60 μmol L−1 avidin after treatment with a 10 fold excess of biotin. The cloud point is shifted back to lower temperatures, indicating that bound avidin is removed from the polymer. The finding that the addition of free biotin did not fully reverse the shift of the cloud point generated by the original avidin addition is expected. The biotin exchange is an equilibrium process and avidin is not removed from the reaction mixture.
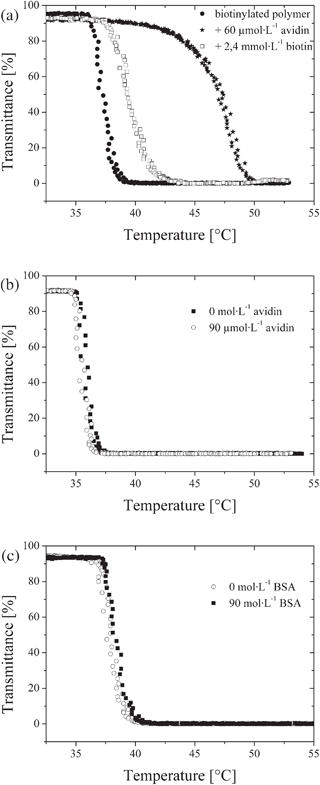 | ||
| Fig. 3 Temperature dependent transmittance of biotinylated polymer in phosphate buffered saline PBS (concentration 2.0 g L−1). (a) Clouding curves of a biotinylated polymer before and after addition of avidin and after subsequent addition of free biotin. (b) Clouding curves of the non-biotinylated polymer before and after addition of avidin. (c) Clouding curves of the biotinylated polymer before and after addition of BSA. | ||
To verify whether the interactions of avidin with the biotinylated polymer are specific, a series of experiments was conducted. First, a thermoresponsive polymer composed of MEO2MA and OEGMA devoid of biotinylated groups was synthesised, and its interaction with avidin was studied under same conditions as for the biotinylated polymer. No shift in cloud point temperature was observed up to high avidin concentrations of 90 μmol L−1 (Fig. 3b). This demonstrates that the shift in the LCST is caused by the protein binding specifically to the functionalised polymer. This finding is not surprising, as the modification of surfaces by oligo- and poly(ethylene glycol) derivatives is a well established method to minimize unspecific adsorption of proteins.3,5,34–36 In the second experiment, possibly non-specific binding of proteins to the polymer due to the attached biotin moieties was investigated using bovine serum albumin (BSA). BSA is well known for its high potential for non-specific interactions,36–38 and is of similar size (Mn = 66.000 g·mol−1) as avidin. Still, treatment of the biotinylated polymer with high concentrations of BSA did not cause any change of the cloud point (Fig. 3c). These control experiments prove that the shift of the phase transition temperature origins from the specific interaction of avidin with the biotinylated polymer.
Conclusion
We have shown that avidin binds specifically to a thermosensitive non-ionic copolymer made of oligo(ethylene glycol) methacrylates, if functionalised by biotin attached via the carboxyl group to the polymer chain. The specific binding of avidin induces a marked shift the LCST of the copolymer. Control experiments demonstrate that the binding of avidin is based on specific interactions. These results provide a proof of concept for responsive sensor-actor systems based on affinity binding.Acknowledgements
The initiative “Spitzenforschung & Innovation in den neuen Ländern” of the German Federal Ministry of Education and Research (BMBF) is acknowledged for financial support (grant 03152201).Notes and references
- C.d. l. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. Chaterji, I. K. Kwon and K. Park, Prog. Polym. Sci., 2007, 32, 1083–1122 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Börner, A. Laschewsky, C. Duschl and J.-F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- E. Wischerhoff, N. Badi, J.-F. Lutz and A. Laschewsky, Soft Matter, 2010, 6, 705–713 RSC.
- D. Roya, J. N. Cambrea and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- I. Tokarev and S. Minko, Soft Matter, 2009, 5, 511–524 RSC.
- Z. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59–62 CrossRef CAS.
- A. S. Hoffman and P. Stayton, Macromol. Symp., 2004, 207, 139–151 CrossRef CAS.
- J. Kopeček, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef CAS.
- J. Jagur-Grodzinski, Polym. Adv. Technol., 2010, 21, 27–47 CAS.
- J. Kim, S. Nayak and L. A. Lyon, J. Am. Chem. Soc., 2005, 127, 9588–9592 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS.
- E. Uğuzdoğan, H. Kayı, E. B. Denkbaş, S. Patır and A. Tuncel, Polym. Int., 2003, 52, 649–657 CrossRef CAS.
- T. Hoare and R. Pelton, Macromolecules, 2007, 40, 670–678 CrossRef CAS.
- Z.-R. Lu, P. Kopečková and J. Kopeček, Macromol. Biosci., 2003, 3, 296–300 CrossRef CAS.
- E. Kokufata, Y.-Q. Zhang and T. Tanaka, Nature, 1991, 351, 302–304 CrossRef.
- K. Li and B. Liu, Polym. Chem., 2010, 1, 252–259 RSC.
- M. Wilchek, E. A. Bayer and O. Livnah, Immunol. Lett., 2006, 103, 27–32 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- Y.-Z. You and D. Oupický, Biomacromolecules, 2007, 8, 98–105 CrossRef CAS.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS.
- F. D. Jochum, P. J. Roth, D. Kessler and P. Theato, Biomacromolecules, 2010, 11, 2432–2439 CrossRef CAS.
- G. N. Grover and H. D. Maynard, Curr. Opin. Chem. Biol., 2010, 14, 818–827 CrossRef CAS.
- B. Le Droumaget and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- X. Jiang, M. Ahmed, Z. Deng and R. Narain, Bioconjugate Chem., 2009, 20, 994–1001 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- Ö. Akdemir, N. Badi, S. Pfeifer, Z. Zarafshani, A. Laschewsky, E. Wischerhoff and J.-F. Lutz, ACS Symp. Ser., 2009, 1023, 189–202 CrossRef CAS.
- N. M. Green, Biochem. J., 1965, 94, 23c–24c CAS.
- M. Colonne, Y. Chen, K. Wu, S. Freiberg, S. Giasson and X. X. Zhu, Bioconjugate Chem., 2007, 18, 999–1003 CrossRef CAS.
- O. Livnah, E. A. Bayer, M. Wilchek and J. L. Sussman, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5076–5080 CAS.
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS.
- I. Szleifer, Curr. Opin. Solid State Mater. Sci., 1997, 2, 337–344 CrossRef CAS.
- W. R. Gombotz, W. Guanghui, T. A. Horbett and A. S. Hoffman, J. Biomed. Mater. Res., 1991, 25, 1547–1562 CrossRef CAS.
- D. J. Vanderah, G. Valincius and C. W. Meuse, Langmuir, 2002, 18, 4674–4680 CrossRef CAS.
- D. Sung, S. Park and S. Jon, Langmuir, 2009, 25, 11289–11294 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, monomer and polymer analysis, calculation for HABA assay. See DOI: 10.1039/c1py00001b |
| ‡ Present address: Institut Charles Sadron, UPR22 CNRS, 23 rue du Loess, BP 84047, F-67034 Strasbourg, Cedex 2, France. |
| This journal is © The Royal Society of Chemistry 2011 |