Multiresponsive polymers: nano-sized assemblies, stimuli-sensitive gels and smart surfaces
George
Pasparakis
a and
Maria
Vamvakaki
*ab
aInstitute of Electronic Structure and Laser (IESL), Foundation for Research and Technology—Hellas (FORTH), P.O. Box 1527, 711 10, Heraklion, Crete, Greece. E-mail: gpasp@iesl.forth.gr; vamvakak@iesl.forth.gr
bDepartment of Materials Science and Technology, University of Crete, P.O. Box 2208, 710 03, Heraklion, Crete, Greece. E-mail: vamvakak@materials.uoc.gr
First published on 2nd March 2011
Abstract
The complex function of living systems is dictated by their inherent cooperative response to multiple external stimuli which induce dynamic changes in their physicochemical properties. Advances in the areas of nano- and bio-technology demand for the development of “smart” synthetic materials that would resemble the living systems in their complex behaviour as a response to applied stimuli. This reversible response directs the formation of hierarchical self-assemblies or stimulates changes in the volume, the shape or the surface characteristics of the system. Progress in this rapidly expanding area can lead to the development of dynamically multiresponsive constructs in the form of polymers, particles, gels or surfaces, for potential use in a wide range of applications such as drug delivery, tissue engineering, self-healing materials, bioseparations, sensors and actuators. This review highlights the recent advances in polymer chemistry to design multiresponsive polymeric materials that recognize independently or synergistically more than one stimulus exhibiting collective responses. Emerging developments, challenges and future trends in this exciting field are also discussed.
 George Pasparakis | George Pasparakis studied Materials Science at the University of Patras (BSc, 2005), before commencing graduate studies at the School of Pharmacy, University of Nottingham, under the supervision of Professor Cameron Alexander. His research project aimed at the construction of artificial cells based on smart glycopolymers that mediate specific cell–polymer interactions. Dr Pasparakis was awarded his PhD in July 2009. He is currently a research associate at IESL-FORTH (Heraklion, Greece), where he expanded his research interests to include photodegradable biomaterials, biophotonics and nanomedicine. His research interests include cell–polymer interactions, drug delivery using smart polymers and soft materials for synthetic biology. |
 Maria Vamvakaki | Maria Vamvakaki received her PhD from the University of Sussex (Brighton, UK) in 1998, working under the supervision of Profs. S. P. Armes and N. C. Billingham. Shortly after, she joined the research group of Prof. C. S. Patrickios at the University of Cyprus (Nicosia, Cyprus) as a postdoctoral fellow. In 2001 she joined the Department of Materials Science and Technology of the University of Crete (Heraklion, Greece) in which she is appointed as an Associate Professor and leads the Materials Chemistry Lab. She is also an affiliated researcher at FORTH-IESL since 2003. Her current research interests include the synthesis of functional and stimuli-responsive polymeric materials and the self-assembly of macromolecules in solution and at a surface. |
Introduction
“Smart” synthetic materials able to respond to external or internal stimuli represent one of the most exciting and emerging areas of scientific interest with a wide range of potential applications.1,2 Mother Nature provides continual inspiration in this field using elegant responsive systems based on macromolecules, such as proteins, polysaccharides and nucleic acids, as structural materials to regulate the most critical biological functions.3During the last two decades, scientists have been trying to mimic nature4 in designing “smart” synthetic materials from various functional molecular building blocks that respond to stimuli such as temperature, pH, ionic strength, light, electric or magnetic field, chemical and biochemical stimuli in order to mediate molecular transport, shape changes, tuning of adhesion and wettability, or to induce signal transduction of (bio-)chemical or physical stimuli into mechanical, optical or electrical responses.5 Biomimetic approaches have been employed in the design, synthesis and engineering of stimuli-responsive polymeric systems, which undergo reversible abrupt phase transitions upon variation of a variable around a critical point and their use in a plethora of applications, including sensors,6 logic operations,7 biomedicine,8,9 tissue engineering and regenerative medicine,10 synthetic muscles,11 “smart” optical or microelectromechanical systems,12membranes,13 electronics14 and self-cleaning surfaces15,16 has been explored. Evidence of the rapidly expanding scientific and technological interest that has been attracted in this field is found in the exponential increase in the number of papers and citations that have been published in this field over the last few years (see Fig. 1). However, despite the significant progress that has been made in the area, mastering highly cooperative interactions to induce huge phase transformations to take place in a cumulative manner by employing minute interactions among the small building blocks is still an obstacle. Moreover, creating synthetic systems capable of responding to multiple stimuli and resulting in one or more responses in a controllable and predictable fashion still represents a significant challenge. Research efforts over the last decade have focused on the synthesis and characterisation of multiresponsive polymers using robust synthetic routes (i.e. “click” chemistries,17–22 orthogonal synthetic strategies,23–26etc.) and controlled polymerisation techniques (i.e. atom transfer radical polymerization (ATRP),27,28 reversible addition–fragmentation chain transfer processes (RAFT),29nitroxide mediated polymerization (NMP),30etc.) aiming to elucidate structure-to-property complexity and controlled function.
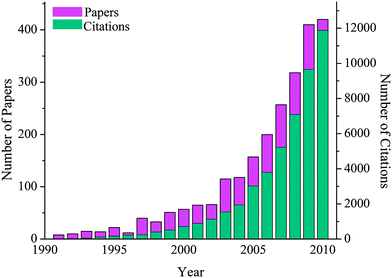 | ||
| Fig. 1 Diagram showing the non-linear increase in the number of papers and citations per year in the field of responsive polymers verifying the rapid development of the field over the last 20 years (Source: ISI Web of Knowledge, keywords: responsive polymers, date of search: 24/01/2011). | ||
This review highlights the scientific progress in the area of multiresponsive polymeric materials in the form of linear or graft copolymers, polymer particles, gels and polymer surfaces. Although there are still many exciting challenges to overcome, the opportunities that stem from this effort might open new avenues in the programmable response of synthetic complex materials in a manner that mimics the delicate subtlety found in natural systems. The selected state-of-the-art examples presented in this review demonstrate the great scope and diversity of these systems in terms of the stimulating mechanism, response time and function.
Linear polymers
So far a vast majority of responsive polymers have been prepared that respond to a single or two different stimuli. In particular, most studies on dual responsive polymers have been focused on pH and temperature changes whereas light, redox and competitive host/guest multiresponsive polymers have also been investigated, however to a lesser extent. Thermoresponsive polymers usually comprise temperature-sensitive monomer repeat units such as N-isopropylacrylamide (NIPA),31 oligo(ethylene glycol) methacrylate,32–362-dimethylamino ethyl methacrylate,37–402-oxazoline41,42 and hydroxypropyl acrylate,43 with the former being the most extensively studied example. PNIPA is a well known temperature responsive polymer which exhibits a solution phase transition temperature in water of around 32 °C, being soluble in the solvent medium below this temperature while it precipitates out of solution as the temperature increases to higher values. This is the so-called lower critical solution temperature (LCST) behaviour and is attributed to the interplay between the intermolecular polymer–water and intramolecular polymer–polymer hydrogen bonding interactions. Above the LCST polymer phase separation occurs, leading to the formation of polymer nanostructures in double hydrophilic PNIPA-based block copolymers or to the shrinkage of PNIPA hydrogels and microgels at elevated temperatures. The pH-sensitive groups of the polymers are typically based on ionizable weak acid44 or base45 moieties. The latter charged units confer to the polymers responsive properties to additional ion-related stimuli such as the ionic strength of the solution and the presence of multivalent counterions. However, the scope of this review focuses more on the recently emerged responsive polymers comprising either materials that respond to two or more stimuli or systems exhibiting cooperative dual responsiveness that leads to increased functional complexity in a synergistic manner.The synthesis of multiresponsive polymers requires not only the rational choice of starting materials but also a suitable synthesis method with respect to the final application, in order to devise linear or graft architectures of complex physicochemical behavior.46 Controlled polymerisation methods have greatly boosted the development of such materials allowing for increased functional complexity without compromising structural precision and fidelity. Also, novel chemistries have been exploited that further expand the polymer chemists' toolbox for new concepts to be introduced in the field of responsive polymers.
A beautiful example of multiresponsive polymer architecture was given by the Thayumanavan group that reported on disulfide linked polymer chains that formed novel block copolymer assemblies with unique responsive properties.47 The first PNIPA block exhibited the well known temperature induced coil-to-globule transition at ca. 35 °C, whereas the second block consisted of a hydrophobic protected poly(2-hydroxyethyl methacrylate) (PHEMA) derivative that can undergo acid hydrolysis and unmask the hydrophilic HEMA segments. It was shown that the protected polymer could self-assemble into micelles at ambient temperatures but flocculated above NIPA's LCST due to the collapse of the PNIPA block and the hydrophobic character of the tetrahydropyran protecting groups of HEMA. However, when mild acidic conditions were introduced in the polymer solution, hydrolysis of the protected HEMA moieties resulted in the disruption of the micelles with subsequent dissolution due to the good solubility of HEMA in water. Besides, the polymer was also found to respond to redox stimuli, due to the disulfide bridge linking the two polymer chains, which was manifestated by simple controlled release experiments of model dye molecules from the disulfide linked polymer micelles. At the same time, Liu et al. also reported on elaborate polymer structures of higher architectural complexity and functionality by synthesizing a coil-centipede like polymer structure with dual-type responsiveness.48 A PEG–Br macroinitiator was used to polymerise glycidyl methacrylate which was further treated with NaN3. This resulted in the ring opening of the glycidyl segments and the introduction of hydroxyl and azido groups along the polymer chain which were used for further derivatisation. Esterification with bromoisobutyryl bromide allowed for an additional ATRP step to grow poly(2-(2-methoxyethoxy)ethyl methacrylate) (PMEO2MA) grafts. The final structure was obtained by “clicking” monoalkyne-capped poly(2-(diethylamino)ethyl methacrylate) (PDEA) polymer chains onto the azide moieties of the precursor polymer. The resulting polymer structure had an interesting coil–rod architecture with an asymmetric centipede-like polymer brush. Owing to the presence of the two responsive polymer chains (PMEO2MA for temperature and PDEA for pH response, respectively), the polymer not only exhibited multiple stimuli responsiveness but also self-assembly into two discrete micellar aggregates, which could be induced by variation of pH and temperature. This study clearly shows how rational design of complex macromolecular architectures can afford nano-assemblies of improved subtlety in their self-assembly properties beyond the classic unimer–micelle transition.
Green chemical pathways have also been explored to obtain responsive polymers of higher complexity. For instance, Ren et al. reported on the synthesis of novel multiresponsive poly(ether amines) by simple nucleophilic addition of amine containing macromonomers to bifunctional epoxy rich moieties.49Polymers of varying monomer ratio were produced by reacting poly(propylene glycol) diglycidyl ether with jeffamine and octadecyl amine macromer which resulted in the formation of poly(propylene glycol) segments randomly linked to either alkyl or jeffamine chains in a linear topology. The polymers were found to exhibit sharp coil-to-globule transition temperatures with onsets ranging from ca. 20 to 65 °C and could self-assemble into micelles encapsulating model fluorescent dyes within the hydrophobic micellar compartments. More interestingly, the cloud points were found to strongly depend on the pH and ionic strength of the solution which further enhanced the responsiveness of the polymers. Similar results have been obtained by the same group when introducing piperazine moieties in the polymer backbone which also exhibits strong pH-responsive properties due to the tertiary amine units.50 In a recent paper the researchers extended the synthetic route to the synthesis of core–shell type microgel particles with organosilica cores coated with poly(ether amine) shells that were found to exhibit excellent T/pH responsive properties.51 This family of polymeric materials could be used in a variety of applications such as drug delivery, rivaling NIPA based systems in that they are potentially less toxic and can be synthesised using green chemistry protocols.
Another, potentially alternative to PNIPA, class of T-responsive polymers is based on hydroxypropyl cellulose (HPC) copolymers that exhibit LCSTs around physiological temperature and can be extracted from natural resources. Ma et al. studied the self-assembly properties of HPC-graft-(2-(dimethylamino)ethyl methacrylate) DMAEMA copolymers in aqueous solutions52 and showed that the LCST onset could be precisely tuned by incorporating ionisable DMAEMA grafts onto the polysaccharides backbone by ATRP, which in turn induced polymer self-assembly into distinct macromolecular assemblies depending on the ionization degree of the DMAEMA segments. The cationic nature of the DMAEMA segments has been also exploited by Xu and coworkers that used an HPC-graft-DMAEMA comb-type polymer to form polymer–nucleic acid polyplexes for gene delivery applications.53In vitro transfection studies showed efficient transfection rates higher than the gold standard PEI (polyethyleneimine)–DNA complexes. Other elegant examples have been demonstrated in the rapidly growing gene therapy field, where polymers that exhibit unique sensitivity in their multiple-type responsiveness are required in order to accurately deliver their payload to the complex intracellular microenvironment. For example, Jiang et al. reported the synthesis and transfection efficiency of novel dual-sensitive nanoparticles.54 PEG was used as a macroinitiator to polymerise a novel cyclic phosphite monomer followed by installation of dipropyltriamine segments on the phosphate backbone. Thiolation was then introduced by using Trauts reagent which afforded the final crosslinkable product. The polymer formed micelles, which could be “locked” by disulfide bond formation in order to improve their stability upon DNA complexation. Interestingly, it was found that the polymer exhibited a unique salt sensitivity which was monitored by DNA decomplexation assays. This resulted in a dual-sensitive DNA release mechanism attributed to the salt-rich environment within the cytosol as well as the disintegration of the SS bonds within the late endosomes. Redox mediated disruption was further exploited by Zhang et al.55 who synthesised novel selenide containing block copolymers that exhibit simultaneous oxidative or reductive response via Se–Se bond cleavage. The polymers were synthesised by polymerizing toluene diisiocyanate in excess of selenide containing diols and the polymers were end capped with two poly(ethylene glycol) (PEG) chains. A triblock copolymer was thus produced which could self-assemble to micelles under aqueous conditions. Interestingly, the micelles exhibited a unique degradation behavior upon addition of either an oxidant (i.e.H2O2) or a reductant (i.e.glutathione) due to the cleavage of the Se–Se bonds. It was found that the micelle disintegration occurred in proportion to the stimulant addition and was very sensitive up to 0.01 mg mL−1 stimulant concentration. Clearly this study enriches redox chemistries in the biomedical context and also demonstrates that these polymers may find useful applications in the field of, for example, drug nanocarriers that degrade upon specific biological events such as inflammation or physiological oxidative stress.
Another interesting strategy for the embodiment of multiresponsive properties in polymers is the exploitation of host–guest chemistries that allow for specific-type responses to small molecules, metal ions, or biomolecules by non-covalent —and hence reversible— binding events. This was demonstrated by Bigot et al.56 who exploited the rich redox chemistry of tetrathiafulvalene (TTF) that exhibits distinct oxidised states (TTF+˙, TTF2+). PNIPA polymer chains were synthesised by RAFT polymerization and subsequently TTF segments were used to end cap the polymer chains by standard esterification routes. The resulting semitelechelic polymer exhibited unique redox enriched thermoresponsive properties owing to the presence of the TTF moieties (Fig. 2). The polymer could form micelles in aqueous solutions which could be disrupted by oxidising the TTF polymer ends upon addition of an oxidising agent (i.e.Fe(ClO4)3). More interesting results were obtained when the researchers elegantly demonstrated the host–guest principle of the TTF moieties with π-rich-electron molecules such as cyclic cyclobis(paraquat-p-phenylene) (CBPQT4+, 4Cl−). It was found that minute addition of CBPQT4+ resulted in efficient micelle disruption, as evidenced by controlled release experiments using Nile red loaded micelles, due to the increase of the hydrophilic nature of the TTF moiety upon complexation with CBPQT4+.
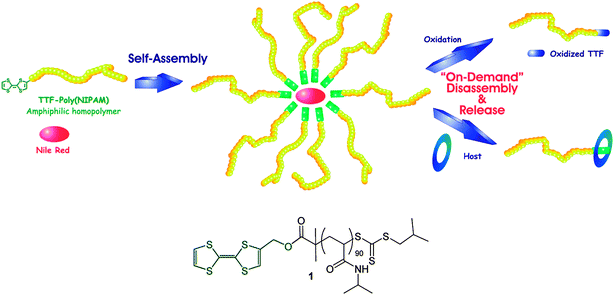 | ||
| Fig. 2 Schematic representation of tetrathiafulvalene end capped PNIPA polymers exhibiting cooperative multiresponsive properties derived from a redox mediated host–guest type self-assembly. Reproduced with permission from ref. 56. | ||
Although the above examples constitute an excellent platform of multiresponsive polymers, in the biomedical context, polymers that respond to biochemically relevant analytes under specific conditions are of paramount importance and need to be developed for a variety of applications such as bioresponsive drug delivery, cell–polymer interactions and cell signaling processes. The Sumerlin group has introduced novel poly(boronic acids) copolymerised with NIPA by facile RAFT polymerisation which resulted in a block copolymer with multiple responsive properties.57Boronic acids are known for their ionization properties at acidic and alkaline regimes, but more importantly for their covalent, albeit reversible, binding to carbohydrate molecules by formation of boronate esters in alkaline pH.58,59 The assembly/disassembly of these polymers can be reversibly controlled by simultaneous pH changes and/or sugar addition, isothermally. This concept was further exploited by the Nolte group60 who presented a simpler sugar binding poly(boronic acid) block copolymer. A pinacol ester protected boronic acid styrene block was polymerised from a PEG–Br precursor by ATRP followed by the removal of the pinacol protecting groups to afford a PEG–poly(boronic acid) block copolymer which could self-assemble into polymersomes. The permeability of the latter could be precisely tuned by application of a pH increase or the addition of boronate binding sugars (i.e.glucose), via a subtle switching of the hydrophobic boronate block to a hydrophilic segment. The principle was elegantly demonstrated by inclusion of model CALB enzymes within the aqueous vesicular compartment and use of the polymersomes as model microreactors that could perform certain bioreactions under strictly confined conditions similar to those found in natural cells.
Another intriguing example of a multi-bio-responsive polymer was reported by the Ulijn group who synthesised novel polyoxazoline based block copolymers.61 Polyoxazolines are known to exhibit LCST close to physiological temperature as well as to be less cytotoxic than the “gold standard” PNIPA. Ring opening polymerization using an alkyne functional initiator was employed to produce alkyne terminated 2-isopropyl-2-oxazoline block. The complementary azide terminated block was prepared by coupling a fluorenylmethoxycarbonyl-tyrosine (FMOC) with 11-azido-3,6,9-trioxaundecan-1-amine by carbodidimide coupling. The two blocks were linked by “click” chemistry to afford the final polymer with one polyoxazoline and one phosphatase-sensitive block, respectively. The polymer exhibited interesting self-assembly properties in aqueous solutions derived either by the application of a temperature stimulus or by the presence of phosphatase which could cleave the phosphate groups of the FMOC containing block with subsequent alternation of the nanoassemblies size (Fig. 3).
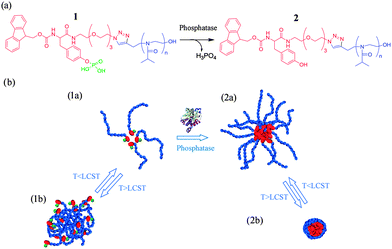 | ||
| Fig. 3 Concerted bio-/thermo-responsive behaviour of phosphatase sensitive block copolymers exhibiting different self-assembly superstructures according to stimuli combination. Reproduced with permission from ref. 61. | ||
These studies clearly demonstrate significant progress in the field of biomimetically inspired responsive polymers that could potentially mimic cellular organelles for example, as artificial membranes that allow for selective flux of small molecules and ions across the cellular compartments, or as dynamic and self-regulated polymer ensembles that can constitute a primitive platform for synthetic biology applications62 based on structure-to-function complexity refinement.
As a final class of multiresponsive polymers we report selected examples of polymers with light responsive properties in concert with other stimuli which provide superior control over a range of conditions. Multiresponsive polymers embedding additional light sensitivity can find numerous applications in sensing technologies, as well as in the biomedical field as exogenously triggered drug carriers, photolabile constructs, etc.
Our group has performed systematic studies on the responsive properties of relatively simple copolymers made of spiropyran derivatives (SP) with DMAEMA moieties synthesised by ATRP.63 It was demonstrated that DMAEMA–SP copolymers exhibited concerted multiresponsive properties to various stimuli, namely, solvatochromism, pH/T responsiveness, as well as light induced conformational changes (Fig. 4). The distinct physicochemical properties of spiropyrans were further extended by Zhao et al.64 who combined spiropyran derivatives with another photoisomerisable azobenzene derivative in an effort to produce photoresponsive polymers comprising two distinct light sensitive monomers on the same polymer backbone, combined with thermoresponsive segments. The researchers systematically probed the effect of a light stimulus on the thermoprecipitation properties of the block copolymers as a function of the azobenzene conformational changes and the spiropyran isomerization. It should be noted that compounds such as spiropyrans are unique in a sense that one can obtain multiple responses to a number of external stimuli (UV/Vis, pH, solvent) from a single molecular structure and hence the use of this class of molecules in the synthesis of intelligent macromolecular ensembles could significantly simplify the preparation process particularly if combined with orthogonal synthetic routes. However, the embodiment of such compounds65 into intelligent polymer assemblies is still rather limited (perhaps due to the elaborate synthetic routes required to isolate them in gram scales) and hence, we feel that there are still many interesting concepts to be exploited in the field of multiresponsive polymers comprising photosensitive moieties, in the immediate future.
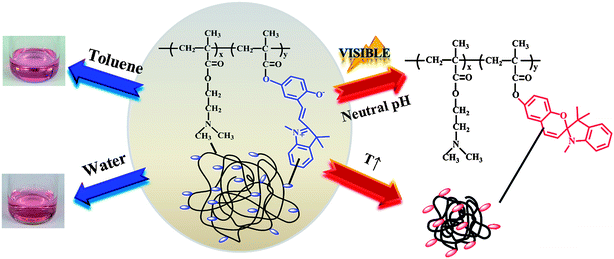 | ||
| Fig. 4 DMAEMA–SP copolymers exhibiting simultaneous thermo/pH responsiveness, solvatochromism as well as light induced conformational changes.63 | ||
Particles
Interesting concepts have been developed in the field of multiresponsive micro/nanoparticles where particles of controlled topology and architecture have started to emerge. There are numerous recent examples demonstrating both all-organic constructs as well as organic/inorganic hybrids that further extend the possible applications of the particles to many research areas. For example, there is active research interest for the development of multiresponsive functional particle entities for biomedical imaging, theranostics, drug delivery and intracellular biosensing.66,67 Particular attention has been also paid recently on the development of core–shell colloidal latexes with temperature and pH responsive properties by the Armes group.68,69In an intriguing study, Gota et al.70 reported novel polymer nanogels that can be used as intracellular thermometers. The nanogels were produced by emulsion polymerization of NIPA copolymerized with a fluorescent methacrylamide derivative in the presence of a crosslinker. Nanosized hydrophilic particles were produced that exhibited unique fluorescence properties depending on the microenvironment conditions. In particular, fluorescence intensity could be finely tuned across a temperature range near the polymer's LCST onset. Also, fluorescence intensity was found to be proportional to potassium concentration but independent of pH changes. It was therefore possible to measure the exact temperature within the cytosol of COS7 cells by monitoring in situ changes in fluorescence intensity with an accuracy of ca. 0.5 °C. In a related study, NIPA based novel microgel particles combining photocleavable 5-(2-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate moieties and two fluorophore monomers, one electron donor (4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole) and one electron acceptor (rhodamine B), were synthesised by precipitation polymerization.71In situ monitoring of the swelling/deswelling properties of the microgels with respect to temperature changes was achieved by a fluorescence resonant energy transfer (FRET) process. Upon UV radiation the photolabile nitrobenzyl groups of the microgels were cleaved leaving behind acrylic acid units that shifted the LCST onset to higher temperatures. This was directly monitored by fluorescence spectroscopy, as the temperature dependent volume transition of the microgels resulted in a marked change of the distance between the donor and the acceptor and hence affected the FRET process. The swelling induced FRET dependence was further exploited by combining NIPA and the donor–acceptor FRET system with metal binding crown ether derivatives for simultaneous ratiometric biosensing of the temperature and the concentration of potassium ions (Fig. 5).72 The beauty of this study stems from the rational design of microgel particles that can lead to unique optical properties derived from the intrinsic multiresponsive (thermo and optical) behaviour of the starting materials.
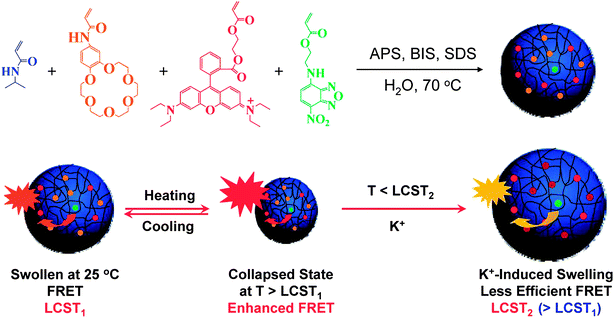 | ||
| Fig. 5 FRET tuning of “smart” microgel particles by temperature and K+ ions. Reproduced with permission from ref. 72. | ||
Another attractive approach for the synthesis of responsive polymer particles uses readily available natural biopolymers that often exhibit superior cytocompatibility and excellent processability. For example, Huang et al. used HPC as a microgel forming biopolymer and exhibited an LCST just above 37 °C, ranging from 41 to 45 °C depending on the degree of substitution of the hydroxypropyl groups.73 The researchers managed to decorate the polymer with thiol moieties via a two step process without affecting significantly the LCST onset of the parent material. Next, the thiol-rich cellulose was used to form stable microgel particles by increasing the temperature above the LCST with simultaneous oxidation of the thiol groups. This concerted thermo/redox property of the resulting material allows not only for dual responsive properties to be studied per se, but can also be used as a mild and safe means to immobilise sensitive protein drugs or biomolecules within the microgels and allow for targeted delivery viaredox or temperature stimuli at the targeted site.
In the last few examples, we present interesting synthetic routes for the synthesis of multifunctional organic/inorganic hybrid particles that exhibit intriguing physicochemical properties derived from the synergistic organic and inorganic component functionalities. For example, recent studies have emerged in the development of polymer coated iron oxide nanoparticles primarily for use as MRI contrast agents but also as magnetic controlled drug delivery agents. Cai et al. synthesised triply responsive magnetic microgel particles,74 starting from Fe2O3 particle seeds that were previously coated with 3-(trimethoxysilyl)-propyl methacrylate segments via a Stöber process. Next, precipitation polymerization was employed to grow a PNIPA–PDMAEMA shell of varying monomer feed compositions around the inorganic core of the particles. The particles had uniform morphology and were systematically characterised in regard to the monomer feed composition, pH/T responsiveness as well as magnetization properties. In particular, the incorporation of the Fe2O3 core further expands the potential applications of these particles in the biomedical field as intelligent contrast agents in MRI, as exogenous activated drug carriers by magnetic fields, etc.
Particles with intriguing optical properties in the multiresponsive context also emerge when “smart” polymers are combined with noble metal nanoparticles exhibiting useful optical properties (i.e. stable photoluminescence, plasmon resonance, etc.). The Marzan group proposed a novel method for the synthesis of multiresponsive microgel particles with a gold core coated with a thin Ni shell which was further coated by a porous NIPA outer shell.75 These particles were produced by precipitation polymerization of the outer PNIPA shell using gold nanoparticle seeds. Subsequently, the thermoresponsive particle precursors were dispersed in a NiCl2 solution containing hydrazine and with the aid of a Pt thin film precoating a thin layer of Ni was grown directly onto the gold core. Interestingly, the Pt layer growth proceeded onto the Au core despite the presence of the PNIPA shell, presumably due to sufficient porosity of the polymer layer that allows for robust modification of the Au surface. Indeed, characterisation data confirmed the robustness of this method and demonstrated that the resulting hybrids exhibited a plasmon resonance peak, LCST at 32 °C and notable magnetization properties at room temperature due to the thin Ni layer. In another study, hybrid multifunctional microgel particles were synthesised for drug delivery and imaging applications.76 First, inorganic Ag nanoparticles were synthesised and coated with a thin polystyrene layer. The as synthesised inorganic particles served as seeds to grow a PNIPA–poly(acrylic acid) microgel layer by precipitation polymerization. The final hybrid particles exhibited pH dependent swelling/deswelling properties due to the ionization of the acrylic acid moieties which were exploited to regulate the release of dipyridamole from the polymeric microgel matrix. Also, the intrinsically photoluminescent inorganic core was used as an imaging probe to monitor the fate of the microgel particles upon intracellular uptake in vitro. The robustness of seeded polymerization from inorganic core particles to obtain multifunctional nanoparticles was further exploited by the same group in order to construct multifunctional microgel particles for the simultaneous optical glucose sensing and the self-regulation of insulin release (Fig. 6).77 This was achieved by polymerization of a gel layer of DMAEMA and vinyl benzene boronic acid onto Ag nanoparticle seeds. Boronic acid is known to bind covalently, albeit reversibly, with cis-diols. The binding is favored at pH 8.9 (close to the boronic acid pKα) where the tetrahedral boronic acid groups favour the diol–boronate ester formation. However, this was circumvented by the presence of the tertiary amine groups of DMAEMA which form B−–N+ pairs that stabilize the boronate ester at physiological pH. Therefore, the researchers observed significant swelling of the microgel layer as a function of glucose concentration (due to hydrogel mesh relaxation) which in turn resulted in a change in the local refractive index around the inorganic core. This gradual refractive index reduction in response to glucose was manifestated by a marked decrease of the photoluminescence intensity of the Ag cores which was used to monitor the glucose concentration in solution. At the same time, the glucose dependent swelling of the microgel particles was used to regulate insulin release from protein loaded microgel particles as a result of the marked change of the insulin diffusion coefficient.
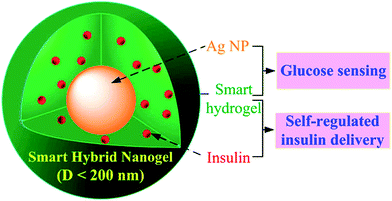 | ||
| Fig. 6 Multifunctional core–shell hybrid nanoparticles for simultaneous optical glucose sensing and self-regulation of insulin release. Reproduced with permission from ref. 77. | ||
It is clear from the above literature examples, that there is indeed significant progress in the construction of multiresponsive particles of varying functionalities aiming at different applications spanning from (bio-)sensing and biophotonics, to theranostics, “lab on a particle” concepts, and intelligent delivery applications.67,78 However, great effort is still required to master the synthesis-to-property principle under confinement in order to implement truly biomimetic nanoparticulate structures that can rival the perfectly balanced natural counterparts.
Polymer gels
Multiresponsive hydrogels are particularly attractive as platforms for the development of intelligent devices and components for many practical applications in friction or permeability control in membranes, drug delivery, tissue engineering and others. So far progress in multiresponsive gels has led to the development of materials that respond to two or three different stimuli. The majority of studies on dual responsive gels have focused on hydrogels that respond to pH and temperature variations79–81 whereas more elaborate examples including gels that are sensitive to light, competitive host/guest, solvent and (bio-)chemical stimuli have been also investigated. The dual temperature and pH responsive hydrogels usually comprise thermosensitive NIPA units and pH-responsive ionizable weak acid or weak base moieties79,80 which form a permanent chemically cross-linked network structure. These materials have been extensively reported in the literature and an extensive reference to their synthesis and properties is out of the scope of the present review.Herein, we will focus on state-of-the-art examples of polymer gels being sensitive to more than two stimuli or displaying a unique cooperative responsive behavior which can be advantageous for many emerging applications in injectable gels, optical sensors, etc.
An intriguing example of such materials are gels that are sensitive to remote stimuli, i.e. light irradiation has attracted great interest lately because it can be applied on patterned surfaces, photonics, and photo-driven devices. Organic molecules exhibiting light responsive behavior include azobenzenes, spiropyrans, spirooxazimes and naphthopyrans with the former being the most extensively studied family of photochromic compounds.82,83 Multiresponsive hydrogels being sensitive to light have been prepared by free radical copolymerization of NIPA with an acrylate spirobenzopyran derivative and N,N-methylene-bis(acrylamide) as the cross-linker.84 These materials were shown to respond to pH/T variations as well as light irradiation and more importantly exhibited a cooperative effect on the gel volume and conductance changes as a function of the applied stimuli. At acidic conditions, the hydrogel showed drastic volume shrinkage, when irradiated with blue light, as a result of proton dissociation of the spiropyran units. However, when the light was switched off the chromophore returned to the protonated open-ring form and the gel expanded. The above rapid response of the material was employed to control mass transfer through a photo- and thermo-responsive gate membrane constructed by grafting the photoresponsive hydrogel to the surface of the membrane pores. The permeability was reversibly controlled by either temperature or light irradiation, independently. However, a complex behavior was found for the flux of an acidic solution as a function of the above stimuli. The solution flow exhibited a two fold increase upon irradiation with blue light at temperatures above the LCST of the polymer and the time scale for recovery was slow, whereas for temperatures below the LCST a smaller increase in flow was observed upon irradiation, which recovered very rapidly when the light was turned off. This effect was attributed to the stabilization of the closed spiropyran form at high temperatures by the surrounding hydrophobic polymer which causes a further collapse of the gel and an increase in the pore size. The precise irradiation of such hydrogels at a micrometre scale can be employed to construct photoresponsive devices which allow controlling mass transfer in microfluidic systems.
An alternative type of multiresponsive materials, which have also attracted great interest, is the so-called supramolecular hydrogels that possess the ability to undergo a rapid sol–gel transition leading to the formation of reversible gels upon the application of suitable stimuli. These gels are obtained by appropriately tuning the interactions in polymer solutions or their mixtures with host–guest complexation molecules or inorganic nanostructures. Alvarez-Lorenzo et al.85 introduced a protocol towards supramolecular multiresponsive gel formation in a mixture of two copolymers each being sensitive to a different stimulus, by triggering one of the copolymers to induce the interaction between the two components and lead to gelation. The method was exploited using mixtures of the photoresponsive poly(N,N-dimethylacrylamide-co-methacryloyloxyazobenzene) (DMA–MOAB) random copolymer with a temperature responsive Pluronic F127 copolymer in water to obtain gels being responsive to both stimuli. Under dark conditions the azobenzene groups of the DMA–MOAB copolymer were in the trans conformation and self-associated to form micellar structures which exhibited minimal interactions with the Pluronic. However, upon irradiation with UV light the azobenzene groups of the DMA–MOAB copolymer underwent a trans to cisisomerization which modified both the air–water interfacial behavior and the self-associative properties of the copolymer, because the cis conformation of the azobenzene groups is relatively more hydrophilic compared to the trans photoisomer. The increase in the hydrophilicity of the photosensitive moieties, upon irradiation, caused the dissociation of the DMA–MOAB copolymer micelles and allowed the unimers to interact with the pluronic micelles leading to mixed micelle formation and a sol–gel transition. As a result of this, a low viscosity liquid solution of the photosensitive copolymer and the Pluronic in the dark could be rapidly transformed to a gel of high viscosity upon irradiation with UV light at body temperature. The light and temperature induced sol–gel transition was also shown to increase the hindrance to diffusion of dye molecules, such as methylene blue, which can be attractive for a wide range of practical purposes, including drug delivery. Competitive inclusion complexes of the azo groups allowed the control of the sol–gel transition of the system upon light irradiation. This was verified by the addition of β-cyclodextrin in the copolymer mixture which hosted the cis azobenzene in the sugar cavities and thereby inhibited their interactions with the pluronic micelles and the gel formation. In another example, Dong and coworkers introduced a novel biomimetic approach for the formation of pH and temperature responsive supramolecular micellar hydrogels.81 Their strategy employed poly(ethylene oxide)–poly(L-glutamic acid) (PEO–PLG) diblock or dendritic block copolymers. Hydrogel formation was induced via a cooperation mechanism of host–guest chemistry and supramolecular complexation between the PEO chains and α-cyclodextrin (α-CD), and hydrogen bonding interactions among the polypeptide blocks. The above hierarchical mechanism led in the first step to the formation of normal or reverse micelles with polypeptide or α-CD/PEO cores, followed by the self-assembly of the micelles to micellar gels upon addition of α-CD or a decrease of the solution pH with acid, respectively. The high mechanical strength and the slow release profiles of actives from the dendritic block copolymer based hydrogels render these materials advantageous for use as scaffolds in tissue engineering or platforms for controlled drug release. However, more complex polymer architectures can lead to rich phase diagrams and provide additional functionalities to the supramolecular gels. ABA and ABC triblock copolymers have been shown to form gels in response to two stimuli which trigger the solution behavior of either two or all three blocks. In a recent study, Zhao et al.87 exploited the use of an ABA type triblock copolymer with dual pH/T responsive properties in surpamolecular gel formation. The polymer was synthesised using a difunctional PEO macroinitiator from which copolymer chains of methoxydi(ethylene glycol) methacrylate and tert-butyl methacrylate were grown; the latter units were hydrolysed in a second step to afford ionizable methacrylic acid moieties. The resulting hydrophilic copolymer exhibited a critical gelatin concentration of ca. 4.6%, which is significantly lower than that reported for AB type diblock polymers. Moreover, the sol–gel transition temperature could be precisely controlled by varying pH and temperature synergistically and full sol–gel–sol cycles were performed in a fully reversible manner. More importantly, the polymer gel phase exhibited an intrinsic hierarchy, that is, the polymer chains associated into flower-type micelles with the PEO blocks forming bridges among the neighboring micelles to yield the three-dimensional network and contribute to the increase of the dynamic storage modulus. The use of triblock copolymer gelators was further extended to a novel ABC type triblock copolymer with exotic multiresponsive properties by Jérôme et al.88 The polymer consisted of a hydrophobic polystyrene (PS) block from which a second poly(vinyl pyridine) (PVP) block was grown. Subsequently, a third hydrophilic poly(ethylene oxide) (PEO) segment was introduced to afford the final PS–PVP–PEO triblock copolymer. The polymer exhibited unique properties, namely, remarkably low gelatin concentration (ca. 8%) as well as a dual responsive gelation behavior due to the exploitation of the ionizable PVP block and the temperature dependent solubility of the PEO block. An additional transition from hard to soft spheres was found upon decreasing the pH, which was attributed to the selective ionization of the PVP block.
Another beautiful example of photoresponsive hydrogels was recently reported by Yu et al.86 who prepared multiresponsive physical gels based on a carboxylic azobenzene polymer (Fig. 7). The material was shown to reversibly switch between the gel and liquid state in response to temperature, light and the solvent polarity. The responsive behavior was induced due to the reversible H-aggregation and hydrogen bond formation leading to packing of the azobenzene moieties at low temperatures and in high polarity solvents such as DMSO. However, the addition of a solvent of lower polarity dissociated the packing structure of the azobenzene groups and disrupted the polymer network. A similar gel–sol phase transition was induced upon UV irradiation which caused the “melting” of the gel within minutes due to the trans–cisisomerization of the azobenzene units. The gel structure was then recovered in the dark in a rather slow isomerization–aggregation process. Finally, the solution temperature had a strong influence on the aggregation state of the azobenzene groups leading to a reversible transition from the H-aggregates to the free state upon heating. The novelty of this work is related to the use of linear homopolymers in order to induce a sol–gel transition in response to a number of different stimuli. This could significantly simplify the concepts in the field of reversible physical gels, and hence, could serve as a new bottom-up approach for the design of gels on demand for numerous applications in intelligent sensors and actuators, injectable gels, supersensitive drug delivery platforms and others.
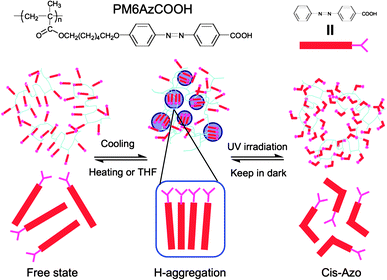 | ||
| Fig. 7 Multiresponsive gels based on a carboxylic azo polymer exhibiting controlled sol–gel transition in response to temperature, light and solvent polarity. Reproduced with permission from ref. 86. | ||
On the other hand, supramolecular gels of rather complex nanostructure are derived by the combination of inorganic components with organic counterparts capable of associative interactions leading to hybrid gel formation. Despite their structural complexity these gels are unique in a sense that their properties and functions are dominated by the synergistic effect of the two functionalities. An intriguing approach to the formation of hybrid supramolecular gels employs reversible metal–ligand interactions to build the self-assembled construction, which is afforded extra optical or magnetic properties by the metal complex. To this end, Beck and Rowan89 synthesised a novel class of hybrid supramolecular gels that exhibit intriguing responsive behavior to heat, metal binding, and light irradiation (Fig. 8). The polymers were prepared from a bifunctional oligoethylene glycol derivative end capped with a bis(2,6-bis(1′-methylbenzimidazolyl)-4-hydroxypyridine) tridentate ligand capable to bind transition metals. The organic ligand formed linear polymers with gel like texture in the presence of selective metal ions at an appropriate organic/inorganic content. The metal type (i.e.Zn, La, Co, Eu) affected the physicochemical properties of the gel resulting in temperature dependent sol–gel transition, thixotropic polymers with mechanoresponsive (shear-thinning) behavior and gels with unique photoresponsive properties. The versatility of this method allows for the development of polymers with tailor made properties by the rational choice of the building blocks and therefore provides access to an important range of multiresponsive materials.
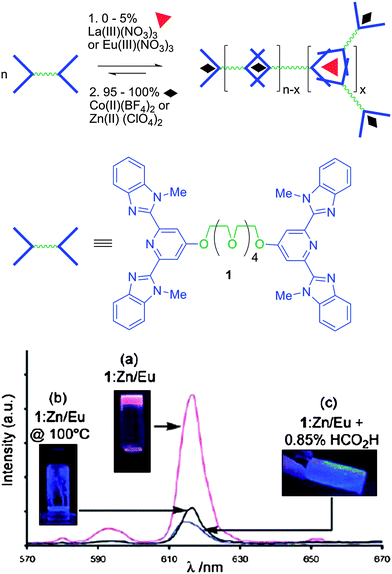 | ||
| Fig. 8 Supramolecular hydrogels responsive to temperature, metals and light formed by coordination of lanthanoid and transition metal ions by a bifunctional oligoethylene glycol tridentate ligand. Reproduced with permission from ref. 89. | ||
Hybrid inclusion complexes (HICs) have also emerged as a potential alternative route for the synthesis of responsive supramolecular hydrogels. This was demonstrated recently by connecting inorganic quantum dot nanoparticles with diblock copolymer chains bearing a temperature responsive block.90 The key point for the hydrogel formation was the combination of two independent cross-links. First an HIC solution was prepared by mixing an azobenzene end functionalized block copolymer, PDMAEMA-b-PNIPA, with β-cyclodextrin-modified CdS (β-CD@QD) quantum dots to give host–guest inclusion complexes of the azobenzene groups of the block copolymers in the CD cavities on β-CD@QD. The HIC comprised a QD core and a block copolymer in the shell with the PNIPA block in the outer layer. Next, the HICs were easily converted to the designed hybrid hydrogels upon heating above the LCST of PNIPA to induce polymer aggregation. The inclusion complex and the domains of the collapsed PNIPA chains served as the two distinct cross-links and rendered the hydrogels excellent dual sensitivity to competitive supramolecular hosts/guests substitutions and temperature variations. Furthermore, the effectiveness of the HIC structure decreased the critical hydrogel concentration to a rather low value compared to other supramolecular stimuli-responsive polymer hydrogels. It is noteworthy that the hydrogel maintained the innate fluorescence behavior of the quantum dots, which indicated their uniform distribution in the material, although the study lacks a detailed investigation of the intriguing optical properties of the gels. The above examples illustrate clearly that supramolecular hydrogels possess unique responsive properties (sol–gel transition, host–guest complexes, etc.) that could have great potential for a variety of applications.
An alternative strategy to the construction of mesostructured multiresponsive hydrogels has emerged, which is based on the polymerization/cross-linking of functional monomers in the interstitial space of colloidal crystal templates arising from the 3-D ordered structure of colloidal particles. Such periodically ordered interconnecting structures are known as inverse opal hydrogels (IOHs) and make use of the periodicity of the colloidal crystal which results in optical diffraction in the wavelength of visible light. The imparted changes in the periodic spacing with the size of the hydrogel, induced in response to the external stimuli, cause a shift in the diffraction wavelength rendering these materials very attractive for use as tunable optical sensors. Braun and coworkers91 were the first to report the synthesis of responsive IOH bearing boronic acid moieties and being sensitive to three different stimuli, glucose concentration, the solution pH and ionic strength. The IOH was created by the copolymerization of 3-acrylamidophenylboronic acid and HEMA in a dried polystyrene colloidal crystal template followed by etching and solvent exchange to obtain the reverse opal film. Diffraction spectra revealed a reversible glucose binding on the IOH which caused the swelling of the hydrogel in the substrate normal direction and was observed as a red shift of the diffracted light as the glucose concentration increased. The kinetics of glucose binding followed a t1/2 dependence, characteristic of a diffusion-limited kinetic response, which corroborate the increase in diffusivity of glucose as the hydrogel swells resulting in a decrease in equilibrium time. More interestingly, the IOH exhibited detectable glucose sensitivity even at high pH values and high solution ionic strength attributed to the dense mesostructure of the hydrogel with high concentration of boronic acid residues. The high sensitivity of IOHs, combined with their ability to incorporate a wide variety of different functional groups at high concentrations, constitutes significant advances in the development of materials for use in highly sensitive and optically active sensors. The construction of multiresponsive IOHs using a simple vertical deposition–evaporation method followed by a capillary-attraction-induced polymerization and the removal of the organic opal template was demonstrated by Gao et al.92Polystyrene microspheres were used as a template in order to embed a face-centered lattice within a polyacrylamide–polyacrylic acid polymer matrix. Hydrogels in the centimetre size range could be easily fabricated that showed remarkable optical sensitivity to multiple stimuli including pH, solvent type (i.e.ethanol, water and their mixtures), as well as small molecule additives (urea, oxalic acid and L-lysine). The reflection spectra were red-shifted upon immersion of the hydrogel in increasing water content ethanol/water mixtures, suggesting the swelling of the gel whereas a marked blue-shifting of the Bragg wavelength at higher ethanol contents was found due to the hydrogel shrinking. The responsive properties of the hydrogel were further exploited upon addition of oxalic acid or urea, while pH changes induced the ionization of the carboxylate segments and caused swelling and red-shifting of the diffraction wavelength. It is demonstrated in the above study that a hydrogel material with remarkable multiresponsive behavior can be relatively easily synthesised to cover large surface areas enabling the use of IOHs in multianalyte optical sensors and activators. Another advantage of these materials is the facile incorporation of a plethora of functionalities providing a broad spectrum of multiresponsive materials on demand for a variety of applications. The intrinsic optical properties of IOHs were extended further by Watanabe et al.93 who prepared a novel dual temperature and light responsive multicolor IOH based on an azobenzene chromophore. A porous PNIPA–poly(4-acryloylamino azobenzene) (PNIPA–PAAB) gel with periodically ordered interconnecting structure was prepared using as a template a colloidal crystal of closest-packed monodispersed SiO2 particles. The gel exhibited an abrupt isotropic volume change when the temperature was altered due to the LSCT behavior of PNIPA and a light-triggered rapid two-state switching between two arbitrary structural colors induced by the isomerization of the incorporated azobenzene units in a temperature controlled environment. When the temperature was changed, structural-color variation of the porous gel was observed, accompanied by a fast and isotropic increase of its volume. A multicolor photochromic behavior controlled by light was also observed in a temperature-controlled environment. The authors envisage the use of this material in writing and erasing colored patterns in a way that mimics natural iridophores of fishes and chameleons.
The examples above illustrate clearly that scientific research in the area of stimuli responsive gels has greatly advanced in terms of the design and synthesis of complex materials with multiresponsive properties. Future trends should focus on further elucidating the microscopic structure of such gels and mastering their cooperative function in order to exploit their exciting applications.
Polymer surfaces
The development of responsive surfaces which can controllably alter their properties on demand upon the application of an external stimulus has attracted the interest of the scientific community due to their wide variety of potential applications, including microfluidic and “lab-on-chip” devices, controlled drug delivery, enzyme immobilization, bioseparation, controlled cell adhesion, sensor development and self-cleaning surfaces. Multiresponsive surfaces are particularly attractive because they allow to mimic the cooperativity encountered in responsive systems found in nature as for example the delivery of a drug under specific conditions of temperature, pH and active concentration. The most extensively employed stimuli in the development of dual responsive surfaces include pH/T variations on grafted polymer chains from a solid substrate. In this context, there are a number of studies on PNIPA grafted chains which provide the temperature responsive component, whereas a polyacid, such as poly((meth)acrylic acid) or a polybase, i.e. poly(4-vinyl pyridine) serves as the grafted pH-responsive counterpart.94–97 Another parameter that strongly influences the surface properties and function is the micro- and nanostructure of the surface. It is generally accepted that microstructured rough surfaces enhance the effects of surface chemistry in terms of wetting characteristics and influence cell attachment and spreading behavior on the surface. Surfaces switching between superhydrophilicity and superhydrophobicity have been developed by grafting P(NIPA-co-AAc) films on a silicon substrate.95 The reversible pH/T dual-responsive behavior as a result of the combined influence of the chemical variation of the surface which alters the hydrogen bonds between the two components and water and of the surface roughness was demonstrated. The potential of these surfaces in a broad range of applications, including microfluidic switching, drug delivery and separation, is proposed. In a related work,98 the same group expanded the robustness of the polymer grafting method to develop a dual-functional nanofluidic device by immobilizing the P(NIPA-co-AAc) brushes onto the walls of a cone-shaped nanopore. The device integrated the ionic gate and the ionic rectifier properties due to the dual pH and temperature response of the grafted polymer. Hence, below the LCST of P(NIPA-co-AAc) the nanopore operated on a low ion conducting state, whereas upon increasing the temperature above the LCST of the copolymer the nanopore switched to a high ion conducting state. The closing/opening of the nanopore was derived from the temperature-triggered conformational transition of the grafted copolymer. At the same time, but independently, the conical nanopore rectified the ionic current in neutral and basic solution but not in acid media. This pH-responsive behavior is attributed to the ionization of the copolymer which, combined with the asymmetrical pore geometry, renders the nanopore a pH-tunable ionic rectifier with nanofluidic properties resembling the ion channels encountered in living systems.Another interesting copolymer architecture for immobilization on solid substrates are block copolymer brushes because they exhibit a rather complex responsive behavior different to that of their homopolymer and random copolymer counterparts.99 The responsiveness of block copolymer grafts is primarily determined by the phase segregation of the two blocks which is, in turn, influenced by the block sequence of the grafted chains and the solvent quality. In a poor solvent for the outer block micelles are formed on the top of the brush with the segregated outer block in the core and the surface grafted block forming the stabilizing corona. If however, the solvent is selective for the outer block, micelles segregated to the grafting surface are formed by the grafted block. To exploit this concept dual pH/T responsive PNIPA-b-polyacid block copolymer brushes were grown from a silicon substrate using a three step process.100 First, HEMA was polymerized from an initiator modified substrate followed by the polymerization of NIPA. The copolymer brushes were next modified to attain the carboxylic acid functionalities by the esterification of the hydroxy groups on the PHEMA block using an excess of succinic anhydride. The dual pH/T responsive properties of the brushes were investigated by height imaging and force-distance measurements using AFM. A switch of the top hydrated layer from the PNIPA block at high pH values and 25 °C, to the PSEMA block when the temperature increased to 50 °C and PNIPA became dehydrated, was reported. At low pH values, PNIPA was on the top layer both at the low and the high temperature range due to the collapsed structure of PSEMA. In a related study, Kilbey et al.97 used surface-initiated photoiniferter-mediated photopolymerization in the presence of tetraethylthiuram disulfide to directly synthesise surface-grafted PMAA-b-PNIPA layers. A lower control over the polymer structure and grafting density was exerted due to the adopted synthetic route however, the block copolymer brushes exhibited the expected response to changes in pH, temperature and ionic strength induced by the customary response characteristics of each component. The PMAA blocks became swollen as the pH was increased, with the maximum degree of swelling observed at pH 5, whereas the PNIPA blocks exhibited a LCST behavior, leading to a broad transition between the swollen and collapsed states. The response of the brushes to changes in salt concentration at constant pH was complex. The presence of ions decreased the LCST of PNIPA and extended it over a wider temperature range. Analogous PNIPA-b-polyamine dual-responsive layers prepared by grafting the polymer on cellulose surfaces exhibited a NIPA/4VP composition dependent pH and temperature response which allowed for further fine-tuning of the wettability of the “intelligent” surfaces.96
An intriguing approach to prepare dual responsive surfaces combined two coating methods, each immobilizing a polymer which exhibits a responsive behavior to a different stimulus.101 First the layer-by-layer (LbL) technique was employed for the facile formation of polyelectrolyte ion permeable layers via the immobilization of two opposite polyelectrolytes. The incorporation of ATRP initiating functionalities in this layer allowed to further grow temperature-responsive brushes on top of the membrane surface resulting in dual responsive coatings; the LbL layer was sensitive to pH and ionic strength variations, whereas the of PNIPA brushes created a thermally responsive top layer. The permeability was controlled through the pH-switchable LbL membrane by altering the net charge of the polyelectrolyte membrane and was combined with the thermally tunable permeability through the temperature-sensitive top layer. The versatility of the applied method can be further exploited to provide different levels of control or stimuli response based on the functionality, composition, and architecture of the bilayer films.
An alternative facile method which employed low cost polymers to prepared dual responsive surfaces switchable between superhydrophobicity and superhydrophilicity as a function of pH and the redox properties of the solution was introduced by Zhu et al.102 The switchable polymer comprised aniline (ANI) and acrylonitrile (AN) moieties and was deposited onto a flat surface in the form of coaxial nanofibers. Pentafluorooctanesulfonic acid (PFOS) was used as a dopant to increase the hydrophobicity of the fibers. The pH dependent wettability of the coaxial nanofibers was attributed to the pH sensitivity of PANI and the doping/dedoping of PFOS, whereas the redox response was due to the three different oxidation states of PANI which vary from 1 (reduced) to 0.5 and 0 (oxidized). However, although the pH induced variations in wettability switched between the superhydrophilic and superhydrophobic states, the contact angle changes upon application of the redox stimulus were limited to ∼60° tuning the surface from superhydrophobic to hydrophobic. This was attributed to the presence of the dopant which increased the hydrophobicity of the material and urges the need for alternative redox responsive materials with more abrupt changes in their physicochemical properties in response to a redox potential.
An elegant example in tuning the surface characteristics exploits the use of synthetic biomolecules in their unique ability to exhibit multiresponsive behavior from a simple molecular structure. This behavior is often dictated by their complex conformational changes leading to hierarchical structure formation. Polypeptides, for example, undergo conformational transitions due to the formation of intra- and/or inter-molecular hydrogen bonds which lead to unfolding/aggregation of the peptide chains. Jiang and coworkers grafted poly-L-lysine chains on a micro/nanostructured substrate which resulted in dual pH/T responsive surfaces.103 The external stimuli induced cooperative conformational changes of the polypeptide chains and switched the wettability of the surface between superhydrophilicity and superhydrophobicity in a reversible manner. The irreversible temperature-dependent β-sheet aggregation of the polypeptide at high pH values led to a hydrophobic state whereas the preservation of the random coil conformation at low pH induced the switch to hydrophilicity. This work presents an excellent example for the design of bio-inspired “smart” multiscale surfaces.
Control over the surface properties exerted by an external remote stimulus has become particularly attractive for the operation of microfluidic devices and the development of surfaces of controlled friction. A photoresponsive azobenzene derivative was copolymerized with N,N-dimethylacrylamide to afford dual temperature and light responsive surfaces.105 Wettability studies verified the increase in the contact angle when the temperature increased above the LCST of the copolymer. However, upon UV irradiation, the azobenzene groups in the polymer switched from the less polar trans form to the more polar cis configuration causing a simultaneous shift of the LCST to higher temperatures and an increase in the hydrophilicity of the surface. This temperature and light responsive wetting behavior is fully reversible upon appropriately tuning the external stimuli.
Multiresponsive surfaces that respond to more than two stimuli are exceptionally advantageous in that they provide extra tools in fine tuning their properties in a complex environment such as the biological systems albeit, more sophisticated and difficult to control in a cooperative manner. Jiang et al.104 extended the wettability variations of a surface in response to three different stimuli, pH, temperature and glucose concentration by grafting a PNIPA–poly(acrylamido phenylboronic acid) copolymer onto a rough silicon substrate (Fig. 9). The surface exhibited a complex cooperative wetting behavior in response to all three stimuli. A switch from superhydrophobicity to superhydrophilicity was induced by either cooling the surface at neutral pH or increasing the pH at low temperature in the presence of glucose or alternatively by exposing the surface to glucose at neutral pH and low temperature. An increase of the LCST of the copolymer with the glucose concentration at neutral and high pH was found which supported the observed changes in wettability. The response of this system to a combination of (bio)chemical stimuli in a synergistic manner renders it very promising for use in diagnostics, drug delivery or cell culture applications. An alternative approach for the development of multiresponsive surfaces that alter their wetting and structural characteristics in a complex manner in response to three different stimuli was proposed by Rodríguez-Hernández and coworkers. They employed homopolymer/block copolymer blends, deposited on a surface by spin coating, to obtain films with thicknesses above 100 nm and thus eliminate possible effects of the support on the surface characteristics.106Polystyrene (PS)/PS-b-PDMAEMA and PS/PS-b-PDEA blends were used and their surface segregation properties were exploited in tuning the surface properties. The polymeric surfaces responded to the exposure of air or water vapor at high temperatures due to compositional variations and the migration of either the hydrophobic or the hydrophilic block to the surface, respectively. Hence, annealing under water vapor enriched the surface on PDMAEMA or PDEAEMA and conferred the surface a pH and temperature responsive character of the amine-based polymer. On the other hand, the polarity of the solvent used for the spin coating process induced surface microphase separation of the block copolymers leading to structuration and the formation of micellar or “donut-like” morphologies on the surface. It is noted that this study presents a facile and cost-effective method for the construction of surfaces of tunable properties, however, its application is limited to flat surfaces and thus wettability changes only from the hydrophilic to hydrophobic range between the two polymer states are anticipated.
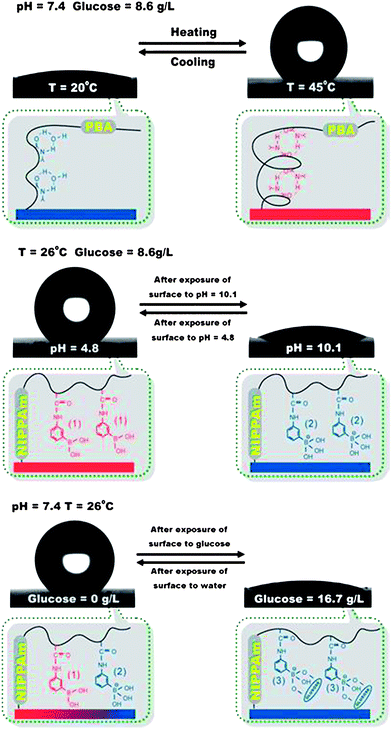 | ||
| Fig. 9 PNIPA-co-PBA surfaces change between superhydrophilicity and superhydrophobicity by pH, temperature and glucose concentration variations. Reproduced with permission from ref. 104. | ||
Another interesting feature of responsive polymer brushes is their ability to tune the nanolubrication between polymer modified surfaces. Interfacial nanotribology is quantitatively affected by conformational changes of the surface bound polymers induced by changes of the external stimuli. PDMAEMA brushes grafted on a gold substrate have been employed to quantitatively study the nanotribological response of chemically end-grafted polyions by a combination of a quartz crystal microbalance with dissipation and atomic force microscopy.107 A pH/T stimuli-induced lubrication behavior between the grafts was measured by force and friction measurements. Reversible transitions were monitored causing an increase of lubrication at low pH due to the repulsive forces between the highly charged, hydrated polymer layer, whereas the friction increased significantly at high pH when the polymer became neutral. Simultaneously, above the lower critical solution temperature the system became attractive and a small friction reduction was found attributed to the nanoscopic flattening of the polymer layer on the gold substrate. This detailed study serves as a potential model to understand interactions in biomacromolecular systems and elucidate lateral forces on stimuli-responsive polymer surfaces which play an important role in a number of applications including flow in microfluidic channels, cell–surface interactions, etc.
Controlling the transport properties through a membrane is a complicated problem that has gained much attention both in technological applications but also in our effort to understand transport phenomena through complex biological membranes. The use of responsive polymer brushes in molecular recognition gate membranes to maintain a specific substance concentration, which prevents toxic substance leakage or transfers chemical signals, has been reported by Yamaguchi et al. who immobilised a PNIPA-co-poly(benzo[18]crown-6-acrylamide) copolymer prepared by plasma graft polymerization on the pores of a polyethylene membrane (Fig. 10).108,109 The crown receptor trapped specific ions which resulted in an increase of the LCST of the copolymer due to the hydrophilic charged species. Hence, a solution free of ions exhibited a high flux through the membrane at temperatures above the LCST when the polymer was collapsed on the pore walls, whereas the flux of a solution containing specific ions (i.e.Ba2+) decreased by about 2 orders of magnitude due to the swelling of the copolymer which was now below the LCST. More importantly the pore size upon swelling and shrinking of the copolymer changed uniformly in all pores, and a clear cut-off value of the solute size was found leading to high performance membranes.
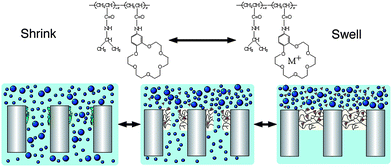 | ||
| Fig. 10 Molecular recognition ion gating membranes based on a grafted NIPA and crown ether copolymer controlling the permeation flux and the solute size in response to temperature and specific solvated ions. Reproduced with permission from ref. 108. | ||
pH-responsive membranes were prepared by Minko et al.110 following a simple route for the formation of a porous polysaccharide layer. An aqueous solution of sodium alginate blended with gelatin in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was spin cast on a silicon wafer and next treated with NaCl to allow for micron size pore formation due to the phase separation of gelatin and alginate. By altering the alginate:gelatin ratio surfaces with average pore size from 200 nm to 1 μm were accessible. The pores of the membrane opened and closed in a pH-dependent manner as evidenced111 by SPM topography imaging. Completely open pores at pH < 4 and completely closed pores at pH > 5 were attributed to the swelling/deswelling behavior of alginate. The versatility of the membrane preparation technique was exploited further by incorporating silver ions within the membrane matrix which provided additional antimicrobial properties. The researchers also immobilised glucose oxidase within the membrane matrix by standard EDC coupling and demonstrated that the enzyme retained its activity. The robustness of the method proposed in this study could constitute a standard platform for multifunctional membranes in a variety of applications such as “smart” coatings, antifouling materials and drug delivery.
:1 ratio was spin cast on a silicon wafer and next treated with NaCl to allow for micron size pore formation due to the phase separation of gelatin and alginate. By altering the alginate:gelatin ratio surfaces with average pore size from 200 nm to 1 μm were accessible. The pores of the membrane opened and closed in a pH-dependent manner as evidenced111 by SPM topography imaging. Completely open pores at pH < 4 and completely closed pores at pH > 5 were attributed to the swelling/deswelling behavior of alginate. The versatility of the membrane preparation technique was exploited further by incorporating silver ions within the membrane matrix which provided additional antimicrobial properties. The researchers also immobilised glucose oxidase within the membrane matrix by standard EDC coupling and demonstrated that the enzyme retained its activity. The robustness of the method proposed in this study could constitute a standard platform for multifunctional membranes in a variety of applications such as “smart” coatings, antifouling materials and drug delivery.
The potential of multiresponsive materials in surface science was manifested recently by Mecerreyes et al.112 who developed a simple material based on a conjugated polymer, poly(3,4-ethylenedioxythiophenes) (PEDOT), bearing imidazolium (Im) ionic liquid moieties and showed that this system was responsive to four different stimuli, namely, temperature, pH, oxidative doping, and the presence of anions. The pH and redox switching of the material including a change in color, oxidation state and wetting characteristics was attributed to the PEDOT component, whereas the temperature and anion responsive behavior was a unique characteristic conferred by the Im groups. The unusual thermochromism was derived from changes of the conjugation length of the PEDOT backbone induced by the steric interactions of the ionic liquid groups, whereas the molecular size of the counter-anions to the Im moieties led to variations in frequency and contact angles in response to the anions. The reversible changes both in solution and in thin films render the material an attractive candidate for potential future applications in optical sensors, cell culture and bioseparation.
Concluding remarks and future trends
Multiresponsive polymers capable of responding reversibly to several external stimuli have attracted particular attention over the last few years. The recent significant progress in the synthesis of well-defined macromolecular architectures and the polymerization of highly functional building blocks using precisely controlled polymerization methods has led to the construction of numerous examples of multiresponsive polymers. These polymeric materials are in the form of linear or graft copolymers, polymer or hybrid (organic–inorganic) particles, physical or chemical gels and polymer surfaces or interfaces. While most reports are related to systems that respond to the combined effect of temperature and pH, materials being sensitive to other physical or (bio-)chemical stimuli such as light irradiation, ions or carbohydrates have been also explored. However, despite the progress in designing and synthesizing such “smart” materials, this area still possesses tremendous challenges and is highly intriguing. Understanding the materials' physico-chemical properties and complex function is an ambiguous goal and a lot of work is required before their implementation in useful applications is possible. The main obstacle is the gap in our ability to control the response of such systems in a cooperative and interdependent manner and to couple the microscopic and macroscopic phenomena. To date, most studies have explored the independent response of the materials to two, three or four different stimuli but lack to investigate the synergistic multiresponsive properties which will reveal the programmable character and dynamic behavior of these complex materials. Another challenge will be the synthesis of new monomers with multi-responsive properties that will allow the straightforward preparation of complex systems in a simple one step process. A problem that should also be addressed is the control exerted over the kinetics of the stimuli-responsive processes which is critical for the use of these systems in certain applications, i.e. switches, artificial muscles and actuators. Synthetic multiresponsive materials are intrinsically static compared to the biological systems which can undergo dynamic changes in their properties and function over time. A possible way to overcome the above challenges would be to follow biomimetic strategies in the design, synthesis and engineering of multiresponsive polymers since nature is a rich source of inspiration for systems that respond in an intelligent manner.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- M. Yoshida and J. Lahann, ACS Nano, 2008, 2, 1101–1107 CrossRef CAS.
- S. Mann, Angew. Chem., Int. Ed., 2008, 47, 5306–5320 CrossRef CAS.
- B. Jeong and A. Gutowska, Trends Biotechnol., 2002, 20, 305–311 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- A. P. de Silva, S. Uchiyama, T. P. Vance and B. Wannalerse, Coord. Chem. Rev., 2007, 251, 1623–1632 CrossRef.
- C. de las Heras Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- J. R. Howse, P. Topham, C. J. Crook, A. J. Gleeson, W. Bras, R. A. L. Jones and A. J. Ryan, Nano Lett., 2005, 6, 73–77.
- R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480–4487 RSC.
- I. Tokarev and S. Minko, Adv. Mater., 2010, 22, 3446–3462 CrossRef CAS.
- B. D. Olsen and R. A. Segalman, Mater. Sci. Eng., R, 2008, 62, 37–66 CrossRef.
- J. A. Howarter and J. P. Youngblood, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS.
- E. Stratakis, A. Mateescu, M. Barberoglou, M. Vamvakaki, C. Fotakis and S. H. Anastasiadis, Chem. Commun., 2010, 46, 4136–4138 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- C. Hein, X.-M. Liu and D. Wang, Pharm. Res., 2008, 25, 2216–2230 CrossRef CAS.
- M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231–1232 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- G. Chen, L. Tao, G. Mantovani, V. Ladmiral, D. P. Burt, J. V. Macpherson and D. M. Haddleton, Soft Matter, 2007, 3, 732–739 RSC.
- O. Kanie, Y. Ito and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 12073–12074 CrossRef CAS.
- K. Ohmori, N. Ushimaru and K. Suzuki, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12002–12007 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- S. Binauld, D. Damiron, L. A. Connal, C. J. Hawker and E. Drockenmuller, Macromol. Rapid Commun., 2011, 32, 147–168 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- V. Sciannamea, R. Jerome and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS.
- Z. M. O. Rzaev, S. Dinçer and E. Pişkin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138–7147 CrossRef CAS.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- M. Vamvakaki, N. C. Billingham and S. P. Armes, Macromolecules, 1999, 32, 2088–2090 CrossRef CAS.
- M. Vamvakaki, G. F. Unali, V. Bütün, S. Boucher, K. L. Robinson, N. C. Billingham and S. P. Armes, Macromolecules, 2001, 34, 6839–6841 CrossRef CAS.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2005, 22, 989–997.
- S.-I. Yamamoto, J. Pietrasik and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 194–202 CrossRef CAS.
- C. Weber, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromolecules, 2009, 42, 2965–2971 CrossRef CAS.
- C. Weber, C. Remzi Becer, W. Guenther, R. Hoogenboom and U. S. Schubert, Macromolecules, 2010, 43, 160–167 CrossRef CAS.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132–5140 CrossRef CAS.
- D. Kuckling, H.-J. P. Adler, K.-F. Arndt, L. Ling and W. D. Habicher, Macromol. Chem. Phys., 2000, 201, 273–280 CrossRef CAS.
- L. Hu, L.-Y. Chu, M. Yang, H.-D. Wang and C. Hui Niu, J. Colloid Interface Sci., 2007, 311, 110–117 CrossRef CAS.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- C. Li, Z. Ge, J. Fang and S. Liu, Macromolecules, 2009, 42, 2916–2924 CrossRef CAS.
- Y. Ren, X. Jiang, G. Yin and J. Yin, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 327–335 CrossRef CAS.
- Y. Ren, X. Jiang and J. Yin, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1292–1297 CrossRef CAS.
- R. Wang, X. Jiang, C. Di and J. Yin, Macromolecules, 2010, 43, 10628–10635 CrossRef CAS.
- L. Ma, R. Liu, J. Tan, D. Wang, X. Jin, H. Kang, M. Wu and Y. Huang, Langmuir, 2010, 26, 8697–8703 CrossRef CAS.
- F. J. Xu, Y. Zhu, F. S. Liu, J. Nie, J. Ma and W. T. Yang, Bioconjugate Chem., 2010, 21, 456–464 CrossRef CAS.
- X. Jiang, Y. Zheng, H. H. Chen, K. W. Leong, T.-H. Wang and H.-Q. Mao, Adv. Mater., 2010, 22, 2556–2560 CrossRef CAS.
- N. Ma, Y. Li, H. Xu, Z. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442–443 CrossRef CAS.
- J. Bigot, B. Charleux, G. Cooke, F. Delattre, D. Fournier, J. Lyskawa, L. Sambe, F. Stoffelbach and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106–2108 RSC.
- G. Pasparakis, A. Cockayne and C. Alexander, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS.
- G. Pasparakis, M. Vamvakaki, N. Krasnogor and C. Alexander, Soft Matter, 2009, 5, 3839–3841 RSC.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- P.-F. Caponi, X.-P. Qiu, F. Vilela, F. M. Winnik and R. V. Ulijn, Polym. Chem., 2011, 2, 306–308 RSC.
- G. Pasparakis, N. Krasnogor, L. Cronin, B. G. Davis and C. Alexander, Chem. Soc. Rev., 2010, 39, 286–300 RSC.
- D. S. Achilleos and M. Vamvakaki, Macromolecules, 2010, 43, 7073–7081 CrossRef CAS.
- Y. Zhao, L. Tremblay and Y. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4055–4066 CrossRef CAS.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900–4921 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- J. Xie, S. Lee and X. Chen, Adv. Drug Delivery Rev., 2010, 62, 1064–1079 CrossRef CAS.
- Q. Yuan, O. J. Cayre, S. Fujii, S. P. Armes, R. A. Williams and S. Biggs, Langmuir, 2010, 26, 18408–18414 CrossRef CAS.
- B. M. Reis, S. P. Armes, S. Fujii and S. Biggs, Colloids Surf., A, 2010, 353, 210–215 CrossRef CAS.
- C. Gota, K. Okabe, T. Funatsu, Y. Harada and S. Uchiyama, J. Am. Chem. Soc., 2009, 131, 2766–2767 CrossRef CAS.
- J. Yin, H. Hu, Y. Wu and S. Liu, Polym. Chem., 2011, 2, 363–371 RSC.
- J. Yin, C. Li, D. Wang and S. Liu, J. Phys. Chem. B, 2010, 114, 12213–12220 CrossRef CAS.
- J. Tan, H. Kang, R. Liu, D. Wang, X. Jin, Q. Li and Y. Huang, Polym. Chem., 2011, 2, 672–678 RSC.
- J. Cai, J. Guo, M. Ji, W. Yang, C. Wang and S. Fu, Colloid Polym. Sci., 2007, 285, 1607–1615 CrossRef CAS.
- A. Sánchez-Iglesias, M. Grzelczak, B. Rodríguez-González, P. Guardia-Girós, I. Pastoriza-Santos, J. Pérez-Juste, M. Prato and L. M. Liz-Marzán, ACS Nano, 2009, 3, 3184–3190 CrossRef CAS.
- W. Wu, T. Zhou, A. Berliner, P. Banerjee and S. Zhou, Chem. Mater., 2010, 22, 1966–1976 CrossRef CAS.
- W. Wu, N. Mitra, E. C. Y. Yan and S. Zhou, ACS Nano, 2010, 4, 4831–4839 CrossRef CAS.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- M. Guenther, D. Kuckling, C. Corten, G. Gerlach, J. Sorber, G. Suchaneck and K. F. Arndt, Sens. Actuators, B, 2007, 126, 97–106 CrossRef.
- D. Matsukuma, K. Yamamoto and T. Aoyagi, Langmuir, 2006, 22, 5911–5915 CrossRef CAS.
- Y. Chen, X.-H. Pang and C.-M. Dong, Adv. Funct. Mater., 2010, 20, 579–586 CrossRef CAS.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS.
- F. Ercole, T. P. Davis and R. A. Evans, Polym. Chem., 2010, 1, 37–54 RSC.
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353–4356 CrossRef CAS.
- C. Alvarez-Lorenzo, S. Deshmukh, L. Bromberg, T. A. Hatton, I. Sández-Macho and A. Concheiro, Langmuir, 2007, 23, 11475–11481 CrossRef.
- D. Chen, H. Liu, T. Kobayashi and H. Yu, J. Mater. Chem., 2010, 20, 3610–3614 RSC.
- T. G. O'Lenick, X. Jiang and B. Zhao, Langmuir, 2010, 26, 8787–8796 CrossRef CAS.
- N. Willet, J.-F. Gohy, L. Lei, M. Heinrich, L. Auvray, S. Varshney, R. Jérôme and B. Leyh, Angew. Chem., 2007, 119, 8134–8138 CrossRef.
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922–13923 CrossRef CAS.
- J. Liu, G. Chen, M. Guo and M. Jiang, Macromolecules, 2010, 43, 8086–8093 CrossRef CAS.
- Y.-J. Lee, S. A. Pruzinsky and P. V. Braun, Langmuir, 2004, 20, 3096–3106 CrossRef CAS.
- J. Y. Wang, Y. Cao, Y. Feng, F. Yin and J. P. Gao, Adv. Mater., 2007, 19, 3865–3871 CrossRef CAS.
- K. Matsubara, M. Watanabe and Y. Takeoka, Angew. Chem., Int. Ed., 2007, 46, 1688–1692 CrossRef CAS.
- T. Peng and Y. L. Cheng, Polymer, 2001, 42, 2091–2100 CrossRef CAS.
- F. Xia, L. Feng, S. Wang, T. Sun, W. Song, W. Jiang and L. Jiang, Adv. Mater., 2006, 18, 432–436 CrossRef CAS.
- J. Lindqvist, D. Nyström, E. Östmark, P. Antoni, A. Carlmark, M. Johansson, A. Hult and E. Malmström, Biomacromolecules, 2008, 9, 2139–2145 CrossRef CAS.
- S. B. Rahane, J. A. Floyd, A. T. Metters and S. M. Kilbey, Adv. Funct. Mater., 2008, 18, 1232–1240 CrossRef CAS.
- W. Guo, H. Xia, L. Cao, F. Xia, S. Wang, G. Zhang, Y. Song, Y. Wang, L. Jiang and D. Zhu, Adv. Funct. Mater., 2010, 20, 3561–3567 CrossRef CAS.
- S. Minko, Polym. Rev., 2006, 46, 397–420 Search PubMed.
- X. Wang, X. Xiao, X. Wang, J. Zhou, L. Li, J. Xu and B. Guo, Macromol. Rapid Commun., 2007, 28, 828–833 CrossRef CAS.
- T. M. Fulghum, N. C. Estillore, C.-D. Vo, S. P. Armes and R. C. Advincula, Macromolecules, 2008, 41, 429–435 CrossRef CAS.
- Y. Zhu, L. Feng, F. Xia, J. Zhai, M. Wan and L. Jiang, Macromol. Rapid Commun., 2007, 28, 1135–1141 CrossRef CAS.
- Y. Guo, F. Xia, L. Xu, J. Li, W. Yang and L. Jiang, Langmuir, 2009, 26, 1024–1028.
- F. Xia, H. Ge, Y. Hou, T. Sun, L. Chen, G. Zhang and L. Jiang, Adv. Mater., 2007, 19, 2520–2524 CrossRef CAS.
- W. Yuan, G. Jiang, J. Wang, G. Wang, Y. Song and L. Jiang, Macromolecules, 2006, 39, 1300–1303 CrossRef CAS.
- A. Bousquet, E. Ibarboure, E. Papon, C. Labrugère and J. Rodríguez-Hernández, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1952–1961 CrossRef CAS.
- N. Nordgren and M. W. Rutland, Nano Lett., 2009, 9, 2984–2990 CrossRef CAS.
- T. Ito, T. Hioki, T. Yamaguchi, T. Shinbo, S.-i. Nakao and S. Kimura, J. Am. Chem. Soc., 2002, 124, 7840–7846 CrossRef CAS.
- T. Yamaguchi, T. Ito, T. Sato, T. Shinbo and S. Nakao, J. Am. Chem. Soc., 1999, 121, 4078–4079 CrossRef CAS.
- V. Gopishetty, Y. Roiter, I. Tokarev and S. Minko, Adv. Mater., 2008, 20, 4588–4593 CrossRef CAS.
- I. Tokarev and S. Minko, Adv. Mater., 2009, 21, 241–247 CrossRef CAS.
- M. Döbbelin, R. Tena-Zaera, R. Marcilla, J. Iturri, S. Moya, J. A. Pomposo and D. Mecerreyes, Adv. Funct. Mater., 2009, 19, 3326–3333 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |