Polymersomes enhance the immunogenicity of influenza subunit vaccine†
Christophe
Barnier Quer‡
a,
Hana
Robson Marsden‡
b,
Stefan
Romeijn
a,
Harshal
Zope
b,
Alexander
Kros
*b and
Wim
Jiskoot
*a
aDivision of Drug Delivery Technology, Leiden/Amsterdam Center for Drug Research, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands. E-mail: w.jiskoot@lacdr.leidenuniv.nl; Tel: +31 71 527 4314
bDepartment of Soft Matter Chemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA, Leiden, The Netherlands
First published on 29th March 2011
Abstract
In this study poly(γ-benzyl-L-glutamate)-K (PBLG50-K) polymersomes are tested as an immune adjuvant for an antigen, influenza hemagglutinin (HA). The polymersomes were prepared according to a solvent removal method and loaded with HA antigensvia adsorption. The immunogenicity of the resulting hybrid assemblies was tested in vivo, resulting in an improvement of the immune response for the influenza antigen co-administered with the polymersomes.
Vaccination against influenza remains the most effective method to prevent infection by the virus and to reduce the associated morbidity and mortality.1 Seasonal influenza vaccines currently in use are mostly subunit formulations, consisting of hemagglutinin antigens (HAs) from a mixture of strains. The downside of these vaccines is their relatively low immunogenicity, which can necessitate their administration with an adjuvant, i.e. a component, added to the antigen to enhance its immunogenicity, although the currently marketed seasonal influenza vaccines do not contain any adjuvant. Well-known examples of adjuvants that are licensed for use in humans are colloidal aluminium salts and emulsions, such as MF59. Adjuvants can act in several different ways, e.g. by creating an antigen depot at the injection site, by protecting the antigen from enzymatic degradation, by improving the delivery of the antigen to dendritic cells (DCs) or by activating DCs.2 Various types of nanoparticles have been shown to be able to act as antigen delivery systems which can combine several of these mechanisms.3,4Polymer as well as lipid-based nanoparticles with HA have been successfully tested, enhancing antigen uptake by the DCs and resulting in enhanced antigen-specific acquired immune responses.5,6Nanoparticles can range in size from 10 to 1000 nm, and some studies have shown that the uptake of particles by DCs and their immune-stimulating effect is dependent on their size.7Nanoparticles can vary in several other properties, such as composition, surface charge and hydrophobicity. The nanoparticles can be loaded with antigens by adsorption, covalent attachment, or encapsulation. However, the elicited immune responses of the current formulations do not offer adequate protection and there is still a need for new alternatives. Polymersomes are self-assembled polymer shells composed of block copolymers.8–10 These block copolymers have amphiphilic properties similar to lipids, but they have much larger molecular weights, and for this reason they have been compared with viral capsids, composed of large polypeptide chains. Depending on the choice of the block copolymer, its molecular weight and biocompatibility, polymersomes can be used as delivery systems with a broad range of tunable properties.11–13 Polymersomes have shown to be stable, in terms of size and structure,14 they enable the encapsulation of both hydrophilic and hydrophobic species and can carry functional moieties, such as structures with cell-penetrating capabilities.15 Moreover, polymersomes based on the degradable di-block polymer polyethylene glycol–polybutadiene functionalized with an HIV-derived Tat peptide successfully enhanced, in vitro, the cellular delivery of nanoparticles to DCs.16 These results highlight the potential use of polymersomes as robust, virus like antigen delivery systems, but they have not been tested for vaccination yet. Recently, we developed a new class of polypeptide-block-peptides which self-assemble into polymersomes.17–19 These particles were shown to be stable for several months. The hydrophilic peptide block is composed of a specific amino acid sequence that is able to form a coiled–coil complex,20,21 allowing for the non-covalent functionalization of the polymersome surface with functional moieties. The ability of this recognition motif was indicated by the development of non-covalent triblock copolymers and model systems for membrane fusion.22,23 The hydrophobic block is composed of poly(γ-benzyl-L-glutamate) and both blocks adopt an α-helical conformation when the amphiphiles are assembled into polymersomes.
The aim of this study was to investigate whether polymersomes can enhance the immunogenicity of a HA subunit vaccine. The polypeptide-block-peptide used in this study was the rod–rod block copolymer PBLG50-K,17 where PBLG50 is the hydrophobic poly(γ-benzyl-L-glutamate) block with an average degree of polymerization of 50, and K is a hydrophilic designed peptide with amino acid sequence G(KIAALKE)3–NH2. This amphiphilic block copolymer has been shown to self-assemble into polymersomes with a size of 250 nm. The HA antigen was from a H3N2 A/Wisconsin strain, which is currently used for seasonal vaccination in combination with HA from two other strains. The association of the antigen with the polymersomes was investigated, and the DC-stimulating capacity in vitro and the immunogenicity in mice of the HA–polymersomes were compared with that of plain HA.
PBLG50-K has been shown previously to assemble into well-defined polymersomes in aqueous buffered solutions.17–19 The polymersome size can be tuned from 200 to 2000 nm with low polydispersity by varying the conditions during the self-assembly process, such as ionic strength and temperature, or the preparation method used.18,19 As previously stated the interaction of nanoparticles with DCs and the resulting immune-stimulating effect is dependent on their size,24 with optimum DC uptake for particles with a diameter of 0.5 micron and below. Therefore for this study, polymersomes at the lower end of this size range were selected. In HEPES sucrose at 20 °C PBLG50-K self-assembled into polymersomes with a hydrodynamic diameter of about 250 nm, a polydispersity index of 0.1 (Fig. 1) and a zeta potential (ζ) of −40 mV. The polymersomes were stable, with no sign of turbidity/sedimentation and no detectable aggregation, as observed with dynamic light scattering (DLS) and electron microscopy, for at least 4 weeks.
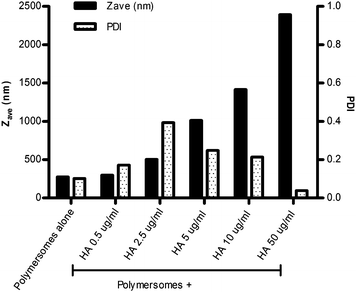 | ||
| Fig. 1 Average hydrodynamic diameter (Zave) and polydispersity index (PDI) of a fixed amount of PBLG50-K polymersomes (100 µg ml−1), mixed with an increasing amount of HA (final concentration ranging from 0.5 to 50 µg ml−1). | ||
The HA proteins are elongated molecules that extend ∼13 nm from the exterior of influenza viruses, being anchored in the viral membrane by means of a hydrophobic domain. This hydrophobic domain causes free HA to aggregate in aqueous solutions. The HA used in this study formed clusters with an average hydrodynamic diameter of about 50 nm in PBS, as measured by DLS. The binding of HA in these clusters is proposed to be relatively weak, as no clear aggregates were observed with transmission electron microscopy (TEM) when samples were stained with either OsO4 (pH 7), or PTA (pH 2 or 7.4) (Fig. 2B inset). Upon the addition of the HA solution to the preformed PBLG50-K polymersomes an immediate particle size increase was observed by DLS. Higher final concentrations (from 0.5 to 50 µg ml−1) of HA resulted in larger HA/polymersome aggregates (Fig. 1). A comparative study with another kind of polymersomes, based on a PBLG-E polymer,18 with a different hydrophilic peptide sequence G(EIAALEK)3–NH2, was conducted and showed no sign of aggregation (data not shown), indicating that the nature of the peptide has a direct impact on the HA/polymersome interaction. TEM revealed that the plain PBLG50-K polymersomes did not aggregate which is in accordance with the DLS data (Fig. 2A). For the polymersome/HA mixtures, clustering was observed, with the HA presumably acting as a non-covalent crosslinker (Fig. 2B and D). The TEM images also revealed that the HA proteins interact with the polymersomes in a relatively weak manner as the shape and size of individual polymersomes did not change.
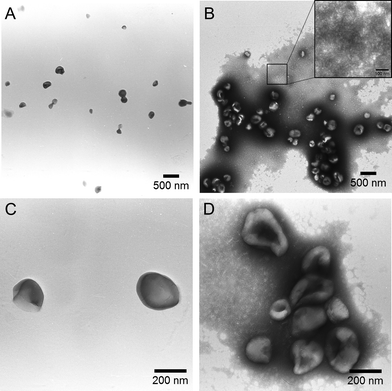 | ||
| Fig. 2 TEM images (PTA staining) of the polymersomes (A and C) and the mixture of polymersome with 10 µg ml−1 HA (B and D). | ||
The association of HA with the polymersomes was further investigated by filtering the suspension through 0.1 µm filters. Under these conditions the PBLG50-K polymersomes were retained on the filter. The filtration of free HA showed a recovery of 88%, whereas for the polymersome/HA complexes it dropped to 28%, showing that most of the HA was associated with the polymersomes.
The adjuvant effect of PBLG50-K polymersomes (mixed with HA antigen) was investigated in an immunization study with mice. In order to study the effect of the polymersomes without any masking from the antigen alone, we used HA doses of 0.5 and 2.0 µg per immunization (corresponding to HA concentrations of 2.5 and 10 µg ml−1 in the formulations). The polymersome concentration was kept constant for all the formulations (100 µg ml−1), resulting in a final HA/polymersome weight ratio of 1/40 and 1/10, respectively.
The two doses of HA were tested in the presence and absence of polymersomes. The HA-specific serum IgG, IgG1 and IgG2a were assessed after the first (prime) and the second (boost) immunization, and hemagglutination inhibition (HI) titers, as a measure for the level of functional antibodies, were measured after the boost. After both the prime and the boost (Fig. 3A and B), PBLG50-K polymersomes significantly enhanced the IgG titers compared to non-adjuvanted HA for the high-dose (2 µg HA) group. In the low-dose group (0.5 µg) there was also a trend toward higher IgG responses for the polymersome formulation as compared to free HA, although the differences were not statistically significant. The IgG1 titers closely followed the total IgG titers, while the IgG2a titers (after prime and boost) were below the detection limit for all groups (results not shown). The HI titer was assessed by measuring the inhibition by the mouse sera of HA-induced red blood cell agglutination. The sera from mice immunized with non-adjuvanted HA showed a dose-dependent HI titer, which was close to the detection limit of the assay for the low-dose group (Fig. 3C). For both HA doses polymersomes acted as adjuvant, as higher HI titers were found, ca. 20 fold for the low HA dose and 8 fold for the high dose, although the latter increase was not statistically significant.
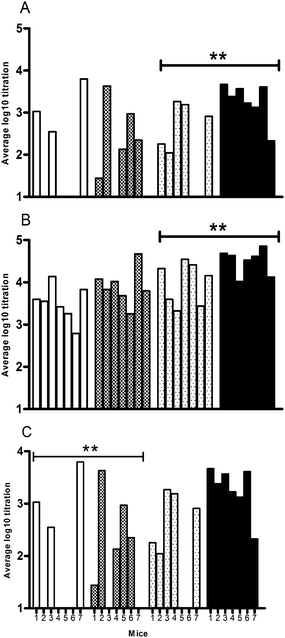 | ||
Fig. 3 The immune response in mice after subcutaneous injection of HA formulations: HA-specific serum IgG titers after prime (A) and boost (B), and HI titers after boost (C). Each bar represents the titer of an individual mouse. The formulations tested are: □ 2.5 µg ml−1 HA,  2.5 µg ml−1 HA + PBLG50-K, 2.5 µg ml−1 HA + PBLG50-K,  10 µg ml−1 HA, ■ 10 µg m−1 HA + PBLG50-K (**: significant difference between the average titers of each group, for p < 0.01). 10 µg ml−1 HA, ■ 10 µg m−1 HA + PBLG50-K (**: significant difference between the average titers of each group, for p < 0.01). | ||
The cytotoxicity of the polymersomes has been tested in vitro. The cell viability was evaluated in Caco-2 cells using the MTT assay. The cells were exposed for 48 hours to a polymersome concentration range of 0.5 to 10 µg ml−1 (the necessary dilution in the cell culture media did not allow the testing of higher polymersome concentrations). The resulting percentages of cell viability showed no sign of toxicity for any of the polymersome concentrations tested (results not shown).
In summary, the immunization study shows an increase of the serum IgG and HI titers, when the antigen is co-administered with the polymersomes. The improvement of the immune response against HA, when associated with the polymersomes demonstrates that the polymersomes can act as an adjuvant. The HA/polymersome hybrid has been characterized with DLS and electron microscopy showing that HA forms complexes with the polypeptide-block-peptide based polymersomes. The HA/polymersome association is presumably a combination of both electrostatic and hydrophobic interactions, arising from the hydrophobic membrane-anchoring domain of the HA, the localized charge on the HA, and the charged corona of the polymersomes. This is confirmed by the difference observed in the DLS study between PBLG-K and PBLG-E. The presence of three protonated lysine residues (at pH 7.4) in K, instead of three glutamic acids in E, had changed the electrostatic interactions between HA and the polymersome surface, resulting in different aggregation behaviors. The mechanism by which polymersomes act as an adjuvant is unknown, but could include a depot effect,5 the ability to target the antigen presenting cells (APCs) with the antigen/adjuvant complexes and enhancement of the antigen uptake by APCs.
As detailed in the introduction, the antigen used in this study is one of the main components of the current subunit seasonal influenza vaccines (which consist of a mixture of HA from different strains). Since neutralizing antibody levels expressed as HI titers are considered to be the main protective immune component for parentally administrated subunit vaccines,25,26 and as this assay is also the test of reference according to the industry standards, the increase of the systemic immune response against HA formulated with PBLG50-K polymersomes, as observed in our study, is a clear improvement compared to the current formulation (HA alone). However, in order to provide a superior level of protection against the influenza infection than HA alone, it has been shown previously that an IgG2a response (indicative of a Th1 immune response) is the strongest isotype in response to viral infection.27 Our formulations induced low IgG2a titers, therefore our future research will be directed at enhancing the IgG2a response of the polypeptide-block-peptide polymersomes by coencapsulation of immuno-modulators.
In conclusion, the work detailed in this paper demonstrates, for the first time, that polymersomes are able to enhance the immunogenicity of an antigen. Moreover, it shows the proof of concept that they can be used as a delivery tool for influenza subunit vaccine with enhanced immune response and no sign of cellular toxicity.
Notes and references
- A. S. Monto, Clin. Infect. Dis., 2009, 48(Suppl. 1), S20–S25 CrossRef.
- J. H. Wilson-Welder, M. P. Torres, M. J. Kipper, S. K. Mallapragada, M. J. Wannemuehler and B. Narasimhan, J. Pharm. Sci., 2009, 98, 1278–1316 CrossRef CAS.
- D. T. O'Hagan and R. Rappuoli, Pharm. Res., 2004, 21, 1519–1530 CAS.
- T. Storni, T. M. Kundig, G. Senti and P. Johansen, Adv. Drug Delivery Rev., 2005, 57, 333–355 CrossRef CAS.
- S. Okamoto, H. Yoshii, T. Akagi, M. Akashi, T. Ishikawa, Y. Okuno, M. Takahashi, K. Yamanishi and Y. Mori, Vaccine, 2007, 25, 8270–8278 CrossRef CAS.
- O. Even-Or, S. Samira, E. Rochlin, S. Balasingam, A. J. Mann, R. Lambkin-Williams, J. Spira, I. Goldwaser, R. Ellis and Y. Barenholz, Vaccine, 2010, 28, 6527–6541 CrossRef CAS.
- J. P. Scheerlinck and D. L. Greenwood, Drug Discovery Today, 2008, 13, 882–887 CrossRef CAS.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS.
- S. F. M. van Dongen, H. P. M. de Hoog, R. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212–6274 CrossRef CAS.
- H. R. Marsden and A. Kros, Macromol. Biosci., 2009, 9, 939–951 CrossRef CAS.
- D. Lensen, D. M. Vriezema and J. C. M. van Hest, Macromol. Biosci., 2008, 8, 991–1005 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- N. A. Christian, M. C. Milone, S. S. Ranka, G. Li, P. R. Frail, K. P. Davis, F. S. Bates, M. J. Therien, P. P. Ghoroghchian, C. H. June and D. A. Hammer, Bioconjugate Chem., 2007, 18, 31–40 CrossRef CAS.
- H. R. Marsden, L. Gabrielli and A. Kros, Polym. Chem., 2010, 1, 1512–1518 RSC.
- H. R. Marsden, J. W. Handgraaf, F. Nudelman, N. A. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2010, 132, 2370–2377 CrossRef CAS.
- H. R. Marsden, C. B. Quer, E. Y. Sanchez, L. Gabrielli, W. Jiskoot and A. Kros, Biomacromolecules, 2010, 11, 833–838 CrossRef CAS.
- H. R. Marsden and A. Kros, Angew. Chem., Int. Ed., 2010, 49, 2988–3005 CrossRef.
- E. H. C. Bromley, K. Channon, E. Moutevelis and D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50 CrossRef CAS.
- H. R. Marsden, N. A. Elbers, P. H. H. Bomans, N. Sommerdijk and A. Kros, Angew. Chem., Int. Ed., 2009, 48, 2330–2333.
- H. R. Marsden, A. V. Korobko, E. N. van Leeuwen, E. M. Pouget, S. J. Veen, N. A. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386–9393 CrossRef CAS.
- C. Foged, B. Brodin, S. Frokjaer and A. Sundblad, Int. J. Pharm., 2005, 298, 315–322 CrossRef CAS.
- N. Hagenaars, E. Mastrobattista, H. Glansbeek, J. Heldens, H. van den Bosch, V. Schijns, D. Betbeder, H. Vromans and W. Jiskoot, Vaccine, 2008, 26, 6555–6563 CrossRef CAS.
- D. Hobson, R. L. Curry, A. S. Beare and A. Ward-Gardner, J. Hyg., 1972, 70, 767–777 Search PubMed.
- F. Nimmerjahn and J. V. Ravetch, Immunity, 2006, 24, 19–28 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00010a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2011 |