DOI:
10.1039/C0PY00422G
(Paper)
Polym. Chem., 2011,
2, 1490-1498
Received
27th December 2010
, Accepted 22nd February 2011
First published on 15th March 2011
Abstract
The residue-specific modification of peptides and proteins is a powerful strategy for preparing biomolecular–synthetic polymer conjugates with advanced properties. This manuscript aims at expanding the present toolbox of residue-selective protein modification reactions and targets arginine, a residue for which selective polymer coupling chemistry has only recently been established. To this end, a protected, α-oxo-aldehyde functionalized ATRP initiator that can be used for the preparation of a variety of α-oxo-aldehyde functionalized polymethacrylates has been developed. Polymerization kinetics for four different methacrylate monomers have been investigated in detail and optimized conditions for the chain-end deprotection to reveal the α-oxo-aldehyde end-group have been elaborated. As a final proof of concept, the residue-specific modification of a model protein, chicken egg white lysozyme (HEWL), at arginine residues has been demonstrated.
Introduction
Synthetic polymers bearing functional chain-ends that are designed to react with proteins are of great interest for the purpose of peptide/protein–polymer conjugation. The most common examples of this are functional derivatives of α-methoxy poly(ethylene glycol) (mPEG) for grafting mPEG onto peptides or proteins.1–3 Polymers bearing reactive chain-ends are described as being α-functional if the reactive functional group is present at the chain-end involved in initiation of polymerization or ω-functional if the functional group is present at the other extremity of the chain. While α-functional polymers with quantitative chain-end functionality are readily accessible via living or controlled polymerization techniques, this is not necessarily the case for ω-functional polymers, for which the functional groups sometimes need to be introduced by post-polymerization modification or by use of functional polymerization terminators.4–16 A very convenient method for producing α-functional polymers is by living radical polymerization.17–20 Atom transfer radical polymerization (ATRP) is a particularly advantageous method for accomplishing this due to the broad functional group compatibility of this polymerization method and consequently the diversity of monomers which can be polymerized.18 Functional initiators for ATRP have been described for the preparation of α-functional polymers that are reactive towards a variety of amino acid residues such as lysine and cysteine, thereby allowing residue-specific conjugation of the respective polymer onto the peptide or protein of interest.20
Although there already exist strategies to specifically modify a number of the 20 most common naturally occurring amino acids, there are still residues for which no or very limited possibilities are available for achieving their residue-specific modification in a peptide or protein. The development of strategies that would allow to increase the number of amino acids that can be specifically modified would enhance the scope of peptide/protein modification and facilitate the engineering of the properties of the final peptide/protein–polymer conjugates. One example of an amino acid residue that has received little attention as a target for residue-specific peptide/protein modification with polymers is arginine. In this study, the preparation of α-functional polymers bearing an arginine-reactive chain-end by ATRP is described. The selective and stable conjugation of ω-functional mPEG bearing an α-oxo-aldehyde group (i.e., a 1,2-di-carbonyl) to arginine residues on a protein has been recently described by our group.21 The selectivity of the reaction of di-carbonyl compounds for arginine residues comes from the ability of the reaction product to outcompete hydrolysis in comparison to the products formed by reaction with other amino acids such as lysine or cysteine. That is, despite the greater reactivity of lysine and cysteine residues towards ketones/aldehydes, di-carbonyl reagents participating in this reaction preferentially modify arginine residues (irreversibly) in favor of lysine or cysteine (reversibly) through equilibrium.21 Herein, the preparation of a functional initiator for ATRP bearing a protected α-oxo-aldehyde group is described. The initiation of polymerization of four methacrylate monomers with widely differing physico-chemical properties by this initiator is described. The four selected monomers are poly(ethylene glycol) monomethyl ether methacrylate (PEGMA), N,N-dimethylaminoethyl methacrylate (DMAEMA), tert-butyl methacrylate (tBuMA) and methyl methacrylate (MMA), which upon chain end (and side-chain when appropriate) deprotection yield hydrophilic, cationic, anionic and hydrophobic polymers, respectively, bearing an α-oxo-aldehyde group at the α-chain-end of the polymer. Finally, as a proof of concept, the coupling of the water soluble polymers to a model protein, chicken egg white lysozyme (HEWL), is demonstrated.
Experimental
Materials
Methylglyoxal 1,1-dimethylacetal (1), cyclohexylamine, anhydrous diethyl ether, calcium chloride, N-bromosuccinimide, carbon tetrachloride, Amberlite IR-120 (H+ form), lithium diisopropylamide (LDA; 1.8 M solution in tetrahydrofuran/heptane/ethylbenzene), tetrahydrofuran (THF), methyl iodide, PEGMA (Mn ≈ 475 g mol−1), methyl methacrylate (MMA), N,N-dimethylaminoethyl methacrylate (DMAEMA), tert-butyl methacrylate (tBuMA), basic alumina, neutral alumina, CuBr, 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA), pentamethyldiethylenetriamine (PMDETA), deuterated solvents, Celite 577 fine, iodine, trifluoroacetic acid (TFA), LiBr, disodium hydrogen phosphate, triethylamine, and chicken egg white lysozyme (HEWL) were obtained from Sigma-Aldrich (Buchs, Switzerland) and used as received. All organic solvents were obtained from Aldrich as reagent grade or better and were used as received, except for THF (inhibitor free) and toluene which were dried through alumina using a solvent purification system, and methyl ethyl ketone which was dried over K2CO3 and distilled.
Equipment
1H- and 13C-NMR spectra were recorded using a Bruker AVANCE-400 spectrometer operating at 400 MHz for protons and chemical shifts referenced to the residual proton signal of the appropriate deuterated solvent. Size-exclusion chromatography was performed on a Waters Alliance GPCV 2000 system equipped with refractive index and differential viscometer detectors. Separation was carried out at 60 °C with TSK-Gel Alpha 2500 + 3000 + 4000 columns using DMF + 0.5 g L−1 LiCl as eluent and a flow rate of 0.6 mL min−1. Size-exclusion chromatography in THF and THF + 5% triethylamine was performed on a Viscotek GPCmax VE-200 system equipped with refractive index detector. Separation was carried out at 45 °C with two TSK-Gel GMHHR-M columns in series and using a flow rate of 1 mL min−1. Molecular weights were determined relative to narrow poly(methyl methacrylate) standards. Analytical HPLC was performed using a Hitachi system equipped with a L-7100 pump, a L-7200 autosampler, a L-7360 column heater and a L-7455 diode array detector. Chromatograms were recorded using a LiChrospher 100 RP-18 (5 μm, 4.5 × 250 mm) column at a flow rate of 1 mL min−1 using gradients of water + 0.1% TFA and acetonitrile + 0.1% TFA. Matrix-assisted laser desorption/ionization time-of-flight mass spectra (MALDI-TOF MS) were obtained with an Axima-CFRTM plus mass spectrometer using dithranol saturated with sodium trifluoroacetate as matrix. 2,5-Dihydroxybenzoic acid was also examined as matrix for the polymers.
Assigned 1H and 13C NMR spectra for 1–5 are available in Fig. S1 in the ESI†.
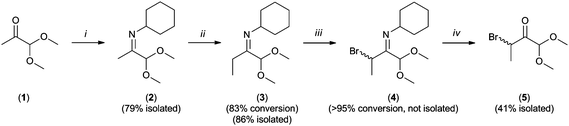
Synthesis of methylglyoxal 1,1-dimethylacetal cyclohexylimine (2).
2 was prepared following a modified protocol from Pichon et al.22 Methylglyoxal 1,1-dimethylacetal (1, 10 g, 85 mmol) was added under inert atmosphere to a flame-dried 100 mL round-bottom flask containing 50 mL anhydrous diethyl ether, cyclohexylamine (9.23 g, 93 mmol) and granular calcium chloride (0.5 g, 4.5 mmol). The contents of the flask were stirred overnight at 45 °C and then the solvent and 1 removed at 70 °C in vacuo (8 mbar) on a rotary evaporator. Methylglyoxal 1,1-dimethylacetal cyclohexylimine was isolated from the residue by distillation as a single fraction (120–125 °C per 8 mbar) to yield 13.31 g (79%) of the imine as a colorless liquid, which was stored frozen as a solid at −30 °C until used.
Synthesis of N-(1,1-dimethoxybutan-2-ylidene)cyclohexanamine (3).
LDA (27.8 mL of a 1.8 M solution, 50 mmol) was added to a 250 mL tri-col flask, 10 mL anhydrous THF added and the flask cooled to −78 °C. 10 g of 2 (50 mmol) dissolved in 10 mL anhydrous THF was added drop wise to this solution over a one hour period, stirred for one hour at −78 °C, warmed to room temperature for one hour and then cooled again to −78 °C. At this point, methyl iodide (3.11 mL in 100 mL anhydrous THF, 50 mmol) was added drop wise over a period of two hours and the solution left to warm gradually overnight. Following this period, the solvent was evaporated on a rotavap (60 °C per 8 mbar) and the residue dissolved in 200 mL diethyl ether, which was washed three times with 50 mL water. The organic phase was dried with sodium sulfate, the solvent evaporated, and the residue distilled as a single fraction between 110 and 130 °C at 6 mbar. The distillate (9.15 g, 86%) contained 83% of 3 and 17% of 2 and was used as is.
Synthesis of 3-bromo-1,1-dimethoxybutan-2-one (5).
5 was prepared by bromination of 3 (2.23 g, 10.5 mmol) in a 100 mL round-bottom flask containing 2.05 g N-bromosuccinimide (11.5 mmol) in 50 mL CCl4. The solution was deoxygenated by bubbling with nitrogen for 15 minutes then stirred under inert atmosphere for 24 h at room temperature. Solids were removed by filtration through a sand plug and 5 g strong-acid ion-exchange resin (Amberlite IR-120) was added to the filtrate, which was then agitated for 30 minutes. The ion-exchange resin was removed by filtration and 5 was isolated by column chromatography using 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ethyl acetate:cyclohexane (v/v) as eluent. The resulting yellow liquid was distilled (95–110 °C/ per 8 mbar) to recover 1 g (45%) of 5 as a very pale liquid which was stored at −30 °C until used. Density: 1.447 g mL−1. m/z (M + Na)+ expected: 232.98, 234.92; found: 233.01, 235.01.
:4 ethyl acetate:cyclohexane (v/v) as eluent. The resulting yellow liquid was distilled (95–110 °C/ per 8 mbar) to recover 1 g (45%) of 5 as a very pale liquid which was stored at −30 °C until used. Density: 1.447 g mL−1. m/z (M + Na)+ expected: 232.98, 234.92; found: 233.01, 235.01.
Polymerization of MMA.
MMA (0.5 mL, 4.7 mmol, distilled prior to use), CuBr (0.67 mg, 0.047 mmol), 5 (6.85 μL, 0.047 mmol) and 0.5 mL toluene were combined in a 10 mL Schlenk tube containing a magnetic stirrer and sealed with a septum. The solution was degassed by flushing with nitrogen for 15 minutes and then immersed in an oil bath set to 90 °C. The ligand, HMTETA (25.3 μL, 0.093 mmol), was then added using a gas-tight syringe. At predetermined times, ∼100 μL of the reaction mixture was withdrawn using a degassed syringe and quenched by dilution/oxygenation in 0.5 mL CDCl3, then frozen in liquid nitrogen and stored at −30 °C. Following analysis of conversion by 1H NMR spectroscopy, the samples in CDCl3 were filtered through neutral alumina, which was flushed with THF until 5 mL filtrate was recovered. The solvent was evaporated in vacuo, the residue taken up in 200 μL THF, precipitated in 2 mL cyclohexane and the solid recovered by centrifugation. The supernatant was removed from the polymer pellet and the process repeated a second time. The polymers were dried in vacuo at room temperature to remove trace solvents. Size-exclusion chromatography was done in THF.
Polymerization of PEGMA475.
PEGMA475 (0.5 mL, 1.1 mmol, filtered through basic alumina and Celite 577), CuBr (3.3 mg, 0.023 mmol), 5 (3.35 μL, 0.023 mmol) and 0.5 mL toluene were combined in a 10 mL Schlenk tube containing a magnetic stirrer and sealed with a septum. This solution was degassed by flushing with nitrogen for 15 minutes and then immersed in an oil bath set to 60 °C. The ligand, PMDETA (9.6 μL, 0.046 mmol), was then added using a gas-tight syringe. At predetermined times, ∼100 μL of the reaction mixture was withdrawn using a degassed syringe and quenched by dilution/oxygenation in 0.5 mL CDCl3, then frozen in liquid nitrogen and stored at −30 °C. Following analysis of conversion by 1H NMR spectroscopy, the samples in CDCl3 were filtered through neutral alumina, which was flushed with THF until 5 mL filtrate was recovered. The solvent was evaporated in vacuo, the residue taken up in 200 μL THF, precipitated in 2 mL diethyl ether in Eppendorf tubes, and centrifuged. The supernatant was removed from the polymer, which precipitated as a viscous liquid droplet at the bottom of the tube, using a syringe and the process repeated a second time. The polymers were dried in vacuo at room temperature to remove trace solvents. Size-exclusion chromatography was done in DMF. Polymerizations performed in MEK and anisole were conducted in exactly the same fashion by replacing the 0.5 mL of toluene by 0.5 mL of the appropriate solvent.
Polymerization of tBuMA.
tBuMA (0.5 mL, 3.1 mmol, distilled prior to use), CuBr (4.4 mg, 0.031 mmol), 5 (4.52 μL, 0.031 mmol) and 0.5 mL toluene were combined in a 10 mL Schlenk tube containing a magnetic stirrer and sealed with a septum. This solution was degassed by flushing with nitrogen for 15 minutes and then immersed in an oil bath set to 75 °C. The ligand, HMTETA (16.6 μL, 0.061 mmol), was then added using a gas-tight syringe. At predetermined times, ∼100 μL of the reaction mixture was withdrawn using a degassed syringe and quenched by dilution/oxygenation in 0.5 mL CDCl3, then frozen in liquid nitrogen and stored at −30 °C. Following analysis of conversion by 1H NMR spectroscopy, the samples in CDCl3 were filtered through neutral alumina, which was then flushed with THF until 5 mL filtrate was recovered. The solvent was evaporated in vacuo, the residue taken up in 200 μL THF and the polymer precipitated in 2 mL 1/1 (v/v) MeOH:H2O in Eppendorf tubes. Following centrifugation, the supernatant was removed from the polymer pellet and the process repeated a second time. The polymers were freeze-dried to remove the residual solvent. Size-exclusion chromatography was done in THF.
Polymerization of DMAEMA.
DMAEMA (0.5 mL, 2.97 mmol, distilled prior to use), CuBr (2.1 mg, 0.015 mmol), 5 (2.2 μL, 0.015 mmol) and 0.5 mL anisole were combined in a 10 mL Schlenk tube containing a magnetic stirrer and sealed with a septum. This solution was degassed by flushing with nitrogen for 15 minutes and then immersed in an oil bath set to 30 °C. The ligand, HMTETA (8.2 μL, 0.03 mmol), was then added using a gas-tight syringe. At predetermined times, ∼100 μL of the reaction mixture was withdrawn using a degassed syringe and quenched by dilution/oxygenation in 0.5 mL CDCl3, then frozen in liquid nitrogen and stored at −30 °C. Following analysis of conversion by 1H NMR spectroscopy, the samples in CDCl3 were filtered through neutral alumina, which was then flushed with THF until 5 mL filtrate was recovered. The solvent was evaporated in vacuo, the residue taken up in 200 μL THF and the polymer precipitated in 10 mL cold pentane. The polymer was recovered by centrifugation at −10 °C. The supernatant was discarded and the residual pentane removed under vacuum. Size-exclusion chromatography was done in THF containing 5% triethylamine.
Investigation of initiator deactivation.
CuBr (4.3 mg, 0.03 mmol), HMTETA (16.3 μL, 0.06 mmol), 5 (4.4 μL, 0.03 mmol) and 1 mL of toluene were combined in a 10 mL Schlenk tube containing a magnetic stirrer and sealed with a septum. This solution was flushed with nitrogen for 15 minutes. The reaction mixture was then immersed in an oil bath preset to 75 °C for 3 hours. The reaction was quenched by exposure to oxygen and dilution in CDCl3.
Optimized polymer deprotection protocols
Trifluoroacetic acid: H2O hydrolysis.
The polymers (either PPEGMA (6, 13.3 mg, Mn ≈ 32800 g mol−1, 0.4 μmol) or PDMAEMA (7, 9.2 mg, Mn ≈ 14600 g mol−1, 0.63 μmol)) were dissolved in 0.5 mL D2O in an NMR tube and 0.5 mL TFA added. The tube was capped and heated to 90 °C for 3 h. For PtBuMA, the polymer (8, 13.5 mg, Mn ≈ 8800 g mol−1, 1.5 μmol) was dissolved in 50 μL DMSO and then 0.5 mL TFA was added to deprotect the tert-butyl side chains. This solution was left at 60 °C for 1 h and then 0.5 mL H2O added and then heated to 90 °C for 3 h. Deprotection was verified by 1H NMR spectroscopy. The deprotected polymers were isolated by size-exclusion chromatography (PD MidiTrap) using water as eluent and recovered by freeze drying (yield: 10, 8.4 mg, 63%; 11, 10.6 mg, 67% (Mn = 25100 g mol−1 as a TFA salt); 12, 2.9 mg, 36% (Mn = 5400 g mol−1 as deprotected polymer)). It was not possible to solubilize PMMA (9) under any conditions which would permit deprotection using this approach.
I2-mediated transacetalization.
The polymers (PPEGMA (6, 13.3 mg, Mn ≈ 32800 g mol−1, 0.4 μmol), PDMAEMA (7, 9.2 mg, Mn ≈ 14600 g mol−1, 0.63 μmol), PtBuMA (8, 13.5 mg, Mn ≈ 8800 g mol−1, 1.5 μmol), or PMMA (9, 10.4 mg, Mn ≈ 12600 g mol−1, 0.82 μmol)) were dissolved in 0.5 mL acetone-d6 in an NMR tube and 3 eq. I2 relative to the polymer end-groups was added. The tube was capped and heated at 90 °C for 3 h. Deprotected PPEGMA (10) was isolated by evaporation of acetone, resuspension in 0.5 mL H2O, purification by size-exclusion chromatography (PD MidiTrap) and recovery by freeze drying (10, 13.3 mg, 100%). The reaction mixture containing PDMAEMA (7) rapidly lost its color and was analyzed as such without further purification. The reaction mixture containing PtBuMA (8) rapidly precipitated. The supernatant was analyzed by 1H NMR spectroscopy and then the contents of the NMR tube were diluted with water, the acetone removed in vacuo, and the small fraction of water-soluble polymer was purified by gel filtration (PD MidiTrap G-25), recovered by lyophilization, and analyzed by 1H NMR spectroscopy. Deprotected PMMA (13) was isolated by silica gel chromatography using 5% methanol in chloroform as eluent (13, 8.3 mg, 80%).
PPEGMA (6, 25 mg, 1.3 μmol, Mn = 19600 g mol−1 SEC, DP 41), PDMAEMA (7, 30 mg, 1.1 μmol, Mn = 26400 g mol−1 SEC, DP 168) and PtBuMA (8, 21.4 mg, 1.7 μmol, Mn = 12800 g mol−1 SEC, DP 90) were deprotected using TFA:H2O (and DMSO when appropriate) and purified as described above. The recovered polymers (10, 20 mg, 1.0 μmol; 11, 39 mg, 0.86 μmol (Mn = 45600 g mol−1 as a TFA salt); 12, 4.2 mg, 0.53 μmol (Mn = 7800 as deprotected polymer)) were dissolved in 600, 500 or 320 μL (for 10, 11, and 12 respectively) of a 0.5 mg mL−1 solution of HEWL in 100 mM phosphate buffer, pH 7.4. Under these conditions, the polymers are in 50 eq. molar excess with respect to HEWL (4.5 eq. relative to arginine residues). The reaction was left to incubate at room temperature. At given time points, aliquots were taken and quenched with an equal volume of 200 mM NH2OH, pH 7, and then analyzed by HPLC. After 14 days, the reaction mixtures were analyzed by SDS-PAGE (14% running gel, 5% stacking gel) and bands revealed by silver staining. Equivalent experiments to those described above were performed in 100 mM borate buffer, pH 9.0, rather than phosphate buffer, pH 7.4.
Results and discussion
Initiator design and synthesis
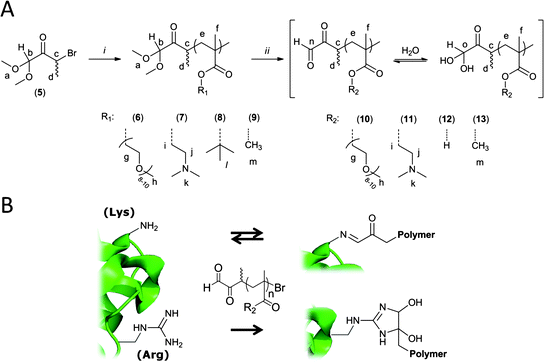
The synthesis of the ATRP initiator 5 is outlined in Scheme 1. It was rationalized that the α-oxo-aldehyde group should be present in a protected acetal form in order to facilitate synthesis and handling of the intermediates owing to their high reactivity. In addition, protection may prevent undesirable coordination of the di-carbonyl to the metal salts used in ATRP, as has already been observed for methyl vinyl ketone.23 The ATRP initiating moiety on 5 is a bromo-ketone group that differs from traditional ATRP initiators, which typically are bromo-esters or bromo-amides. The successful polymerization of MMA from a bromo-ketone initiator has only recently been shown by Sivaram and co-workers.24,25 Polymerization from a bromo-ketone has the advantage that the α-oxo-aldehyde functional group will be directly connected to the main chain of the polymer via a stable C–C bond. For instance, this differs from the α-functional polymer bearing an aldehyde group prepared by Tao et al., for which the aldehyde is connected to the polymer main-chain via an ester bond.26 The presence of a methyl group on the carbon bearing the bromine atom on 5 was found to be necessary, as its omission (see compound 15 in the ESI†) lead to irreproducible initiation of polymerization of MMA under a variety of examined conditions (Fig. S2†). Hydration of the ketone on initiator 5, like that observed for 1 (Fig. S1†), was not observed by 1H NMR spectroscopy in CDCl3 and therefore this phenomenon is not likely to interfere with ATRP polymerization in non-aqueous media.
The synthesis and isolation of 2 proceeded smoothly and in good yield (Scheme 1). The alkylation of the lithium enamide of 2 with methyl iodide was always incomplete and the isolation of 3 from the mixture was not possible because of the degradation of the imine concurrent with column chromatography. Bromination of the reaction mixture containing 2 and 3 proceeded readily at room temperature. It was found that heating this reaction mixture, as reported by others,27 leads to significant browning and reduced yield. In addition, hydrolysis of the imine on 4 was performed using a strong acid ion-exchange resin due to highly complicated reaction mixtures obtained when using conditions reported by others.27 The strong-acid resin used herein was easily removed by filtration. 5 was isolated from its non-methylated analog (15, see ESI†) by column chromatography and was conveniently purified by distillation.
ATRP of PEGMA, DMAEMA, tBuMA and MMA
The polymerization of PEGMA, DMAEMA, tBuMA and MMA using 5 as initiator (Scheme 2) was reproducible and linear semi-logarithmic kinetic plots were obtained (Fig. 1 and S3–S5†). In all cases, linear increases of Mn,SECversus conversion and narrow molecular weight distributions were observed. The good accord between Mn,NMR and Mn,SEC indicated that polymerization was being uniquely initiated by 5. This is supported by the single distribution observed for 9 by MALDI-TOF MS (Fig. S12†) as discussed later. In addition, well-defined polymers with different lengths could be prepared (Table 1). In all cases, it was found, however, that initiator efficiency was not unitary. This has been observed by others for bromo-ketone initiators24 as well as for bromo-esters bearing acetals,26 and is a common feature for the initiation of ATRP. The possible mechanism of deactivation of 5 was examined by conducting ATRP in the absence of monomer for 3 h at 75 °C. NMR and MS evidence showed that one possible route for deactivation of the initiating radical species (i.e., 5 activated by Cu(I)) occurs via hydrogen abstraction from either solvent or monomer to form 1,1-dimethoxybutan-2-one (m/z expected: 132.08, found: 132.12; 1H NMR found in Fig. S6†). Initiator coupling was not observed under the conditions examined.
Table 1 Characteristics of selected polymers bearing a protected α-oxo-aldehyde chain-end (6–9) prepared by ATRP
| Monomer |
[M]:[I]:[Cu]:[L] |
Conversion (%) |
M
n (theory)/g mol−1 |
M
n (NMR)/g mol−1 |
M
n (SEC)/g mol−1 |
M
w/Mn (—) |
I
eff
(—) |
Time/min |
Solvent |
|
I
eff calculated as Mn (theory)/Mn (NMR).
|
| MMA |
100:1:1:2 |
82% |
8200 |
10700 |
12500 |
1.22 |
0.77 |
60 |
Toluene |
| MMA |
50:1:1:2 |
88% |
4400 |
8100 |
9000 |
1.14 |
0.54 |
60 |
Toluene |
| MMA |
20:1:1:2 |
84% |
1700 |
6000 |
5200 |
1.11 |
0.28 |
60 |
Toluene |
| PEGMA |
33:1:1:2 |
93% |
14500 |
20700 |
19100 |
1.17 |
0.70 |
60 |
Anisole |
| PEGMA |
50:1:1:2 |
58% |
13800 |
28400 |
22800 |
1.26 |
0.48 |
40 |
Anisole |
| PEGMA |
33:1:1:2 |
95% |
14900 |
19400 |
19200 |
1.20 |
0.77 |
60 |
Toluene |
| PEGMA |
33:1:1:2 |
92% |
14400 |
29500 |
27700 |
1.21 |
0.49 |
60 |
MEK |
|
tBuMA |
100:1:1:2 |
75% |
10700 |
17700 |
22100 |
1.10 |
0.60 |
120 |
Toluene |
|
tBuMA |
50:1:1:2 |
80% |
5700 |
10000 |
11500 |
1.31 |
0.57 |
60 |
Toluene |
|
tBuMA |
30:1:1:2 |
82% |
3500 |
7800 |
9300 |
1.31 |
0.45 |
40 |
Toluene |
| DMAEMA |
200:1:1:2 |
40% |
12500 |
17000 |
15600 |
1.12 |
0.73 |
120 |
Anisole |
| DMAEMA |
250:1:1:2 |
40% |
15800 |
19800 |
18100 |
1.12 |
0.80 |
180 |
Anisole |
| DMAEMA |
300:1:1:2 |
40% |
18900 |
23400 |
20600 |
1.14 |
0.80 |
240 |
Anisole |
![Polymerization of MMA with [M0] : [I] ratios of 100 (squares), 50 (circles), and 20 (triangles) in toluene at 90 °C. (Top) Evolution of experimental Mn,SEC (filled symbols) and Mw/Mnversus conversion in comparison to theoretical values (open symbols). (Bottom) Semi-logarithmic kinetic plots of monomer conversion. Kinetic plot determined by 1H NMR spectroscopy in CDCl3.](/image/article/2011/PY/c0py00422g/c0py00422g-f1.gif) |
| | Fig. 1 Polymerization of MMA with [M0]:[I] ratios of 100 (squares), 50 (circles), and 20 (triangles) in toluene at 90 °C. (Top) Evolution of experimental Mn,SEC (filled symbols) and Mw/Mnversus conversion in comparison to theoretical values (open symbols). (Bottom) Semi-logarithmic kinetic plots of monomer conversion. Kinetic plot determined by 1H NMR spectroscopy in CDCl3. | |
The polymerization of PEGMA proceeded readily in MEK, toluene and anisole. However, a slight shouldering of the molecular weight distributions was observed at higher masses. This phenomenon has been observed by others for the polymerization of this monomer by both ATRP and RAFT polymerization and has been ascribed to chain coupling reactions.28,29 This shoulder was not significantly affected by the nature of the solvent nor was it significantly improved by lowering the temperature or diluting the polymerization medium (data not shown). Nevertheless, despite this small shoulder, molecular weight distributions remained narrow and linear polymerization kinetics were observed. In general, linear first-order kinetics and relatively narrow molecular weight distributions were also obtained for the polymerization of DMAEMA with [M0]:[I] ratios above 100 (Table 1 and Fig. S4 and S7†). Conversely, PDMAEMA polymers prepared in reactions with [M0]:[I] ratios of 100 and below had relatively broad molecular weight distributions (Fig. S7†), but remained monomodal. Both MMA and tBuMA were readily polymerized under the conditions examined and yielded monomodal polymers with low dispersities. As the target molecular weights decreased, the polydispersities of PtBuMA slightly increased, though remained comparable to values reported in the literature for this monomer.30
End-group deprotection
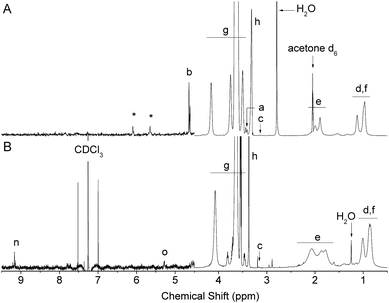
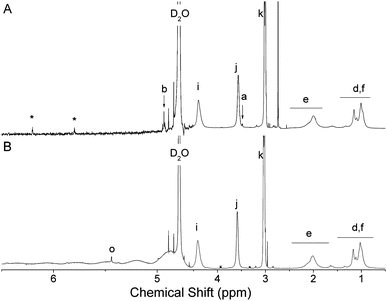
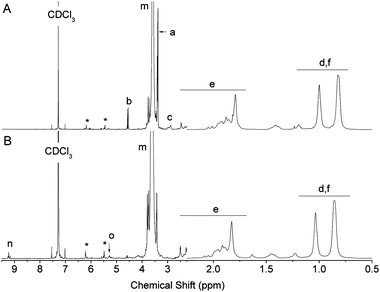
The deprotection of the acetal on the α-terminus of polymers 6–9 can be accomplished in a number of manners. In a previous study, mPEG bearing a protected α-oxo-aldehyde group has been successfully deprotected by both I2-mediated transacetalization in acetone and by hydrolysis using a strong acid ion-exchange resin at elevated temperatures.21 When the ion-exchange approach was investigated herein for the deprotection of 6–8 (9 is not water soluble), precipitation occurred and the polymer could no longer be re-solubilized in water. Deprotection of polymers 6 and 7 was therefore achieved in H2O:TFA at 90 °C, which was a protocol adapted from Tao et al. for the deprotection of an acetal protected α-functional poly(methacrylate) prepared by ATRP.26 While at room temperature no deprotection occurred, at 90 °C quantitative deprotection of 6 and 7 was evidenced by the disappearance of the peaks a and b corresponding to the protected species and the appearance of peaks n and o characteristic for the α-oxo-aldehyde and its monohydrate, respectively, on polymers 10 and 11 (Fig. 2 and 3). The 1H NMR spectrum of 10 revealed 31% anhydrous (9.2 ppm) and 23% monohydrated (5.3 ppm) α-oxo-aldehyde chain-ends (Fig. 2) while the corresponding spectrum of 11 showed 27% monohydrate (5.4 ppm) α-oxo-aldehyde chain-end (Fig. 3). It should be noted that while in both cases quantitative deprotection was observed by the disappearance of peaks a and b, the integration of the peaks n and o corresponding to the different hydrated forms of the α-oxo-aldehyde never accounted for 100% owing to the acidity of the protons and their exchange with the deuterated NMR solvent, as previously observed.21 As 8 was not soluble in H2O, TFA, or mixtures of the two, it was first dissolved in DMSO to which TFA was added to deprotect the tert-butyl side chains on the polymer. Following this step, the polymer remained soluble in the reaction medium following addition of an equal volume of water. The resulting deprotected polymer (12) showed no residual tert-butyl groups and 15% of the monohydrate (5.4 ppm) α-oxo-aldehyde end-group (Fig. 4).
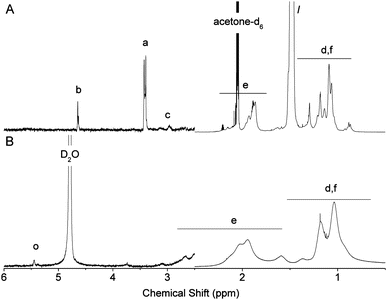 |
| | Fig. 4
1H NMR spectra of PtBuMA (8, Mn,NMR 8800 g mol−1) before (A) and after (B) full deprotection to reveal the α-oxo-aldehyde chain-end and carboxylic acid side chains (12). Peak assignments are given in Scheme 2. The signal intensity increased above 2.5 ppm to better visualize polymer end-groups. | |
The deprotection of 6–9 by I2-mediated transacetalization in acetone was also performed owing to the solubility of all polymers in this solvent. Quantitative deprotection of 6 and 9 was achieved by this method. The 1H NMR spectrum of 10 revealed 6% anhydrous (9.7 ppm) and 4% monohydrated (5.3 ppm) α-oxo-aldehyde chain-ends (Fig. S8†) while the corresponding spectrum of 13 showed 24% anhydrous (9.2 ppm) and 28% monohydrate (5.3 ppm) α-oxo-aldehyde chain-ends (Fig. 5). The reaction mixture containing 7 rapidly became discolored and no deprotection was evident by 1H NMR spectroscopy (Fig. S9†). The reaction of iodine with tertiary amines has been previously described.31 In addition, deprotection of 8 by this method was hampered by the partial deprotection of the tert-butyl ester side-groups of the polymer, which resulted in polymer precipitation. This deprotection was observed by the appearance of tert-butanol and 2-methyl-1,2-propene in the reaction vessel (Fig. S10†). While analysis of the water-soluble fraction of this reaction mixture by 1H NMR spectroscopy revealed significant side-group deprotection, incomplete deprotection of the acetal was also observed (40% residual acetal, Fig. S10†). Overall, this approach is not appropriate for the deprotection of 7 and 8. The SEC chromatograms of all polymers (6–13) before and after deprotection did not show any significant evidence for side-chain or main-chain degradation, except for 8 and 12, for which the removal of the tert-butyl side chains concurrent with deprotection significantly changes the Mn of the polymer (Fig. S11†).
MALDI-TOF mass spectrometry was performed on 6–13 (low molecular weight samples isolated very early following initiation) in order to verify the nature of the end-groups on the polymers. Unfortunately, despite extensive efforts, only 9 and 13 yielded spectra of sufficient quality for end-group analysis (Fig. S12†). The spectrum of 9 clearly shows a single distribution indicating that 5 is the sole species initiating ATRP. The spectrum of 10 shows deprotection of the acetal group on 9 to yield the α-oxo-aldehyde group, present uniquely as a di-hydrate under the conditions used for MALDI-TOF MS. The conditions used to deprotect 9 for MALDI-TOF analysis were slightly milder in order to simultaneously visualize the hemiacetal intermediate. Under conditions used for quantitative deprotection, no hemiacetal was evident by 1H NMR spectroscopy (Fig. 5). These results, including the successful conjugation of 10–12 to a protein (vide infra), indicate that the polymers do indeed bear the desired α-oxo-aldehyde end-group.
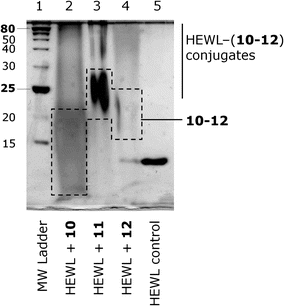
In order to demonstrate that the water-soluble polymers 10–12, which bear an α-oxo-aldehyde group at their α-terminus, are reactive towards arginine residues on proteins, these polymethacrylates were incubated with HEWL at both pH 7.4 and 9 in 4.5-fold excess with respect to the number of arginine residues present on the protein (i.e., 50-fold molar excess with respect to the protein). These conditions have previously been shown to be effective for the modification of HEWL with mPEG bearing an analogous α-oxo-aldehyde group.21 At given time points, the reaction was quenched with NH2OH, which scavenges excess reagent and cleaves any potential protein–polymer conjugates formed at lysine residues. Fig. 6 shows that the reaction proceeds smoothly over 7 days, as monitored by the disappearance of native HEWL by HPLC (raw chromatograms supplied in Fig. S13†). As previously observed for the reaction of HEWL with mPEG bearing an α-oxo-aldehyde group, the reaction was faster at pH 9 owing to the greater nucleophilicity of arginine residues at this pH.21 Following complete disappearance of native HEWL (14 days), the reaction mixture was analyzed by SDS-PAGE (Fig. 7 and S14†), which confirmed the absence of native HEWL (or traces thereof) and the appearance of a broad band for the conjugates at higher molecular weight. The bands corresponding to the non-conjugated polymers were identified based on a control experiment performed in the absence of protein (Fig. S14†). The selective reaction of mPEG bearing an α-oxo-aldehyde group with arginine residues on HEWL has previously been demonstrated.21 One key criterion for demonstrating the selectivity of the modification at arginine versus lysine residues was resistance to displacement by hydroxylamine. In all cases herein, the reaction was quenched with hydroxylamine prior to analysis by HPLC or SDS-PAGE, providing good indirect indication for modification of HEWL at arginine residues.
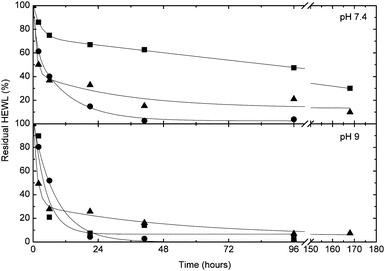 |
| | Fig. 6 Kinetics of reaction of HEWL with 50 molar equivalents of reactive polymers 10 (squares), 11 (circles) and 12 (triangles) at pH 7.4 (top) and pH 9 (bottom). Residual HEWL quantified by HPLC following quenching of the reaction at given time points with hydroxylamine to eliminate polymers conjugated to HEWL at lysine residues. | |
Conclusions
In this study, the synthesis of α-functional polymers bearing an arginine-reactive α-oxo-aldehyde functional group by ATRP has been shown. This has been accomplished by polymerization of PEGMA, DMAEMA, tBuMA and MMA from a protected α-oxo-aldehyde functionalized ATRP initiator. In all cases, the polymerization was controlled and well-defined polymers with a wide variety of physical properties ensued. Conditions for the deprotection of the polymers to reveal the reactive α-oxo-aldehyde group have been established and the conjugation of the water-soluble polymers to a model protein has been shown. The approach presented herein extends the versatility of arginine-specific modification of proteins from mPEG to a wide range of polymers that can be produced by ATRP.
Acknowledgements
Financial support from the NCCR Nanoscale Science, the Fonds Québecois de la Recherche sur la Nature et les Technologies (Quebec, Canada; postdoctoral fellowship to M.A.G.) and the Alexander von Humboldt-Foundation (postdoctoral fellowship for F.R.W.) is gratefully acknowledged.
References
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Polym. Chem., 2010, 1, 1352–1373 RSC.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591–2611 RSC.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243–254 CrossRef CAS.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- H.-A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253–262 CrossRef CAS.
- D. R. W. Hodgson and J. M. Sanderson, Chem. Soc. Rev., 2004, 33, 422–430 RSC.
- R. E. Connor and D. A. Tirrell, Polym. Rev., 2007, 47, 9–28 Search PubMed.
- A. J. Link, M. L. Mock and D. A. Tirrell, Curr. Opin. Biotechnol., 2003, 14, 603–609 CrossRef CAS.
- J. C. M. van Hest, Polym. Rev., 2007, 47, 63–92 Search PubMed.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45–53 RSC.
- H. G. Börner and H. Schlaad, Soft Matter, 2007, 3, 394–408 RSC.
- D. W. P. M. Löwik, L. Ayres, J. M. Smeenk and J. C. M. van Hest, Adv. Polym. Sci., 2006, 202, 19–52.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- M. A. Gauthier and H.-A. Klok, Biomacromolecules, 2011, 12, 482–493 CrossRef CAS.
- N. Pichon, A. Harrison-Marchand, L. Toupet and J. Maddaluno, J. Org. Chem., 2006, 71, 1892–1901 CrossRef CAS.
- A. Mittal, S. Sivaram and D. Baskaran, Macromolecules, 2006, 39, 5555–5558 CrossRef CAS.
- A. Mittal, D. Baskaran and S. Sivaram, Macromol. Symp., 2006, 240, 238–244 CrossRef CAS.
- A. Mittal and S. Sivaram, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4996–5008 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- N. G. De Kimpe and M. T. Rocchetti, J. Agric. Food Chem., 1998, 46, 2278–2281 CrossRef CAS.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772–4779 CrossRef CAS.
- R. Hoogenboom, D. Fournier, J. M. Kranenburg and U. S. Schubert, Polym. Prepr., 2007, 48, 177–178 CAS.
- R. Krishnan and K. S. V. Srinivasan, Eur. Polym. J., 2004, 40, 2269–2276 CrossRef CAS.
- G. Bowmaker and S. Hannan, Aust. J. Chem., 1971, 24, 2237–2248 CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.